

Abstract
Aim:
To performe a time-dependent topographical delineation of protein-drug interactions to gain molecular insight into the supremacy of Ortho-7 over HI-6 in reactivating tabun-conjugated mouse acetylcholinesterase (mAChE).
Methods:
We conducted all-atom steered molecular dynamics simulations of the two protein-drug complexes. Through a host of protein-drug interaction parameters (rupture force profiles, hydrogen bonds, water bridges, hydrophobic interactions), geometrical, and orientation ordering of the drugs, we monitored the enzyme's response during the release of the drugs from its active-site.
Results:
The results show the preferential binding of the drugs with the enzyme. The pyridinium ring of HI-6 shows excellent complementary binding with the peripheral anionic site, whereas one of two identical pyridinium rings of Ortho-7 has excellent binding compatibility in the enzyme active-site where it can orchestrate the reactivation process. We found that the active pyridinium ring of HI-6 undergoes a complete turn along the active site axis, directed away from the active-site region during the course of the simulation.
Conclusion:
Due to excellent cooperative binding of Ortho-7, as rendered by several cation-π interactions with the active-site gorge of the enzyme, Ortho-7 may be a more efficient reactivator than HI-6. Our work supports the growing body of evidence that the efficacy of the drugs is due to the differential bindings of the oximes with AChE and can aid to the rational design of oxime drugs.
Keywords: acetylcholinesterase structure, nerve gas, oxime drugs, protein-drug complexes, steered molecular dynamics simulation
Introduction
Serine hydrolase, acetylcholinesterase (AChE, acetylcholine acetylhydrolase, E C 3.1.1.7) is a pivotal enzyme that terminates nerve impulses at cholinergic synapses found at neuromuscular junctions. The AChE enzyme, anchored to the surface of the postsynaptic muscle membrane inside the synaptic space, operates close to the diffusion controlled limit and rapidly hydrolyses the cationic neurotransmitter acetylcholine into acetate and choline ions1, 2, 3. The high second-order rate constant [>108 (mol/L)−1·s−1]1, 4 of the AChE catalytic hydrolysis activity is a hallmark of evolutionary enzyme perfection. The serine residue of AChE is activated by the adjacent histidine and glutamic acid residues, which together form the enzyme's catalytic triad5. The AChE monomer has an ellipsoidal shape with dimensions of ∼45 Å×60 Å×60 Å. The most striking feature of the AChE structure is a deep and narrow gorge, ∼20 Å long, which perforates almost half-way into the enzyme1. The base of the gorge consists of two subsites: the esteratic subsite comprising the active catalytic triad and an anionic subsite responsible for binding the quaternary trimethylammonium tail group of acetylcholine (choline-binding pocket)5. In the case of Mus musculus AChE (mAChE), the catalytic triad consists of Ser 203, Glu 334, and His 447 and the anionic subsite is made up of Trp 86, Glu 202, and Tyr 337. In the third domain, a hydrophobic region is contiguous with or near the esteratic and anionic loci and is responsible for binding aryl substrates and active site ligands. Moreover, a fourth domain in the enzyme binds cationic ligands. This fourth domain is over 20 Å away from the active site, located at the rim of the active gorge and has been called the peripheral anionic site (PAS)6, 7, 8 and comprises aromatic residues Tyr 72, Tyr 124, Trp 286, and Tyr 341 and an acidic residue Asp 74. The four domains of the enzyme act together to produce the complex reaction dynamics9, 10, 11, 12, 13, 14, 15, 16, 17 responsible for effective binding with ligands and eventual access to the catalytic triad domain.
For the ligand to enter the active site gorge, the structural fluctuations in the enzyme play an important role in opening the gorge every few picoseconds, leading to fractal gating motion18 that allows the passage of the substrate. Upon successful entry into the gorge, the substrate must move through the morphologically inhomogeneous pore surface to reach the active triad. The diffusion through such an inhomogeneous pore is reminiscent of subdiffusive fractal diffusion19, 20, 21 and is the rate-determining step for the catalytic activity of the enzyme. In addition, water molecules in the gorge must give way to the ligand as it moves through this tortuous path22. The arrangement of the active triad (serine, glutamic acid, and histidine) is crucial for its enzymatic activity. Mutations in any of these residues to, for example, alanine leads to a dramatic decrease in the catalytic activity by at least 3300-fold9.
In fact, the active triad is the primary target for controlled inhibition of AChE in acetylcholine deficient patients, such as in Alzheimer's disease or in myasthenia gravis, which is characterized by fluctuating muscle weakness and fatigue due to a loss of receptors. In such patients, treatment with cholinesterase inhibitors or immunosuppressants has been proposed23, 24. The active triad of the AChE enzyme, on the other hand, is also the primary target of lethal organophosphorous (OP) compounds such as tabun, sarin, soman, and VX, which are known as nerve agents. Destruction of OP-related nerve agents by several nucleophiles is a continuing area of research25, 26. OPs are also used for pest control27. Intentional (in a suicidal attempt28) or unintentional (through trace dietary contaminants29 or through chemical warfare30) OP intoxication is a major threat to human kind. While intoxication by OPs accounts for up to 80% of pesticide-related hospitalization31, the possibilities of terrorist attacks (like the Iraqi weaponized sarin attack against the Kurdish32 or the Tokyo subway sarin attack33) with nerve agents looms large, and thus the discovery of effective OP antidotes is necessary and urgent34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46. Thus, fundamental studies16, 17 elucidating the passage of molecules through the AChE gorge into the active triad of the enzyme is of paramount importance for the discovery of new drugs.
The toxic effect of OPs on AChE occurs through a SN2-type nucleophilic attack on OPs by the active site serine, which creates a covalent acyl-enzyme intermediate (Scheme 1)47. The inhibited enzyme subsequently fails to hydrolyze the neurotransmitter acetylcholine resulting in over-stimulation of cholinergic receptors48. The inhibited enzyme further undergoes an aging reaction that leads to a permanent alkyl-phosphate adduct bound to the active serine. Aged enzymes cannot be liberated by any known chemical means. However, prior to aging, the inhibited enzyme can be reactivated by strong nucleophiles, such as oximes. The medical management of an OP victim includes drugs such as atropine (a muscarinic receptor antagonist that artificially maintains the respiration capacity), diazepam (or similar anticonvulsant drug, commonly administered to treat seizure) and oxime reactivators34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46 (which liberate the inhibited enzyme). Since aging of an inhibited enzyme occurs within minutes to hours and results in the permanent disability of the enzymatic activity of AChE, the victim should be administered these drugs as soon as possible49.
Scheme 1.

AChE inhibition by OP compound, tabun. Aging of the tabun conjugated enzyme and its oxime-drug based reactivation.
However, not all oximes are equally potent drugs that can liberate inhibited enzymes. For example, the widely used compounds bispyridinium oxime, obidoxime, monopyridinium oxime and pralidoxime (2-PAM) are considered to be ineffective for certain nerve agents and numerous new oximes have been synthesized in past decade50. It is now accepted that while various bispyridinium oximes are effective reactivators at high concentrations, no compounds are effective against all tested nerve agents. For example, HI-6 is a potent reactivator of AChE inhibited by nerve agents GD and GF37, but it is impotent against tabun conjugation36. Similarly, oximes like HLö-7 are more efficient reactivators of sarin and VX-inhibited AChE. The potency of HI-6 to reactivate tabun-inhibited AChE is very low and does not reach the reactivating efficacy of obidoxime or Ortho-736, 43. Currently, strategies for the development of effective oximes can be broadly classified into two categories: the kinetic approach37, 39, 41, 42, 43 and a structural approach35, 36, 38, 40. Kinetics studies on oxime function have revealed that reactivation proceeds via a two-step reaction that consists of the formation of a fully reversible Michaelis-type enzyme-inhibitor-oxime complex followed by the splitting of the complex into a phosphorylated oxime and reactivated enzyme. The efficacy of the reactivator is attributed to the nucleophilicity of the oximes and the decay rate of the intermediate complex, and is dependent on the structure of the oxime and of the OP moiety, as well as the architecture of the enzyme. The availability of a high-resolution three-dimensional structure of AChE from various species has allowed detailed structural studies determining the mechanisms of reactivation of the enzyme. Crystallographic experiments with mAChE using inhibitors and different oximes have provided insight into the structural aspects involved in the interactions between the active center of the gorge, the inhibitors and the oximes35, 36. Such studies provide important data on the structural requirements of effective reactivators and thus complement kinetic experiments.
Apart from the kinetic and structural approaches, one can think of a third avenue to explore the dynamical structure-activity relationship of oxime-AchE complexes-potent drug discovery. What interests us most is the recent end-point reactivation experiments36 where it has been shown that given their similar skeletal structure (Figure 1), Ortho-7 is 45 times more efficient than HI-6 in reactivating the tabun-conjugated AChE enzyme. Recent X-ray crystal structures of bispyridinium oximes HI-6 and Ortho-7 in complex with mAChE36 have revealed that one of two pyridinium rings binds reversibly to the AChE molecule at its peripheral site (PAS pyridinium ring) and the other is directed towards the active catalytic site (active pyridinium ring). The 4-carboxylamide-pyridinium ring (the PAS pyridinium ring) of HI-6 has been found to be sandwiched by the indole ring of Trp 286 and the phenol ring of Tyr 124 driven by strong cation-π interaction between the PAS pyridinium ring and the side chains of Trp 286 and Tyr 124. While the PAS of AChE holds this pyridinium ring tightly, the central linker of HI-6 is directed into the active-site gorge, and the 2-hydroxy-iminomethylpyridinium ring (active pyridinium ring) is directed towards the active catalytic site36. Similar to HI-6, the PAS pyridinium ring of Ortho-7 also forms a cation-π interaction with the side chains of Tyr 72 and Trp 286. The heptyl linker of the two pyridinium rings of Ortho-7 lies along the active gorge, but the second pyridinium ring (active pyridinium ring), unlike that of the HI-6, forms a near parallel cation-π with the phenyl ring of Tyr 337 and a T-shaped cation-π interaction with the indole ring of Trp 8636. The differential binding of the two drugs in PAS and in the active-site gorge of AChE has been attributed to the difference in the efficacy of two drugs in reactivating the tabun conjugated enzyme.
Figure 1.

Chemical structure of the two oxime drugs. (A) HI-6, [1-(2-hydroxy-iminomethylpyridinium)-1-(4-carboxyamino)-pyridinium dimethylether]. and (B) Ortho-7, [1,7-heptylene-bis-N,N'-2-pyridiniumaldoxime]. The carbon, nitrogen, and oxygen atoms are numbered for referral purpose. The two pyridinium rings, one directed towards the active triad region and one directed towards the PAS region of the AChE enzyme, are indicated.
In this work, we pose several questions. How long (duration) does the differential binding of the two drugs in the PAS and in the active-site gorge of AChE last during unbinding from the active-site gorge? What are the roles of water molecules, buried inside the gorge22 in protein-drug binding? What roles do PAS residues play in the drug interaction8? What role does the structural heterogeneity of the active-site gorge space, such as its tortuosity, or chemical heterogeneity, such as the distribution of aromatic and polar residues lining the gorge's inner surface play during binding/unbinding of the drug? The answers to these questions may substantiate the experimental findings of the comparative efficacy of the two drugs. With this in mind, we studied the unbinding of the two protein-drug complexes through steered molecular dynamics simulations (SMD)51, 52, which mimics the experimental atomic force microscopy (AFM) measurements53, 54, 55.
Materials and methods
Conventional molecular dynamics simulation
All simulations, CMD as well as SMD, reported here were performed by the fully parallel version of the software, GROMACS 3.3.256 using GROMOS 96 force field57. Before we performed the simulations, the structures of the two protein-drug complexes (PDB ID 2GYV and 2GYU)36 were repaired, and the simulating systems were prepared (see Supporting Information). The isothermal isobaric simulation protocol comprises three major steps: energy minimization, position restrained run and finally the production run. Energy Minimization: To start, the protein-drug complexes were put in a cuboid box with dimensions that kept the protein outer surface at least 10 Å away from the box wall. This was to ensure that no wall effect appeared in the simulated results. The system was then solvated with simple point charge (SPC) water molecules and five electropositive sodium ions were added to each of the simulating systems to attain electroneutrality. The system was then found to be composed of the following: mAChE.HI-6 complex dissolved in 33 179 water molecules in a system volume 1080 nm3 with a density of 1011.87 g/L and a total of 104 957 atoms; mAChE.Ortho-7 complex dissolved in 31 718 water molecules in a system volume 1032.08 nm3 with a density of 1016.57 (g/L) and a total of 100 576 atoms.
In the energy minimization step, a flexible SPC water model was used instead of a rigid model; this flexible model allowed the steepest descent minimization technique to be followed, and energy minimization was thought to converge when the maximum force in the system was smaller than 100 kJ·mol−1·nm−1. Position Restrained Run: The purpose of the position restrained run was to “soak” the water and the drug into the protein-drug complex. A linear constraint solver algorithm58 was used to restrain the atom positions, and a simulation time step 2 fs was used to integrate the equation of motion for all atoms. The solvent and solute were separately coupled to temperature reservoirs of 300 K using the Berendsen temperature coupling method with coupling times of 0.1 ps. First, the water molecules were heated to this temperature for a 300 ps simulation run, while the protein-drug complex remained fixed. Then, the simulation was restarted for heating the protein molecule to 300 K for 300 ps with the already equilibrated water molecules. Finally, the drug molecule was heated in a similar fashion for 100 ps. Pressure was restrained to 1 atm using the Berendsen method with a coupling time of 0.5 ps. Long-range electrostatic interactions were handled by Particle-Mesh Ewald electrostatics59 with the real-space cut-off fixed at 12 Å and the highest magnitude of wave vectors used in reciprocal space controlled by Fourier spacing with a parameter held at 1.2 Å. Grid dimensions were controlled with this Fourier spacing and with interpolation order 4. Production Run: Finally, for both the complexes, 5 ns CMD runs were performed for the corresponding systems comprising the drug, protein, water and ions with the same set of simulating conditions as in position restrained runs.
Time-dependent root mean square deviation, a statistical measure of the magnitude of deviation of protein structure in solution from its crystal structure, determined from the CMD data, showed that for Cα atoms and all the atoms of the protein, the deviation was within 3 Å, and the structure appeared to be stabilized within 2 ns. We performed SMD simulation with these stabilized structures obtained after 2 ns of CMD run as the starting point. SMD results were found to be insensitive with respect to the starting structure of simulation.
Steered molecular dynamics simulation
By definition, SMD is an acceleration protocol, implemented in conventional molecular dynamics (CMD) to expedite conformational changes through the application of external force in a macromolecular system, which is otherwise prohibitively slow for monitoring, even by modern computers. Using SMD, one can overcome slow natural time scales, explore putative conformational pathways and explain single-molecule experiments60. As adopted in our context, the unbinding/binding events of the drug molecules from the tortuous landscape of the active-site gorge of the enzyme are ultraslow in nature20, 21. Application of a time-dependent external force on the drug, as depicted in Figure 2, facilitates the unbinding process. The SMD investigations proceed through three stages. First, we must generate a hypothesis for the conformational transition (reaction coordinate). In our case, the hypothesis was the 20 Å long active-site gorge of the protein, through which the drug molecules bind/unbind into the active catalytic site of the protein. Second, we must apply forces that induce the hypothetical process during reasonable wall clock time. As shown in Figure 2, a pull force was applied on the center of mass of the drug molecule. Finally, we must analyze results through the concepts of protein-drug rupture force profile, hydrogen bonding and hydrophobic interactions between them, water-bridges connecting the protein and the drug etc such that the energetic barriers and specific unbinding events unfold to evaluate the hypothesis and determine the reasons behind the relative efficacy of the two drugs investigated.
Figure 2.

Unbinding of a drug molecule from the active-site gorge of mAChE (the active serine of which is shown in blue colored space filling representation) through the application of an external time-dependent harmonic force in the Z-direction as depicted by a spring. The artificial spring, characterized by a force constant, is attached to the center of mass of the drug molecule (shown by space filling representation) and is pulled with a constant velocity until it completely dislocates from the gorge.
SMD simulation51, 52 is accomplished by restraining the drug with a harmonic pulling potential,
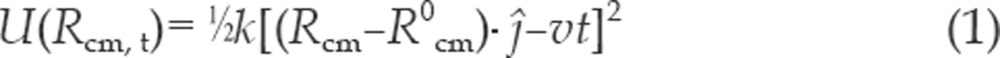 |
in addition to the normal molecular force field operating on the system. In the present study, we took the pulling potential operative on the center of mass of the drug molecule whose sojourn is identified as R0cm at the start of the simulation and Rcm at time t. In Figure 2, this pulling potential is represented by a spring whose stiffness is k. The minimum of the pulling potential is moved with a constant velocity v away from the active-site gorge in a given direction shown by the normalized vector ĵ in Equation 1. The SMD method requires the predetermination of the unbinding pathway, which is not always possible a priori, and one normally selects a straight path51, 61, chosen by guesswork based on available structural information of the receptor. The accuracy of the guesswork can then be judged by the efficiency of the pulling, such that the drug molecule moves along a favorable pathway with minimal collisions with the surrounding residues in the pulling direction. Based on the available structural information of the AChE gorge, we chose a straight line path for pulling (arrow in Figure 2) that leads from the bottom of the gorge through the gorge opening (PAS residues), which we called the positive Z-axis. To verify that the ligand would exit the binding site along our supposed exit route within the 5 ns simulation time, we simulated the unbinding of both of the drugs with a force directed in the negative Z direction, and to the left and right with respect to Z-axis. The result showed that drug molecules did not exit the gorge through any of these paths within the stipulated 5 ns time. We must note that one idea would be to design a simulation method so that the drug molecule finds its own route of exit and is independent of any guesswork. Such methods have only recently started to emerge62.
While the drug molecule is pulled away from the protein gorge, the center of mass of the protein is held in space to prevent it from drifting. This fixing ensures that the protein molecule is free to undergo internal motion and its constituent amino acid residues can respond to changes in the environment as the drug molecule progressively departs, but the protein molecule cannot undergo translational drift. SMD results that mimic AFM experiments53, 54, 55 normally apply a pulling force and a pulling velocity much larger than an AFM experimental setup. In other words, the order of magnitude of timescales in which these studies are performed varies, namely, nanosecond (SMD) versus milliseconds (AFM). As a result, SMD results cannot be readily extrapolated to produce the AFM observations63, 64. However, the aim of the present study was to obtain the mechanical pattern65 during protein-drug interactions, which are indicative of the main residues responsible for binding/unbinding. SMD results can also provide qualitative information on the rotational flexibility of the oxime drug inside the gorge and its proximity to the active catalytic triad, both of which are essential for the reactivation (see Scheme 1) of the nerve agent-inhibited enzyme by the oxime anion that is bound to the pyridinium ring. We will consider only a qualitative comparison between two sets of SMD data, one for mAChE.HI-6 and the other for mAChE.Ortho-7 complexes under identical simulating conditions when k=280 pN/Å and v=0.005 Å/ps. In a recent study16, this value of spring stiffness and pulling velocity was determined for conditions that produce the lowest protein-drug complex rupture force requiring the lowest amount of mechanical work, a necessary condition that foretells that the nonequilibrium effects51, 62 are minimized. This pulling velocity also ensures that within 5 ns simulation time, the drug molecule completely moves away from the 20 Å long active-site gorge of the protein. In order to verify the dependence of the SMD results on the spring stiffness and the pulling velocity, we conducted a much longer simulation (∼45 ns) of the 2GYU complex in which the spring stiffness and pulling velocity were each lowered by a factor of 10. Although the results provided information on water bridges, hydrophobic interactions etc, the qualitative feature of the unbinding transition remained unaltered. The stress that the enzyme experiences during the egress of the drug (see Figures 3 and 4) is measured through,
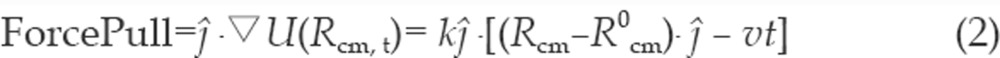 |
While many results presented below are based on the analysis tools of GROMACS56 and LIGPLOT66 programs, some tools were developed in-house.
Figure 3.

Pulling force and center of mass distance (A), water bridge (B), and hydrophobic atom pair numbers (C) between the drug and the enzyme shown as a function of time during the exit of the drug, Ortho-7 from mAChE gorge. In (B), the grey-colored bars indicate events in which a given amino acid residue of the enzyme that forms a water bridge with Ortho-7 also forms direct hydrogen bonds between themselves. The plots in parts (A) and (C) are prepared by 10 point adjacent averaging of the SMD results. The double-sided arrows in (A) represent the fall in the force profile from an intermittent peak and subsequent rise in center of mass distance between the enzyme and the drug.
Figure 4.

Same as Figure 3 but for the drug, HI-6.
Results
One can naively forecast that the binding of a drug to the protein is mediated by ion-ion interactions, hydrogen bonding, a variety of protein-drug non-bonded interactions, hydrophobicity, and shape and orientation complementation. But given the similar skeletal structure of the two drugs, it is hard to understand their relative binding/unbinding characteristics. This is because at the outset both the bispyridinium drugs appear to score equally in these grounds except for some subtle differences revealed in their crystal structures36. The protein-drug complexes under conditions that are relevant to biological situations, that is, the system dissolved in water, are not rigid. They are dynamic entities that can unveil new structural determinants that contribute to drug interactions. Therefore, we attempted a detailed time-dependent topographical delineation of protein-drug interactions, which can dissect the relative contributions of these interactions in the two protein-drug complexes.
Force profiles, water bridges, hydrodgen bonds and hydrophobic interactions during pulling SMD
SMD pulling simulations provide computed force profiles and atomistic details of the particular stress responses as the drug molecule leaves the enzyme gorge. These data are of enormous importance, as the force profiles exhibit a complex pattern of the barriers that can be related to a variety of interatomic interactions, such as breaking of direct hydrogen bonds (DHB), water bridges (WB) or hydrophobic interactions (HI) between the drug and the enzyme. Figures 3 and 4 present the results for the two complexes, mAChE.Ortho-7 and mAChE.HI-6, respectively. Note that it is called WB when the protein and the drug molecules are connected with each other through shared hydrogen bonds, mediated via one intervening water molecule. In Figures 3(A) and 4(A), along with the force profiles, we also present the progressive separation of the drug from the drift-fixed center mass of the enzyme. The fluctuating force profiles in these figures are a hallmark of protein-drug interactions. As the drug molecule progressively egresses from the protein gorge, old DHB and WB break, while newer ones form, both of which differ in their strength. Furthermore, the old hydrophobic alignment vanishes, and the drug molecule searches for new complements between its hydrophobic surface and that of the gorge residues. The peaks and valleys of force profiles result from the breaking and making of interactions.
The fluctuating force profiles in Figures 3(A) and 4(A) result from the multidimensional energy landscape offered by the protein gorge-drug interactions. This tortuous landscape is composed of a multitude of barriers and isolated traps caused by molecular interactions or by steric compulsion. Once trapped in such a barrier, the force required to pull the drug molecule out increases. Once out of the barrier, the center of mass separation between the drug and enzyme suddenly increases. Such transitions are marked in these figures by double-sided blue arrows. The maximum of the force profile, called the rupture force, occurs at ∼2345 ps for mAChE.Ortho-7 complex (Figure 3) and was measured to be 615 pN. The same data for the mAChE.HI-6 complex can be seen in Figure 4 and are given as ∼3000 ps and ∼740 pN. Therefore, the pulling of the drugs out of the 20 Å gorge with a constant force of 615 pN, as in the case of Ortho-7, would cost 177 kcal/mol of energy; for HI-6 with a constant force of 740 pN, this requires 213 kcal/mol. Thus, the binding free energy of HI-6 to the enzyme gorge is larger than Ortho-7. Based on this criterion, one can argue that HI-6 would continually be held by the gorge more effectively, causing more effective reactivation of the oxime conjugated enzyme. This idea is counterintuitive to the experimental observation that Ortho-7 is a superior drug to HI-6 in enzyme reactivation36. However, as we shall present below, this energy criterion alone is not sufficient to support the experimental observation.
Water plays a major role in protein structural relaxation as well as in drug molecule entry and binding22. One can easily imagine that a dehydrated protein undergoes strong electrostatic interactions between its polar residues resulting in a rigid protein structure, which may not allow the drug molecule to enter or leave its active site. On the other hand, water breaks these intra-protein specific interactions through the formation of protein-water hydrogen bonds. Thermal mobility of water, in turn, facilitates protein conformation fluctuations, resulting in the access of the drug molecule to its active site. The drug molecule may even bind to a protein residue through common hydrogen bonds with the oxygen atom of an intervening water molecule. In part B of Figures 3 and 4, we presented the temporal occurrences of such WB. In these panels, we also showed those protein-drug DHB (grey bars) between a given amino acid residue and a drug atom, which are also found to form protein-water-drug WB. This results in a triangular arrangement of the hydrogen bond network. For example, at 942 ps Gly 122 donates a hydrogen atom (attached to the amino group) to a water molecule, the water molecule in turn donates a hydrogen atom to the N27 atom of Ortho-7 molecule forming the WB. At the same time, the N27 atom of the drug molecule donates the attached hydrogen atom to the nitrogen atom of Gly 122, making the DHB and completing the triangular hydrogen network. See supporting information, S-1 and S-2 for a view of some typical snapshots of the SMD unbinding transitions, where these hydrogen bond networks formed by DHB and WB are presented for the two complexes. A list of residues that form these isolated hydrogen bond networks along with protein-drug DHB and their temporal occurrences are depicted in Figures 5 and 6 for the two drug complexes.
Figure 5.

Number of direct hydrogen bonds as a function of time between amino acid residues of mAChE and Ortho-7 during the process of leaving the active-site gorge. The amino acid residues and atom numbers of the drug molecule with which it forms hydrogen bonds are mentioned. The grey-colored bars are indicative of the simultaneous formation of water bridges between the protein and the drug with an intervening molecule of water in addition to a direct hydrogen bond, which is otherwise plotted here. Note that Ser 203 is found to form only water bridges and no direct hydrogen bond.
Figure 6.

Same as Figure 5 but for the drug molecule, HI-6.
The residues mentioned in Figures 5 and 6 and many other residues are also found to form WB, although they do not form a triangular-type hydrogen bond network. A detailed analysis of such WB (not all of them are shown in Figures 5 and 6) between the two protein-drug complexes clearly reveals that both drugs form WB with the help of oxygen and nitrogen atoms attached to their two pyridinium rings. Interestingly, the PAS residues in the mAChE.Ortho-7 complex make and break WB in a systematic fashion. For example, the O10 and N9 atoms attached to the peripheral pyridinium ring initially form WB with PAS residues. Subsequently as time progresses, the progressive egress of the drug molecule from the active-site gorge makes the O28 and N27 atoms attach to the active pyridinium ring of the drug molecule exposed to PAS residues causing them to form WB at the outer rim of the gorge. By contrast, the PAS residues in the mAChE.HI-6 complex are water bridged with the O3 and N4 atoms attached to the peripheral pyridinium ring for about 3500 ps. This long duration of intermittent interaction between the PAS residues and the carboxylamide group of HI-6 is reminiscent of excellent complementation between the peripheral pyridinium ring of the HI-6 drug molecule and PAS of the protein.
Apart from the PAS residues, other residues, such as Ser 203, or the residues of the anionic subsite located at the base of the gorge, Glu 202 and Tyr 337 in the mAChE.Ortho-7 complex form WB with the O28 and N27 atoms of Ortho-7. However, these residues are never found to participate in WB formation in the mAChE.HI-6 complex. Similarly, residues like Phe 295 and Gln 291, which form WB with O3 and N4 atoms of HI-6, are never found to form WB in the case of the Ortho-7 complex. WBs between Ser 203, Glu 202, Tyr 337 of mAChE and O28 atom of Ortho-7 are quite extensive: they range in a time scale of 1 to 2000 ps with intermittent making and breaking. This is also true in the case of Phe 299 and Gln 291 in mAChE.HI-6 complex. It should be noted that the same water molecule is found to participate in WB formation between any of the residue-drug partners mentioned above. This can be explained by the fact that the water molecules in the gorge lose their mobility22 and are thus “trapped” between the protein and drug. Importantly, the probability of formation of WB between the O28 atom of the active pyridinium ring of Ortho-7 and the catalytically active Ser 203 or, for that matter, the Glu 202 and Tyr 337 for such a long duration, signifies complementation between the active pyridinium ring of Ortho-7 and the active-site locality of the gorge.
In panel C of Figures 3 and 4, the temporal profiles of the hydrophobic interactions during the egress of the drug from the active-site gorge of the protein are presented. While in Figures 5 and 6 some of the residues involved in DHB are mentioned, in Table 1, the complete lists of residues responsible for DHB and HI along their numbers during the 5 ns simulation run are presented for the two complexes. As the drug molecule egresses from the gorge, the hydrophobic contact between them is diminished. On average, the HI experienced by HI-6 is larger than for the Ortho-7 molecule (compare panel D of Figures 3 and 4). It is well known that removal of the hydrophobic surface area of the drug molecules from water and its subsequent attachment to a hydrophobic region of the protein molecule enhances protein-drug binding. As seen in Table 1, barring Tyr 341, all other PAS residues have a significantly larger number of HI with the HI-6 molecule than with Ortho-7. This is clearly indicative of a situation in which the complementarity between the drug's hydrophobic surface and the PAS of the enzyme is particularly high and leads to higher protein-drug binding energy in the case of the mAChE.HI-6 complex (which is also supportive of the comparative rupture force profiles of the two complexes discussed earlier). Protein-drug DHB plays a crucial role in binding and especially because of their directional property often decides the relative geometry or orientation of bound drugs. The N4 and O3 atoms attached to the peripheral pyridinium ring of HI-6 form DHB with all PAS residues and also extensively with the Glu 285 and Ser 298 during the course of the simulation, occasionally with total DHB numbers as high as six. In comparison, DHB in mAChE.Ortho-7 are not that extensive, and the maximum number of DHB is rarely found to be as high as three. The majority of DHB in this complex are formed between the O28 atom, attached to the active pyridinium ring of Ortho-7 with non-PAS residues such as Glu 292, Gly 121, Gly 342, Ser 293, and Gly 122. Interestingly, during the 5 ns simulation, the oxygen atom of the ether bridge of HI-6 did not form either WB or DHB.
Table 1. Residues of mAChE forming hydrophobic interactions (HI), hydrogen bonds (HB) with Orhto-7 and HI-6 and their total numbers during SMD simulation.
| Residues | Number of HIs (2GYV) | Number of HIs (2GYU) | Number of HBs (2GYV) | Number of HBs (2GYU) |
|---|---|---|---|---|
| TYR341 | 22332 | 4839 | 49 | 1 |
| TRP86 | 11141 | 443 | ||
| ASP74 | 8691 | 11135 | 6 | 50 |
| GLU292 | 7768 | 9072 | 61 | 6 |
| TRP286 | 7575 | 34705 | 1 | 231 |
| TYR72 | 4496 | 11092 | 37 | 42 |
| TYR124 | 3634 | 12721 | 78 | 67 |
| GLY121 | 3236 | 76 | 57 | – |
| TYR337 | 2684 | 104 | 35 | – |
| GLY342 | 1111 | 342 | 240 | 22 |
| GLU202 | 961 | – | 14 | – |
| SER125 | 934 | – | – | – |
| LEU 76 | 870 | 1440 | 2 | – |
| PHE297 | 656 | 2049 | – | |
| ILE294 | 455 | 1 | – | – |
| SER293 | 372 | 2 | 126 | 323 |
| THR 75 | 211 | 546 | 25 | 22 |
| PHE338 | 195 | 166 | 8 | – |
| ILE365 | 157 | 719 | – | – |
| GLY120 | 128 | – | – | – |
| GLY126 | 123 | – | – | – |
| GLU285 | 102 | 998 | – | 1183 |
| VAL 73 | 52 | – | – | – |
| HIS447 | 49 | – | – | – |
| ASN 87 | 28 | 19 | – | – |
| SER298 | 16 | 40 | – | 1791 |
| GLY448 | 12 | – | – | – |
| PRO344 | 12 | 34 | – | – |
| VAL343 | 11 | – | 4 | – |
| HIS 287 | 8 | – | – | – |
| GLY122 | 7 | 23 | 98 | – |
| LEU130 | 7 | – | – | – |
| GLN291 | 6 | 63 | – | 373 |
| ARG296 | 1 | 330 | – | 371 |
| PRO 78 | 1 | – | – | – |
| PHE295 | - | 499 | 3 | 37 |
| LEU289 | - | 157 | – | – |
| PRO 88 | - | 42 | – | – |
In Figure 7, we plotted the free energy of DHB formation between the protein residues and the drug molecules. The free energies were evaluated following the theoretical recipes provided by van der Spoel et al67. As can be seen, whenever a common protein residue of the two complexes forms a DHB with the two drug molecules, the free energy of their formation is higher for the HI-6 molecule. Moreover, in almost all the cases, HI-6 participates in both forward (hydrogen donor) and backward (hydrogen acceptor) hydrogen bonding with the protein molecule. This is not the case with Ortho-7, further justifying its measured lower binding energy to the protein gorge in comparison with HI-6 (177 vs 213 kcal/mol).
Figure 7.

Free energy of direct protein-drug hydrogen bonding of the two complexes (circles: mAChE.Ortho-7, squares: mAChE.HI-6). Black- and grey-colored symbols represent forward hydrogen bonding (where the drug molecule is the hydrogen donor) and backward hydrogen bonding (where the drug molecule is the hydrogen acceptor), respectively.
From an analysis of WB, hydrogen bond networks, DHB and HI we showed that while the peripheral pyridinium ring of HI-6 is strongly held by the PAS of the enzyme due to excellent complementarity, the Ortho-7 molecule scores poorly in this regard. As generally visualized, for the oxime drug to be effective, the active pyridinium ring of the drug needs to be in close proximity and be properly oriented to exert a nucleophilic attack on the phosphorus atom of the enzyme-inhibitor complex (Scheme 1), or in other words, to the active serine Ser 203 moiety of the unreacted enzyme. In this regard, the active pyridinium ring of Ortho-7 due to its enhanced interactions in the active-site region of the enzyme scores well above the HI-6 molecule. In particular, as presented below, this instantaneous structure-activity relationship of the active pyridinium ring of Ortho-7 leads to a relative geometry or orientation of the bound drug that might be considered conducive for nucleophilic attack.
Temporal spatial distances and orientations of the drug molecules during their egress from the active-site gorge of mAChE
As mentioned in the Introduction, the X-ray crystal structure of mAChE·Ortho-7 complex36 reveals that upon entry to the active-site gorge of mAChE, the Ortho-7 molecule induces a structural change in enzyme side chains such that its peripheral pyridinium ring forms cation-π interactions with the Tyr72 and Trp286 PAS residues. Similarly, the active pyridinium ring of Ortho-7 forms a cation-π interaction with the phenyl ring of Tyr 337 and a T-shaped cation-π interaction with the indole ring of Trp 86. Accordingly, we monitored the relevant minimal distance in the SMD simulation and the results are presented in panels A and B of Figure 8, respectively. It can be clearly seen that the aforementioned cation-π interaction involving the peripheral pyridinium ring stands out for ∼1 ns, after which the corresponding minimal distance increases substantially leading to a weaker interaction. On the other hand, for the active pyridinium ring (Figure 8B), the minimal distance shows a plateau up to ∼2.5 ns. This signifies that the cation-π interaction prevailing between the active pyridinium ring and residues at the active-site region is strong such that the oximate oxygen O28 of Ortho-7 is tightly sandwiched by Tyr 337 and Phe 338, which can temporarily overcome the stress exerted by the SMD spring.
Figure 8.

Time-dependent minimal distance between the (A) peripheral pyridinium ring of Ortho-7 and the PAS residues, Tyr 72 (grey line) and Trp 286 (black line), (B) active-site pyridinium ring of Ortho-7 and the phenyl ring of Tyr 337 (grey line) and the indole ring of Trp 86 (black line), (C) peripheral pyridinium ring (carboxylamide-pyridinium) of HI-6 and the PAS residues, Tyr 124 (grey line) and Trp 286 (black line), and (D) active-site pyridinium ring of HI-6 and Tyr 337 (grey line) and Phe 338 (black line) during the process of the drugs leaving the active-site gorge of mAChE.
Similar to this, the main features of the X-ray crystal structure36 of the mAChE.HI-6 complex are as follows: a strong cation-π interaction between the peripheral pyridinium ring of HI-6 and the Tyr 124 and Trp 286 PAS residues, non-bonded contacts between the active pyridinium ring of HI-6 with the Tyr 337 and Phe 338 residues. The minimal distances between these pyridinium rings and protein residues are shown in panels (C) and (D) of Figure 8, respectively. As can be seen in these figures, the PAS interactions can withstand SMD pulling for about 4 ns, such that the corresponding minimal distance is observed as a plateau (Figure 8C). By contrast, interactions involving the active pyridinium ring cannot survive the mechanical stress, and the corresponding minimal distance increases from the very beginning of the SMD simulation (Figure 8D). It is interesting to note that Trp 286 forms cation-π interactions with the peripheral pyridinium ring for each of the two drugs. In addition, in Table 1 it can be seen that Trp 286 forms a larger number of HI and DHB in the case of the HI-6 complex than in the Ortho-7 complex. These three interactions together bind the HI-6 drug in the PAS region more efficiently than in the case of Ortho-7. Similar to this, Tyr 337 participates in the binding of the drugs in the active-site region of the enzyme. From Table 1, one can see that Tyr 337 forms a larger number of HI and DHB in the case of the Ortho-7 complex. Once again, these interactions are collectively responsible for binding the Ortho-7 molecule more strongly in the protein's active-site region as compared to the HI-6 complex. Therefore, the results presented in Figure 8 convincingly show that the interaction between the peripheral pyridinium ring of HI-6 and the PAS residues are in excellent complement with each other; in the case of the mAChE.Ortho-7 complex, this complementarity holds true in the active-site region of the enzyme with the active pyridinium ring of Ortho-7.
In order to delineate the time dependent topography of protein-drug interactions, we monitored the center of mass (COM) distances, presented in Figures 9 and 10. In Figure 9, we plotted the COM distance between the peripheral pyridinium ring of the two drugs and the hydrophobic aromatic residues of the PAS. Along with the data in Table 1, a clear cooperative trend emerges between the degree of HI and the COM distances. For example, the PAS residues, Tyr 72, Tyr 124 and Trp 286 form a larger number of HI with HI-6 than with Ortho-7, whereas Tyr 341 forms a larger number of HI with Ortho-7 than with HI-6. The COM distances between the peripheral pyridinium ring of HI-6 and Tyr 72, Tyr 124, Trp 286, in turn, exhibit a long plateau at about 0.5 nm (with intermittent fluctuations in and around this value) from 0 to ∼3500 ps. Throughout this time scale, however, the COM distances between these three PAS residues and the peripheral pyridinium ring of Ortho-7 show a trend soon after the pulling stress is present. For peripheral pyridinium ring of Ortho-7, this plateau of COM distance with respect to Tyr 341, occurs at about 0.75 nm and continues to hold until about 2200 ps. We must note that Tyr 341, one of the key residues that determine the shape of the gorge entry shows significant mobility of its side chains8. In particular, the concerted motion of the two loops, Cys 69-Cys 96 and Val 340-Gly 342 that bears Tyr 341, controls the open and close transitions of the gorge. This larger mobility of the side chains of Tyr 341 is responsible for larger COM distances than other PAS residues.
Figure 9.

Center of mass distance between the peripheral pyridinium moiety of the drugs during their exits from the active-site gorge, and the hydrophobic aromatic residues of the PAS of mAChE, as indicated in the plots. Grey lines are the results for the SMD simulation of the mAChE.Ortho-7, and the black lines are for mAChE.HI-6 complexes.
Figure 10.

Center of mass distances between the APR of the drugs and the hydrophobic aromatic residues (other than those at the PAS) of mAChE as indicated in the plots. In all the plots, the SMD pulling results shown by grey lines are for the mAChE.Ortho-7, and the black lines are for the mAChE.HI-6 complexes.
In Figure 10, we present the COM distances between the non-PAS hydrophobic aromatic residues lining the gorge wall and the active pyridinium rings of the two drugs. These aromatic residues offer fewer HI to both of the bispyridinium drugs as compared to the aromatic PAS residues (see Table 1). It can be inferred from the figure that, on a relative basis, out of the two active pyridinium rings of Ortho-7 and HI-6, the former is preferentially more directed towards the inner part of the gorge. This observation corroborates our minimal van der Wall distance calculation (Figure 8): namely the minimal distance between the active pyridinium ring of Ortho-7 and the phenyl ring of Tyr 337, between the active pyridinium ring of Ortho-7 and the indole ring of Trp 86, between the active pyridinium ring of HI-6 and Tyr 337, between the active pyridinium ring of HI-6 and Phe 338. In short, the COM distance calculations reinforced our previous observation that while the hydrophobic surface compatibility between the PAS residues and the peripheral pyridinium ring of HI-6 is superior to Ortho-7, the active pyridinium ring of the latter compound finds excellent binding complementarity in the active-site region of the enzyme compared with the former drug compound.
Discussion
The preferential binding of the two drug molecules, one (HI-6) at the PAS region and the other (Ortho-7) at the active-site region of the protein, might have major consequences on the protein-drug structure-activity relationship. One such consequence is the proximity of the drug's reactive group to the active-site region of the protein molecule, where it is supposed to orchestrate a nucleophilic attack in order to liberate the OP conjugated protein. Figure 11 shows a plot of the COM distance between the active pyridinium rings of the two drugs and the active serine moiety (Ser 203) during the exit of the drug from the gorge. It can be seen from the figure that throughout the exit time, the active pyridinium ring of Ortho-7 is closer to COM of Ser 203 than for the HI-6 compound. Interestingly, during the first 900 ps of unbinding, the pyridinium ring of Ortho-7 moves closer to Ser 203. This proximity may be attributed to the cooperative mechanical response of the enzyme towards the egress of the drug molecule. COM separation then suddenly jumps to ∼1.25 nm in the next 200 ps, due to the rupture of several DHB, HI, WB. In the next 2500 ps, the plot for Ortho-7 flattens out, and the average COM separation becomes close to 1.1 nm. The plot for HI-6 instead shows a monotonous increase in COM separation with intermittent barriers. The mechanical response of the enzyme in its active-site region and the presence of a large and effective barrier (until ∼2500 ps) towards the exit of the active pyridinium ring of the Ortho-7 drug molecule may underlie its efficacy in reactivating the OP inhibited protein.
Figure 11.

Center of mass distances between Ser 203 and the active pyridinium moiety of the drugs, as marked in the plot, during their exit from the active-site gorge of mAChE.
To probe further into the relative efficacy of the two drugs on a geometric perspective, in Figures 12 (mAChE.Ortho-7) and 13 (mAChE.HI-6) the temporal spatial separation between the nitrogen atoms of the two pyridinium rings and the active Ser 203 are plotted. Similarly, the relative orientations and separations of the entire drug molecule are presented at regular intervals to delineate the proximity and orientations of oximate oxygen atoms of the two complexes with respect to Ser 203. The efficacy of oxime reactivators depends on their affinity and reactivity to the OP-inhibited enzyme. Both the bispyridinium oxime drugs studied here have strong positive charges on each of their pyridinium rings and, therefore, the electrostatic interactions of these two drugs with the negatively charged anionic site at the active center and the PAS of AChE can be regarded as spontaneous. Their reactivity, on the other hand, is derived from the nucleophilicity of the oxime anion that is bound to the active pyridinium rings of the two drugs. However, both their affinity and reactivity depend on a host of other physical features, such as, steric compatibility, protein-drug binding energy, electrostatic effects, HI, DHB, WB with the water molecules that lubricate the gorge, the shape and the size of the whole molecule, as well as the orientation of the functional groups towards the active center of the enzyme. Based on a host of such physical interactions above, we have shown that the active pyridinium ring of Ortho-7 (and not HI-6) preferentially binds to the active site region. However, the active pyridinium ring of both drugs and the functional group that contains the oximate oxygen are identical. Yet, as can be seen in Figures 12 and 13, the relative geometry and orientation of the bound drug molecules are dramatically different. While the distances of the two nitrogen atoms of the two pyridinium rings of Ortho-7 from Ser 203 COM increase systemically, HI-6 showed a cross-over midway during the egress. As a result, while oximate oxygen O28 of Ortho-7 is always oriented towards the Ser 203 moiety, the active pyridinium ring of HI-6 turns and the oximate oxygen O1 of HI-6 is oriented away from the active serine during their egress. Therefore, for both proximity and orientation, which are the two primary requirements for an efficient nucleophilic attack on OP-conjugated enzyme, HI-6 is expected to unbind.
Figure 12.

Temporal spatial distance during SMD pulling between the center of mass of active-site Ser 203 and the N20 atom (grey line) and the N2 atom (black line) of the two pyridinium rings of Orhto-7 that was initially close to active triad and the peripheral site, respectively. Relative orientations and separations of the drug molecule with respect to Ser 203 (shown by space filling representation) are also shown at regular intervals. The grey circle encircling O28 atom of the drug is the oximate oxygen. See Supplementary information S-1 for the snapshots of protein-drug hydrogen bonds and water bridges at the time steps indicated in the figure by arrows.
Figure 13.

Temporal spatial distance during SMD pulling between the center of mass of active-site Ser 203 and the N2 atom (grey line) and the N3 atom (black line) of the two pyridinium rings of HI-6 that was initially close to active triad and the peripheral site, respectively. Relative orientations and separations of the drug molecule with respect to Ser 203 (shown by space filling representation) are also shown at regular intervals. The grey circle encircling O1 atom of the drug is the oximate oxygen. See Supplementary information S-2 for the snapshots of protein-drug hydrogen bonds and water bridges at the time steps indicated in the figure by arrows.
To reinforce the structure-activity issue concerning the relative geometry and orientation of the enzyme-bound drugs we performed an all-atom superposition of the two protein-drug complexes at several time steps during the simulation. The superposition of the two structures (excluding the water molecules) is performed in two steps. First, a translation of the HI-6 complex at the center of the reference (Ortho-7 complex) is performed. Next the structure is rotated to get the eigenvalue of rotation to derive the minimal root mean square distance. In Figure 14, we present some of these superposed structures. Clearly, it can be seen from the figure that at ∼1500 ps (until about 3500 ps) the oximate oxygen of HI-6 in association with its active pyridinium ring, and the central chain linker takes a complete turn so that it is directed away from the active serine. In comparison, the oximate oxygen of Ortho-7 is directed towards the same serine moiety. At 4500 ps the active pyridinium ring of HI-6 once again takes a complete turn. Although the oximate oxygen still faces the active serine, the distance is too far (Figures 5A and 14) to be effective for a nucleophilic attack at the OP inhibited serine.
Figure 14.

Results of all-atom superimposed structures of the two complexes, mAChE.HI6 and mAChE.Ortho-7. Shown at different times are the active site Ser 203 and the two drugs. The Ser 203 residue of the two complexes is shown in space-filling representation, green for mAChE.Ortho-7 and gold for mAChE.HI6 complex. Ortho-7 drug is displayed in licorice representation and HI-6 is displayed in ball-and-stick representation. The oximate oxygen atoms of the two drugs (Ortho-7: O28, HI-6: O1) are also shown.
The excellent complementarity between the PAS residues and the peripheral pyridinium ring of HI-6 is due to the carbamide group, instead of the oxime group (as in Ortho-7), which effectively clamps the drug molecule to the rim of the gorge. This, in addition to a shorter central chain linker (compare with Ortho-7) between the two pyridinium rings of HI-6, results into a highly flexible pyridinium ring of the drug molecule directed towards the active-site region of the enzyme. The preferential binding of HI-6 at the PAS of the enzyme could also be another source of major concern. It is well known that the affinity of reactivators for AChE is the most important step for their reactivating efficacy. Since each of the two pyridinium rings of HI-6 contains a net positive charge, their affinity towards the PAS region can be considered to be the same. One can now easily visualize a random event when the peripheral pyridinium ring is first bound to the PAS, while the active pyridinium ring of HI-6 with the attached oxime group, instead of being inside the gorge, is left outside the gorge. Because of excellent PAS complementarity, this situation can have serious implications. The drug molecule would then, instead of reactivating the OP-conjugated enzyme, clog the gorge entrance, inhibiting both entry and exit to the active-site, like other PAS inhibitors8, 15. On the other hand, the Ortho-7 molecule is centrosymmetric and because of its relatively poor binding compatibility at the PAS is free from such a situation.
We investigated the dynamics of the enzyme's mechanical response towards unbinding of the two drugs from its active-site gorge at an all-atom level. Through this approach, we unveiled what could be the reasons behind the relative efficacy of the two drugs given the subtlety in their seemingly similar chemical structures. The present study was not intended to obtain an exact force profile pertaining to the drug unbinding process. Rather, the purpose had been the exploration of structure-activity relations between the two oxime complexes on a comparative basis in a common simulation platform. Scrutiny of various dynamical interaction parameters clearly shows that the two drug molecules bind differently with the same enzyme. Namely, the peripheral pyridinium ring of HI-6 binds to the PAS region at the gorge entrance with excellent complementarity, while the active pyridinium ring of Ortho-7 have been found to be suitably fitted in the active-site region. Tyr 124 and Trp 286 residues of PAS of the enzyme have been found to clamp the peripheral pyridinium ring of HI-6 for a sufficiently long time (about 3500 ps) through a strong cation-π interaction and a host of other aforementioned protein-drug interaction parameters. The PAS preferential bindings of HI-6 have been found to have major consequences. In particular, HI-6 binding energy with the enzyme is found to be larger than that in the case of Ortho-7. Moreover, during the egress while the peripheral pyridinium ring is held tightly by the PAS residues, the active pyridinium ring of HI-6 is found to undergo a complete turn along the gorge axis, directed away from the active triad of the enzyme. Also, as has been argued, the PAS binding compatibility of HI-6 may even render it to act like other PAS inhibitors instead of a drug-reactivating agent. The active pyridinium ring of Ortho-7, on the other hand, forms a parallel cation-π interaction with the phenyl ring of Tyr 337 and a T-shaped cation-π interaction with the indole ring of Trp 86. This, along with the other interactions (as mentioned above) monitored in this work, makes the oximate oxygen of Ortho-7 always directed towards the active triad region. One of the principal motivations against oxime drug design is the cooperative binding of the bispyridinium compounds in the active-site gorge such that the proximity and orientations of the drug molecule, necessary for an effective nucleophilic attack can be attained. In a dynamical situation, as has been dealt with here, on both these grounds Ortho-7 is found to be a more suitable choice, which is in support of the experimental findings and could function as a more effective antidote against tabun intoxication.
Conclusions
The recent end-point reactivation experiments have shown that given their similar skeletal structure, Ortho-7 is far more efficient than HI-6 in reactivating the tabun-conjugated AChE enzyme. Based on X-ray crystallographic measurements, the experimental results are attributed to the differential binding of oxime drugs with the enzyme36. However, since the protein-drug complexes are dynamical entities, our effort in this work was focused on the time-dependent topographical delineation of their differential bindings. In particular, our quest was the delineation of dynamics of enzyme's mechanical response towards unbinding of the two drugs from its active-site gorge at an all-atom level through force-field based classical steered molecular dynamics simulations. Through this approach, we tried to unveil the moot reasons behind the relative efficacy of the two drugs given the subtlety in their seemingly similar chemical structures, and we have answered several questions related to protein-drug interactions, which we raised in the introduction. The 5 ns force probe simulation began with the recently reported X-ray crystal structures of the two complexes. We chose the pulling spring constant and the pulling velocity of the drugs, in accordance with the recent state of the art force probe simulation protocol. The present study was not intended to obtain the exact force profile pertaining to the drug unbinding process. Rather, the purpose was the exploration of structure-activity relations between the two oxime complexes on a comparative basis in a common simulation platform.
The peaks and valleys of the force profiles signify the enzyme's response during the drug's egress and can be attributed to the breaking and making of direct hydrogen bonds, water bridges, hydrogen bond networks, and hydrophobic interactions. Scrutiny of these interaction parameters clearly shows that the two drug molecules bind differently with the same enzyme. Namely, the peripheral pyridinium ring of HI-6 binds to the PAS region at the gorge entrance with excellent complementarity, while the active pyridinium ring of Ortho-7 has been found to be suitably fitted in the active-site region. Tyr 124 and Trp 286 residues of PAS of the enzyme have been found to clamp the peripheral pyridinium ring of HI-6 for a sufficiently long time (about 3500 ps) through a strong cation-π interaction and a host of other protein-drug interaction parameters. The PAS preferential binding of HI-6 was found to have major consequences. In particular, HI-6 binding energy with the enzyme was found to be larger than that for Ortho-7. Moreover, during the egress, while the peripheral pyridinium ring is held tightly by the PAS residues, the active pyridinium ring of HI-6 is found to undergo a complete turn along gorge axis, directed away from the active triad of the enzyme. To accommodate such a large rotation of HI-6 inside the gorge, the protein's structure has to undergo a substantial change as manifested by the larger gorge proper radius. Moreover, the PAS binding compatibility of HI-6 may even render it to act like other PAS inhibitors, instead of as a drug-reactivating agent. The active pyridinium ring of Ortho-7, on the other hand, forms a parallel cation-π interaction with phenyl ring of Tyr 337 and a T-shaped cation-π interaction with the indole ring of Trp 86. This, along with the other interactions (as mentioned above) monitored in this work, makes the oximate oxygen of Ortho-7 always directed towards the active triad region. One of the principal motivations against oxime drug design is the cooperative binding of the bispyridinium compounds in the active-site gorge such that the proximity and orientations of the drug molecule, necessary for an effective nucleophilic attack can be attained. However, neither the protein nor the drugs are static structures. On both of these grounds conducive for nucleophilic attack, Ortho-7 is found to be a more suitable choice, a result that supports the experimental findings; Ortho-7 could function as a more effective antidote against tabun intoxication. Before we conclude, one remark is warranted. The starting crystal structures, and consequently the present work on the oxime complexes, involve the use of non-phosphorylated enzyme. The chemical environment and, therefore, the interactions the drug molecules will experience in the OP conjugated enzyme will be certainly different from those that are reported here. In that spirit, the present structural dynamics of the non-phosphorylated AChE-oxime complexes can be treated as a precursor to the phosphorylated counterpart.
Supporting information
Preparation of initial crystal structures, snapshots of the protein-drug interactions for the two complexes showing hydrogen bonds, water bridges, triangular hydrogen bond network in association with water bridges (S-1, S-2), and trajectory profiles of the drug molecules during their egress from the active site gorge presented in the form of movies (S-3, 4, 5, 6) are available.
Author contribution
Manoj K KESHARWANI performed research. Bishwajit GANGULY contributed new tools and wrote the paper. Amit DAS performed research and analysed data. Tusar BANDYOPADHYAY designed research, performed research, analysed data and wrote the paper.
Abbreviations Used
mAChE, Mus musculus acetylcholinesterase; HI-6, 1-(2-hydroxy-iminomethylpyridinium)-1-(4-carboxyamino)-pyridinium dimethylether dichloride; Ortho-7,1,7-heptylene-bis-N,N'-2-pyridiniumaldoxime dichloride; PAS, Peripheral anionic site; CMD, Conventional molecular dynamics; SMD, Steered molecular dynamics;
PDB ID codes: 2GYU, 2GYV
Acknowledgments
Financial support from DAE-BRNS, Mumbai, India is gratefully acknowledged (vide DAE-Sanction No 2007/37/19/BRNS). Manoj K KESHARWANI is thankful to UGC, New Delhi, India for the supporting fellowship. Tusar BANDYOPADHYAY is grateful to Prof T MUKHERJEE and Swapan K GHOSH for their constant encouragement during the course of the work. Manoj K KESHARWANI and Bishwajit GANGULY are thankful to Dr P K GHOSH for his support to this work.
Supplementary Information
Preparation of initial structures
References
- Quinn DM. Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states. Chem Rev. 1987;87:955–79. [Google Scholar]
- Kandel ER, Schwartz JH, Jessell TM.Principles of neural science. 3rd ed. Norwalk, CT: Appleton & Lange; 1991
- Barnard EA.In the peripheral nervous system. Hubbard JI, Editor. New York: Plenum Publishers; 1974. p201–6.
- Voet D, Voet JG.Biochemistry. 2nd edition. Wiley & Sons; 1995
- Kraut J. Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem. 1977;46:331–58. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- Radic Z, Reiner E, Taylor P. Role of the peripheral anionic site on acetylcholinesterase: inhibition by substrates and coumarin derivatives. Mol Pharmacol. 1991;39:98–104. [PubMed] [Google Scholar]
- Barak D, Kronman C, Ordentlich A, Ariel N, Bromberg A, Marcus D, et al. Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core. J Biol Chem. 1994;269:6296–305. [PubMed] [Google Scholar]
- Bourne Y, Taylor P, Radic Z, Marchot P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 2003;22:1–12. doi: 10.1093/emboj/cdg005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kua J, Mccammon JA. Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: an ab initio qm/mm study. J Am Chem Soc. 2002;24:10572–7. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- Bui JM, Tai K, Mccammon JA. Acetylcholinesterase: enhanced fluctuations and alternative routes to the active site in the complex with fasciculin-2. J Am Chem Soc. 2004;126:7198–205. doi: 10.1021/ja0485715. [DOI] [PubMed] [Google Scholar]
- Shen T, Tai K, Henchman RH, Mccammon JA. Molecular dynamics of acetylcholinesterase. Acc Chem Res. 2002;35:332–40. doi: 10.1021/ar010025i. [DOI] [PubMed] [Google Scholar]
- Kua J, Zhang Y, Mccammon JA. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J Am Chem Soc. 2002;124:8260–7. doi: 10.1021/ja020429l. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kua J, Mccammon JA. Influence of structural fluctuation on enzyme reaction energy barriers in combined quantum mechanical/molecular mechanical studies. J Phys Chem B. 2003;107:4459–63. [Google Scholar]
- Tai K, Shen T, Börjesson U, Philippopoulos M, Mccammon JA. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys J. 2001;81:715–24. doi: 10.1016/S0006-3495(01)75736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K, Shen T, Henchman RH, Bourne Y, Marchot P, Mccammon JA. Mechanism of acetylcholinesterase inhibition by fasciculin: a 5-ns molecular dynamics simulation. J Am Chem Soc. 2002;124:6153–61. doi: 10.1021/ja017310h. [DOI] [PubMed] [Google Scholar]
- Niu C, Xu Y, XuY, Luo X, Duan W, Silman I, et al. Dynamic mechanism of e2020 binding to acetylcholinesterase: a steered molecular dynamics simulation. J Phys Chem B. 2005;109:23730–8. doi: 10.1021/jp0552877. [DOI] [PubMed] [Google Scholar]
- Xu Y, Shen JH, Luo XM, Silman I, Sussman JL, Chen KX, et al. How does huperzine a enter and leave the binding gorge of acetylcholinestearse? Steered molecular dynamics simulations. J Am Chem Soc. 2003;125:11340–9. doi: 10.1021/ja029775t. [DOI] [PubMed] [Google Scholar]
- Shen TY, Tai K, McCammon JA. Statistical analysis of the fractal gating motions of the enzyme acetylcholinesterase. Phys Rev E. 2001;63:041902. doi: 10.1103/PhysRevE.63.041902. [DOI] [PubMed] [Google Scholar]
- Metzler R, Klafter J. The random walk's guide to anomalous diffusion: a fractional dynamics approach. Phys Rep. 2000;339:1–77. [Google Scholar]
- Bandyopadhyay T. Single-file diffusion of subdiffusive particles. Eur Phys Lett. 2008;81:16003-p1–p6. [Google Scholar]
- Bandyopadhyay T. Single-file diffusion through inhomogeneous nanopores. J Chem Phys. 2008;128:114712. doi: 10.1063/1.2894839. [DOI] [PubMed] [Google Scholar]
- Henchman RH, Tai K, Shen T, McCammon JA. Properties of water molecules in the active site gorge of acetylcholinesterase from computer simulation. Biophys J. 2002;82:2671–82. doi: 10.1016/S0006-3495(02)75609-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallak M, Giacobini E. Physostigmine tacrine and metrifonate: the effect of multiple doses on acetylcholine metabolism in rat brain. Neuropharmacology. 1989;28:199–206. doi: 10.1016/0028-3908(89)90093-2. [DOI] [PubMed] [Google Scholar]
- Kaur J, Zhang MQ. Molecular modelling and qsar of reversible acetylcholinesterase inhibitors. Curr Med Chem. 2000;7:273–94. doi: 10.2174/0929867003375254. [DOI] [PubMed] [Google Scholar]
- Kesharwani MK, Khan MA, Bandyopadhyay T, Ganguly B.Importance of hydrogen bonding on efficient destruction of organophosphorus nerve agent gv: a dft study with simple and α-nucleophiles. New J Chemistry 2010; in press.
- Khan MA, Kesharwani MK, Bandyopadhyay T, Ganguly B. Solvolysis of chemical warfare agent VX is more efficient with hydroxylamine anion: A computational study. J Mol Graph Model. 2009;28:177–82. doi: 10.1016/j.jmgm.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Bertolote JM, Fleischmann A, Eddleston M, Gunnell D. Deaths from pesticide poisoning: a global response. Br J Psychiatr. 2006;189:201–3. doi: 10.1192/bjp.bp.105.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M, Phillips MR. Self poisoning with pesticides. Br Med J. 2004;328:42–4. doi: 10.1136/bmj.328.7430.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr DB, Bravo R, Weerasekera G, Caltabiano LM, Whitehead RD Jr, Olsson AO, et al. Concentrations of dialkyl phosphate metabolites of organophosphorus pesticides in the US population. Environ Health Perspect. 2004;112:186–200. doi: 10.1289/ehp.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs TC, Maynard RL, Sidell FR.Eds. Chemical warfare agents: toxicology and treatment. New York: Wiley; 1996. p243
- Taylor P.Anticholinesterase agents. In: Hardman JG, Limbird LE, Eds. Goodman & Gilman's the Pharmacological Basis of Therapeutics, 10th ed. New York: McGraw-Hill; 2001. p175–91.
- Newmark J. The birth of nerve agent warfare: lessons from syed abbas foroutan. Neurology. 2004;62:1590–6. doi: 10.1212/01.wnl.0000124519.85516.50. [DOI] [PubMed] [Google Scholar]
- Lee EC. Clinical manifestations of sarin nerve gas exposure. JAMA. 2003;290:659–62. doi: 10.1001/jama.290.5.659. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EFR, Aracava Y, Fawcett WP, Maristela O, Randall WR, et al. Effective countermeasure against poisoning by organophosphorus insecticides and nerve agents. Proc Natl Acad Sci USA. 2006;103:13220–5. doi: 10.1073/pnas.0605370103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekström FJ, Åstot C, Pang YP. Novel nerve-agent antidote design based on crystallographic and mass spectrometric analyses of tabun-conjugated acetylcholinesterase in complex with antidotes. Clin Pharm Ther. 2007;82:282–93. doi: 10.1038/sj.clpt.6100151. [DOI] [PubMed] [Google Scholar]
- Ekström F, Pang Y-P, Boman M, Artursson E, Akfur C, Börjegren S. Crystal structures of acetylcholinesterase in complex with hi-6, ortho-7 and obidoxime: structural basis for differences in the ability to reactivate tabun conjugates. Biochem Pharm. 2006;72:597–607. doi: 10.1016/j.bcp.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Woreka F, Thiermanna H, Szinicza L, Eyerb P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem Pharm. 2004;68:2237–48. doi: 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Čalić M, Šinko G, Bosak A. Structure-activity approach in the reactivation of tabun-phosphorylated human acetylcholinesterase with bispyridinium para-aldoximes. Arh Hig Rada Toksikol. 2007;58:201–9. doi: 10.2478/v10004-007-0013-7. [DOI] [PubMed] [Google Scholar]
- Worek F, Aurbek N, Koller M, Becker C, Eyer P, Thiermann H. Kinetic analysis of reactivation and aging of human acetylcholinesterase inhibited by different phosphoramidates. Biochem Pharm. 2007;73:1807–17. doi: 10.1016/j.bcp.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Kuca K, Musilek K, Paar M, Jun D, Stodulka P, Hrabinova M, et al. Targeted synthesis of 1-(4-hydroxyiminomethylpyridinium)-3-pyridiniumpropane dibromide — a new nerve agent reactivator. Molecules. 2007;12:1964–72. doi: 10.3390/12081964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Tonga M, Maxwell DM, Saxena A. Comparison of oxime reactivation and aging of nerve agent-inhibited monkey and human acetylcholinesterases. Chemico-Biol Inter. 2008;175:261–6. doi: 10.1016/j.cbi.2008.04.034. [DOI] [PubMed] [Google Scholar]
- Vesela S, Kuca K, Jun D. Efficacy and dosing of antidotes applied to daphnia intoxicated by nerve agent tabun. Environment Toxicol Pharmacol. 2008;26:283–9. doi: 10.1016/j.etap.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Kuca K, Cabal J, Kassa J, Jun D, Hrabinová M. A comparison of the potency of the oxime HLö-7 and currently used oximes (HI-6, pralidoxime, obidoxime) to reactivate nerve agent-inhibited rat brain acetylcholinesterase by in vitro methods. Acta Med (Hradec Kralove) 2005;48:81–6. [PubMed] [Google Scholar]
- Stojiljković MP, Jokanović M. Pyridinium oximes: rationale for their selection as causal antidotes against organophosphate poisonings and current solutions for auto-injectors. Arh Hig Rada Toksikol. 2006;57:435–43. [PubMed] [Google Scholar]
- Eddleston M, Szinicz L, Eyer P, Buckley N. Oximes in acute organophosphorus pesticide poisoning: a systematic review of clinical trials. QJM. 2002;95:275–83. doi: 10.1093/qjmed/95.5.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonijevic B, Stojiljkovic MP. Unequal efficacy of pyridinium oximes in acute organophosphate poisoning. Clin Med & Res. 2007;5:71–82. doi: 10.3121/cmr.2007.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstedt B. Pharmacology of organophosphorus cholinesterase inhibitors. Pharmacol Rev. 1959;11:567–688. [PubMed] [Google Scholar]
- Eddleston M, Mohamed F, Davies JOJ, Eyer P, Worek F, Sheriff MHR, et al. Respiratory failure in acute organophosphorus pesticide self-poisoning. QJM. 2006;99:513–22. doi: 10.1093/qjmed/hcl065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmark J. Therapy for nerve agent poisoning. Arch Neurol. 2004;61:649–52. doi: 10.1001/archneur.61.5.649. [DOI] [PubMed] [Google Scholar]
- Reiner E, Simeon-Rudolf V. Pyridinium, imidazolium and quinuclidinium compounds: toxicity and antidotal effects against the nerve agents tabun and soman. Arh Hig Rada Toksikol. 2006;57:171–9. [PubMed] [Google Scholar]
- Grubmüller H, Heymann B, Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–9. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr Opin Str Biol. 2001;11:224–30. doi: 10.1016/s0959-440x(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Morfill J, Neumann J, Blank K, Steinbach U, Puchner EM, Gottschalk KE, et al. Force-based analysis of multidimensional energy landscapes: application of dynamic force spectroscopy and steered molecular dynamics simulations to an antibody fragment-peptide complex. J Mol Biol. 2008;381:1253–66. doi: 10.1016/j.jmb.2008.06.065. [DOI] [PubMed] [Google Scholar]
- Marszalek PE, Lu H, Li H, Carrion-Vazquez M, Oberhauser AF, Schulten K, et al. Mechanical unfolding intermediates in titin modules. Nature. 1999;402:100–3. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- Heymann B, Grubmüller H. Dynamic force spectroscopy of molecular adhesion bonds. Phys Rev Lett. 2000;84:6126–9. doi: 10.1103/PhysRevLett.84.6126. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Hess B, Van Der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–17. [Google Scholar]
- van Gunsteren WF, Billeter S, Eising AA, Hünenberger PH, Krüger P, Mark AE, et al. Biomolecular simulation: the gromos96 manual and user guide. Zürich: Vdf Hochschulverlag AG an der ETH Zürich, 1996.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comp Chem. 1997;18:1463–72. [Google Scholar]
- Darden T, York D, Pedersen L. Particle Mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–92. [Google Scholar]
- Nome RA, Zhao JM, Hoff WD, Scherer NF. Axis-dependent anisotropy in protein unfolding from integrated nonequilibrium single-molecule experiments, analysis, and simulation. Proc Natl Acad Sci USA. 2007;104:20799–804. doi: 10.1073/pnas.0701281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers W, Schulten K. Investigating a back door mechanism of actin phosphate release by steered molecular dynamics. Proteins. 1999;35:262–73. [PubMed] [Google Scholar]
- Yang K, Liu X, Wang X, Jiang H. A steered molecular dynamics method with adaptive direction adjustments. Biochem Biophys Res Commun. 2009;379:494–8. doi: 10.1016/j.bbrc.2008.12.099. [DOI] [PubMed] [Google Scholar]
- Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72:1568–81. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller H.Proteins as molecular machines: force probe simulations Computational soft matter: from synthetic polymers to proteinsAttig N, Binder K, Grubmüller H, Kremer K, Eds. NIC Series200423401–21. [Google Scholar]
- Izrailev S, Stepaniants S, Isralewitz B, Kosztin D, Lu H, Molnar F, et al. Computational molecular dynamics: challenges, methods, ideas. In: Deufhard P, Hermans J, Leimkuhler B, Mark AE, Reich S, Skeel RD, eds. Computational science and engineering. Berlin: Springer; 1998. p39–65.
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- van der Spoel D, van Maaren PJ, Larsson P, Tîmneanu N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J Phys Chem B. 2006;110:4393–8. doi: 10.1021/jp0572535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Preparation of initial structures