

Abstract
Aim:
To investigate the protective effects of rosiglitazone (RGZ) against the neuronal toxicity induced by advanced glycation end products (AGEs) and the underlying mechanisms.
Methods:
Neuroblastoma cell line SH-SY5Y was used. Cell viability and apoptosis were assessed using MTT assay and flow cytometry, respectively. Superoxide dismutase (SOD) and catalase activities were measured using biochemical methods. Intracellular reactive oxygen species (ROS) were monitored using 2',7'-dichlorodihydro-fluorescein diacetate (DCFH-DA). Secreted β-amyloid1–42 (Aβ1–42) level was assessed by ELISA. The expression of mRNA of Bcl2, Bax, Caspase3, Aβ precursor protein (APP), β-site APP-cleaving enzyme 1 (BACE1), and insulin degrading enzyme (IDE) were measured using quantitative real-time PCR (Q-PCR), and their protein levels were examined using Western blot.
Results:
RGZ (0.1–10 μmol/L) significantly increased the cell viability that was reduced by AGEs (1000 μg/mL). RGZ (10 μmol/L) significantly ameliorated AGEs-triggered downregulation of SOD and catalase, and production of ROS. It also reversed Bcl2 downregulation, Bax upregulation and Caspase3 expression caused by AGEs. Moreover, it significantly attenuated AGEs-induced Aβ secretion and APP protein upregulation. RGZ did not affect BACE1 expression, but induced IDE expression, which promoted degradation of Aβ. All the effects were blocked by the specific PPARγ antagonist GW9662 (10 μmol/L).
Conclusion:
RGZ protects the euroblastoma cells against AGEs-induced injury via its anti-oxidative, anti-apoptotic and anti-inflammatory properties that seems to be mediated by PPARγ activation. The results suggest a beneficial role for RGZ in the treatment of Alzheimer's disease.
Keywords: Alzheimer's disease, advanced glycation end products, rosiglitazone, apoptosis, β-amyloid, PPARγ, SH-SY5Y cells
Introduction
Although the pathogenesis of sporadic Alzheimer's disease (AD) is not clearly understood, it is likely dependent on several age-related factors1. Diabetes is one of the most important risk factors for AD, and the multiple mechanisms connecting the two diseases have been proposed1. The pathological complications of diabetes affect several organ systems including the brain2, which undergoes changes that could increase the risk of cognitive disorders including AD3. However, details of the association between diabetes and dementia have not yet been fully elucidated. Hyperglycemia is accompanied by accelerated formation of advanced glycation end-products (AGEs), which have been associated with increased amyloid deposition and neurofibrillary tau tangle formation4. Moreover, AGEs have been demonstrated to cause oxidative stress and inflammatory responses as well as cytotoxicity in neuronal cells5, 6, 7, 8, 9, 10. An emerging body of evidence also suggests that AGEs decrease mitochondrial activity and lead to energy depletion11, 12.
The central pathological event in AD is the progressive accumulation of amyloid-β (Aβ) in the brain. Aβ derives from sequential cleavage of the β-amyloid precursor protein (APP) by β- and γ-secretase. β-secretase (β-site APP-cleaving enzyme 1, BACE1) cleaves the ectodomain of APP, producing an APP C-terminal fragment. This fragment is further cleaved within the transmembrane domain by γ-secretase, resulting in the release of a family of Aβ peptides with different C-terminal variants, predominantly Aβ40 and Aβ4213. In addition, the level of Aβ is determined by the balance between its generation and turnover14. Aβ is degraded by endopeptidases, among which insulin-degrading enzyme (IDE) is predominant. IDE mediates much of the degradation of soluble monomeric Aβ13.
Peroxisome proliferator-activated receptors (PPARs) are a family of three related nuclear receptors (α, γ, and δ) that act as transcription factors in the regulation of genes responsible for lipid and energy metabolism. PPARγ is of particular importance for lipid and carbohydrate metabolism, and participates in the regulation of serum glucose levels and insulin sensitivity14. Two thiazolidinedione agonists of PPARγ, pioglitazone and rosiglitazone (RGZ), are widely prescribed for the treatment of type 2 diabetes mellitus14. There is now an extensive body of evidence that has demonstrated the efficacy of these agonists in reducing neuronal cell loss in in vitro models of neurotoxicity, in vivo models of cerebral ischemia-reperfusion injury, Parkinson's disease, and amyotrophic lateral sclerosis15, 16, 17, 18, 19, 20, 21, 22, 23. In particular, recent animal and clinical trials of RGZ have shown significant improvement in memory and cognition in animal models of AD, and AD patients, respectively24. Thus, PPARγ represents an important new therapeutic research target for the treatment of AD. However, the mechanisms mediating this potential beneficial effect remain to be fully elucidated.
AGEs may contribute to the etiology of many disease processes, including AD15, by accumulating on β-amyloid plaques and exerting chronic oxidative stress via receptor-mediated mechanisms. Accordingly, in this study AGEs were used to induce neuronal toxicity in SH-SY5Y neuroblastoma cells in vitro to investigate the protective effects of RGZ. The antioxidative and anti-apoptotic properties of RGZ were also evaluated. The investigation therefore provides further insights into the mechanisms whereby RGZ exerts a beneficial effect in AD.
Materials and methods
Chemicals
Fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), trypsin-ethylenediaminetetracetic acid (EDTA), and antibiotics for cell culture were from Gibco/BRL Life Technologies (Grand Island, NY, USA). RGZ maleate tablets were purchased from SmithKline Beecham Pharmaceuticals (West Sussex, UK). GW9662, an inhibitor of PPARγ, was purchased from Cayman Chemical (Ann Arbor, MI, USA). All other chemicals and reagents, unless otherwise noted, were obtained from Sigma Chemical (St Louis, MO, USA).
Preparation of AGE-BSA
AGE-bovine serum albumin (BSA) was prepared by incubating 20 g/L BSA with 0.5 mol/L glucose at 37 °C for 3 months under sterile conditions, as described previously16, 17, 18. Control non-glycated BSA was incubated under the same conditions except for the absence of glucose. At the end of the incubation period, preparations were dialyzed against phosphate buffered saline (PBS) for 48 h while stirring to remove unincorporated glucose. The protein concentration was determined by Lowry assay. AGEs protein-specific fluorescence determinations were performed by measuring emission at 440 nm and excitation at 370 nm using a fluorescence spectrophotometer (F-3000, Hitachi, Japan).
Cell culture
The neuroblastoma cell line SH-SY5Y was grown in DMEM and Ham's F12 nutrient mixture (DMEM/F12; 1:1) routinely supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin, and incubated at 37 °C in a humidified atmosphere of 5% CO2. All experiments were carried out 24–36 h after cells were seeded. During AGEs studies, the growth medium was treated with 1000 μg/mL AGE-BSA or non-glycated BSA in the presence or absence of 10 μmol/L RGZ or 10 μmol/L of the PPARγ antagonist GW9662 for 24 h.
Cell viability assay
Methylthiazoletetrazolium (MTT) was dissolved in PBS at a concentration of 5 mg/mL. After 48 h incubation, 25 μL of the MTT solution was added to each well of 96-well plates and incubated for 4 h at 37 °C in a humidified atmosphere of 5% CO2. At the end of the incubation period, the medium were discarded using a suction pump. The extraction buffer of 20% w/v sodium dodecyl sulfate (SDS) in 50% of N,N-dimethylformamide in demineralized water (50:50, v/v) was prepared at pH 4.7. The absorbance was determined at 570 nm. The A570 was taken as an index of cell viability and the activity of mitochondria. The net absorbance from the plates of cells cultured with the control medium (not treated) was considered as 100% cell viability and mitochondrial activity.
Measurement of the activity of superoxide dismutase (SOD) and catalase (CAT)
The SH-SY5Y cells were washed twice with PBS and lysed in 200 μL of lysis buffer. The supernatants were obtained by centrifugation at 14 000×g at 4 °C for 10 min, and antioxidant enzyme activity assays were performed on the resulting supernatants. The protein content was determined by Lowry's method. The superoxide dismutase (SOD) activity was determined by mixing the reaction mixture with the samples. After adding one unit of SOD and 0.005 unit xanthine oxidase, the absorbance was read at 550 nm for 5 min. The SOD activity was calculated as:
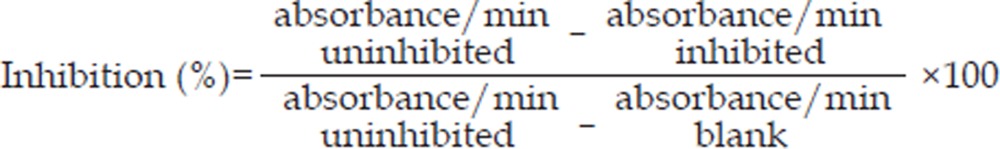 |
Catalase activity was determined by adding 995 μL of 30 mmol/L H2O2 to 5 μL of cell lysate. The disappearance of H2O2 was monitored at 240 nm. The catalase activity was calculated as:
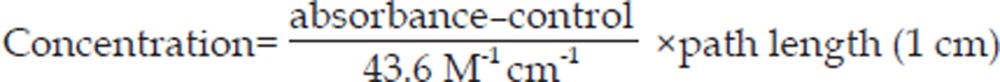 |
Measurement of intracellular reactive oxygen species (ROS)
Intracellular reactive oxygen species (ROS) were monitored by using the fluorescent probe 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA). Intracellular H2O2, or low molecular weight peroxides, oxidize DCFH-DA to the highly fluorescent compound dichlorofluorescein (DCF). SH-SY5Y cells were seeded in 96-well plates and were incubated with increasing concentrations of AGEs and/or RGZ for 24 h. Cells were incubated with 10 μmol/L DCFH-DA at 37 °C for 30 min, then washed twice with PBS, and finally the fluorescence intensity of DCF was measured in a microplate-reader at excitation wavelength 485 nm and emission wavelength 538 nm.
Apoptosis assay
Apoptosis was measured according to the manufacturer's instructions (Bioscience Research Reagents, Temecula, CA, USA). Briefly, SH-SY5Y cells were collected by trypsin-EDTA, washed with PBS, resuspended in 100 μL of binding buffer and stained with 3 μL of fluorescein isothiocyanate-labeled Annexin-V and 2 μL of propidium iodide (PI). After 15 min of incubation, the cells were immediately analyzed by flow cytometry.
Quantitative real time polymerase chain reaction (Q-PCR)
The total RNA was extracted from the cells and converted to cDNA, according to the manufacturer's instructions. The total RNA (3 μg) was converted to cDNA by reverse transcriptase using a SuperScriptTM II Reverse Transcriptase kit. Q-PCR of the cDNA samples was performed with an iCycler iQTM Real-Time Detection System. Primer Sequences for quantitative PCR are indicated in Table 1. Five microliters of cDNA template was amplified in a mixture of 12.5 μL of iQ SYBR Green Supermix, primer, and sterile water in a final volume of 25 μL. For all experiments, the negative controls used water instead of cDNA for the PCR reaction. The data were normalized by subtracting the difference of the threshold cycle values between the target gene of interest and the β-actin housekeeping gene.
Table 1. Primer sequences and PCR reaction condition used in this study.
| Gene name | Gene Bank ID | PCR primers (5′–3′) | Tm (°C) | Length of PCR product |
|---|---|---|---|---|
| APP | NM_001136016 | Forward: GAGACACCTGGGGATGAGAATG | 60 | 122 bp |
| Reverse: GCTTGACGTTCTGCCTCTTCC | ||||
| BACE1 | NM_012104 | Forward: TACCAACCAGTCCTTCCGC | 60 | 116 bp |
| Reverse: CCGTGGATGACTGTGAGATG | ||||
| Bax | NM_004324 | Forward: AGTGTCTCAAGCGCATCGG | 60 | 141 bp |
| Reverse: CCCAGTTGAAGTTGCCGTC | ||||
| Bcl2 | NM_000633 | Forward: ATGTGTGTGGAGAGCGTCAAC | 60 | 135 bp |
| Reverse: CAGTTCCACAAAGGCATCCC | ||||
| Caspase3 | NM_032991 | Forward: AGAACTGGACTGTGGCATTG | 60 | 164 bp |
| Reverse: CACAAAGCGACTGGATGAAC | ||||
| IDE | NM_004969 | Forward: GCCGAAGCCTTGTCTCAACT | 60 | 79 bp |
| Reverse: CAAATAGGCCATGTTACAGTGCAA | ||||
| β-actin | NM_001101 | Forward: CCCAGCACAATGAAGATCAAGATCAT | 60 | 101 bp |
| Reverse: ATCTGCTGGAAGGTGGACAGCGA |
Western blot
After treatments, the cells were collected and homogenized in lysis buffer containing a mixture of protease inhibitors. Cell extracts, prepared by centrifugation at 16 000×g, were resolved by SDS-PAGE. Proteins were then transferred onto a nitrocellulose membrane, and blotted with the appropriate primary antibodies: polyclonal rabbit anti-Bcl2 (Santa Cruz), polyclonal rabbit anti-cleaved Caspase3 (Cell Signaling, Beverly, MA, USA), polyclonal rabbit anti-Bax (Santa Cruz), monoclonal mouse anti-BACE1 (Chemicon), polyclonal rabbit anti-APP (Zymed, San Francisco, CA, USA), polyclonal goat anti-IDE (Santa Cruz), polyclonal mouse anti-β-actin (Santa Cruz). Anti-mouse and anti-rabbit as well as anti-goat secondary antibodies were coupled to horseradish peroxidase (Santa Cruz). The detection was performed by using an ECL Plus kit (Amersham Biosciences, Milan, Italy). Relative expressions of proteins were quantified by densitometric scanning of the X-ray films with a GS 700 Imaging Densitometer (Bio-Rad) and a computer program. β-actin was used for standardization of the samples.
Aβ detection
Aβ1-42 were detected using a human β-amyloid1–42 ELISA kit (BioSource) and conditioned medium. The kit protocol was followed to prepare samples. Briefly, the plate was washed four times with wash buffer. The standard peptide was resuspended and serial dilutions were prepared. Dilutions of the samples to be tested were also prepared. Standards or samples (100 μL) were added to the appropriate wells of the pre-coated microtiter plate and incubated for 2 h at room temperature. They were washed four times with wash buffer. The diluted detection antibody (rabbit polyclonal Aβ1–42, 100 μL) was placed into the wells and incubated for 2 h at room temperature. The samples were then washed four times with wash buffer. One hundred microliters of the HRP-conjugated anti-rabbit antibody was added to the wells and incubated for 2 h at room temperature. The wells were washed four times with wash buffer and then 100 μL of chromogen solution was added to each well. The wells were incubated for 30 min at room temperature, protected from light, and then 100 μL of stop solution was added to each well. Optical density was measured at 450 nm. The results were calculated from a standard curve. The data represent the means from experiments done in triplicate.
Statistical analysis
The statistical significance of the differences was determined using one-way ANOVA followed by the Scheffe F test. Differences in which the probability (P)-value<0.05 were considered statistically significant. All graphs represent the mean±standard deviation (SD) for three separate experiments.
Results
RGZ ameliorates AGEs-induced neuronal insult
To determine the cytotoxic effects of AGEs at various concentrations for 24 h in SH-SY5Y cells, MTT assays were performed to assess cell viability. As shown in Figure 1A, AGEs treatment at a range of 100–2000 μg/mL resulted in a reduction of cell viability from up to 14% to 25% in a concentration-dependent manner. Therefore, AGEs at 1000 μg/mL was used to induce neuronal insults in the following experiments. In order to investigate the protective effect of RGZ, SH-SY5Y cells were treated with 1000 μg/mL AGEs and RGZ with or without the PPAR-γ inhibitor GW9662, for 24 h. As shown in Figure 1B, RGZ significantly attenuated the decrease in AGEs-induced neuronal viability in a concentration-dependent manner. In contrast, GW9662 treatment reversed the protective effect of RGZ on AGEs-induced neuronal insult (Figure 1D), whereas GW9662 alone did not affect the cell viability (Figure 1C). These results suggest that RGZ protecting from AGEs-induced neurotoxicity in SH-SY5Y cells is associated with PPARγ.
Figure 1.

Effects of RGZ on the cell viability of SH-SY5Y cells treated with AGEs. (A) Cytotoxicity of AGEs in SH-SY5Y cells. SH-SY5Y cells grown to confluence were incubated with AGEs or a non-glycated control at concentration of 0, 100, 250, 500, 1000, and 2000 μg/mL. After incubation for 24 h, cell viability was quantified by MTT assay. Note that AGEs decreased cell viability in a concentration-dependent manner. (B) RGZ ameliorated AGEs-induced neuronal insult in SH-SY5Y cells. SH-SY5Y cells were treated with 1000 μg/mL AGEs in the absence or presence of 0.1, 1.0, 10 μmol/L RGZ for 24 h. (C) GW9662 did not affect cell viability of SH-SY5Y cells. SH-SY5Y cells were treated with 0, 5, 10, or 20 μmol/L GW9662 alone for 24 h. (D) RGZ ameliorated AGEs-induced neuronal insult in SH-SY5Y cells was eliminated by GW9662, an antagonist for PPARγ. SH-SY5Y cells were treated with 1000 μg/mL AGEs and 10 μmol/L RGZ with or without GW9662 for 24 h. Data are shown as mean±SD (n=3 independent experiments). cP<0.01 compared with the control group. fP<0.01 compared with AGEs-treated group. iP<0.01 compared with AGEs+RGZ-treated group.
RGZ inhibits oxidative stress induced by AGEs in SH-SY5Y cells
To determine the effects of RGZ on oxidative stress induced by AGEs in SH-SY5Y cells, ROS production and the activities of antioxidant enzymes were measured. As expected, RGZ significantly attenuated AGEs-induced ROS production (Figure 2A), and the reduction of the activities of SOD as well as catalase (Figure 2B and 2C, respectively). Similarly, the PPARγ antagonist GW9662 eliminated the upregulatory effects of RGZ on the activities of SOD and catalase in this process. But reduction of ROS induced by RGZ was not blocked by PPARγ antagonist GW9662. These results indicate that the cytotoxic effect of AGEs on SH-SY5Y cells may be mediated by oxidative stress, while RGZ ameliorated AGEs-induced downregulation of SOD and CAT activities in a PPARγ-dependent manner.
Figure 2.

Effects of RGZ on the activities of SOD and catalase as well as the production of ROS in AGEs-treated SH-SY5Y cells. SOD and CAT activity as well as ROS levels were determined in SH-SY5Y cells after incubation with AGEs (1000 μg/mL) for 24 h with or without 10 μmol/L RGZ and 10 μmol/L GW9662. (A) ROS production. (B) SOD activity. (C) Catalase activity. Data are shown as mean±SD (n=3 independent experiments). cP<0.01 compared with the control group. fP<0.01 compared with AGEs-treated group. iP<0.01 compared with AGEs+RGZ-treated group.
RGZ prevents cell apoptosis induced by AGEs in SH-SY5Y cells
Cell apoptosis was evaluated by Annexin V and propidium iodide (PI) double-staining. As shown in Figure 3, treatment with AGEs for 24 h (Figure 3B) resulted in a 10% increase in apoptosis of SH-SY5Y cells when compared to controls (Figure 3A), 11.48% vs 1.36% apoptotic cells. In contrast, RGZ treatment (Figure 3C) attenuated cell apoptosis up to 4% when compared to AGEs treatment alone (Figure 3B). The anti-apoptotic effect of RGZ was blocked by GW9662 (Figure 3D), indicating the involvement of PPARγ in this process.
Figure 3.

Effects of RGZ on AGEs-induced apoptosis in SH-SY5Y cells. SH-SY5Y cells were incubated with AGEs (1000 μg/mL) for 24 h with or without 10 μmol/L RGZ and 10 μmol/L GW9662. (A) The non-glycated control for 24 h. (B) The glyco-AGE (AGEs) 1000 μg/mL for 24 h. (C) AGEs (1000 μg/mL) with RGZ (10 μmol/L) for 24 h. (D) AGEs (1000 μg/mL) and RGZ (10 μmol/L) as well as GW9662 (10 μmol/L) for 24 h. Annexin fluorescence intensity was determined by flow cytometric analysis (FACS).
Effects of RGZ on Bcl2 and Bax as well as Caspase3 expression in AGEs-treated SH-SY5Y cells
The apoptosis-relevant molecules Bcl2 and Bax, as well as Caspase3, were determined by Q-PCR and Western blot, respectively. As shown in Figure 4, exposure to AGEs resulted in an upregulation of pro-apoptotic Bax (Figure 4B and 4E) and a downregulation of anti-apoptotic Bcl2 (Figure 4A and 4D) both in mRNA and protein levels as well as activation of Caspase3 (Figure 4C and 4F). RGZ treatment markedly reversed the effect of AGEs on these molecules. Similarly, PPARγ antagonist GW9662 eliminated the anti-apoptotic effects of RGZ in AGEs-treated SH-SY5Y cells. These observations suggest that RGZ could suppress the AGEs-induced apoptosis via PPAR-γ activation.
Figure 4.

Effects of RGZ on Bcl2 and Bax as well as Caspase3 expression in AGEs-treated SH-SY5Y cells. SH-SY5Y cells were treated with 1000 μg/mL AGEs for 24 h, with or without incubation of 10 μmol/L RGZ and 10 μmol/L GW9662. (A) The mRNA of Bcl2. (B) The mRNA of Bax. (C) The mRNA of activated Caspase3. (D) The protein expression of Bcl2. (E) The protein expression of Bax. (F) The protein expression of activated Caspase3. Data are shown as mean±SD (n=3 independent experiments). bP<0.05, cP<0.01 compared with the control group. eP<0.05, fP<0.01 compared with AGEs-treated group. hP<0.05, iP<0.01 compared with AGEs+RGZ-treated group.
Effects of RGZ on Aβ1–42 secretion and APP expression as well as BACE1 activity in AGEs-treated SH-SY5Y cells
To evaluate whether the effect of RGZ on AGEs-induced neuronal insults is related to APP processing, the mRNA and protein levels of APP and BACE1, as well as Aβ1–42, were determined. It showed that exposure to AGEs significantly increased the mRNA and protein levels of APP (Figure 5A and 5C) and BACE1 (Figure 5B and 5D), as well as Aβ1–42 (Figure 6A). However, RGZ treatment only attenuated AGEs-induced upregulation of APP proteins (Figure 5C), and not APP mRNA levels (Figure 5A). There were no changes in mRNA and protein levels of BACE1 after treatment with RGZ when compared to AGEs treatment alone (Figure 5B and 5D). In addition, RGZ treatment resulted in a notable reduction of APP protein and Aβ1–42 in SH-SY5Y cells (Figure 5C and 6A). This effect was abrogated by the PPARγ antagonist GW9662 (Figure 5C and 6A), suggesting a role for PPARγ in the amelioration of AGEs-induced APP protein regulation and Aβ1–42 secretion by RGZ.
Figure 5.

Effects of RGZ on the expression of APP and BACE1 in AGEs-treated SH-SY5Y cells. SH-SY5Y cells were treated with 1000 μg/mL AGEs for 24 h, with or without incubation of 10 μmol/L RGZ and 10 μmol/L GW9662. (A) The mRNA expression of APP. (B) The mRNA expression of BACE1. (C) The protein expression of APP. (D) The protein expression of BACE1. Data are shown as mean±SD (n=3 independent experiments). bP<0.05 compared with the control group. eP<0.05 compared with AGEs-treated group. hP<0.05 compared with AGEs+RGZ-treated group.
Figure 6.

Effects of RGZ on the secretion of Aβ1–42 and mRNA as well as protein levels of IDE in AGEs-treated SH-SY5Y cells. SH-SY5Y cells were treated with 1000 μg/mL AGEs for 24 h, with or without incubation of 10 μmol/L RGZ and 10 μmol/L GW9662. (A) The level of Aβ1–42 was measured by ELISA. (B) The mRNA expression of IDE. (C) The protein expression of IDE. Bars show the mean±SD (n=3 independent experiments). bP<0.05, cP<0.01 compared with the control group. eP<0.05, fP<0.01 compared with AGEs-treated group. hP<0.05, iP<0.01 compared with AGEs- and RGZ-treated group.
Effects of RGZ on IDE expression in AGEs-treated SH-SY5Y cells
To confirm the assumption that the attenuation of Aβ secretion by the PPARγ agonist RGZ is mediated by IDE, the mRNA and protein levels of IDE were also determined in SH-SY5Y cells following exposure to RGZ. As shown in Figure 6B and 6C, there were no significant changes in either mRNA or protein levels of IDE in SH-SY5Y cells treated with AGEs alone. However, RGZ treatment resulted in an upregulation of IDE indicated by an increase in its mRNA and protein levels (Figure 6B and 6C). In contrast, PPARγ antagonist GW9662 significantly eliminated the effect of RGZ on the mRNA and protein levels of IDE (Figure 6B and 6C). These results indicate that PPARγ is involved in the regulation of IDE expression by RGZ in AGEs-induced SH-SY5Y cells.
Discussion
Accumulating evidence indicates that AGEs formation and oxidative stress are important pathways leading to neuronal cell death in AD4, 19, 20. The production of AGEs has not been established in vitro. However, a recent study showed that extensively-modified, non-physiological AGEs formed under in vitro conditions may exert the same biological effects as AGEs-rich serum obtained from diabetic patients21. Therefore, AGE-BSA was used in this study.
PPARγ agonists have been proposed as an alternative for the treatment of AD because of their anti-inflammatory and anti-oxidative effect24. In this study, we investigated the protective effect of the PPARγ agonist RGZ on AGEs-induced injury in human neuroblastoma SH-SY5Y cells. SH-SY5Y cells were chosen for their expression of PPARγ, and thus are a valuable cell model to study PPARγ-mediated effects on neuronal cells22. A novel finding of this study is that RGZ at relatively low concentrations protects against AGEs-induced cytotoxicity in SH-SY5Y cells through anti-oxidation and anti-apoptosis effects, as well as reducing Aβ production.
The antioxidant activity of RGZ may contribute to its neuro-protective action23. In the present study, RGZ protected neuronal cells from AGEs-induced injury via its antioxidant activity. Treatment with RGZ at a concentration of 10 μmol/L was capable of reducing AGEs-induced oxidative stress by upregulating the antioxidant enzyme activities of SOD and catalase (Figure 2B and 2C). SOD and CAT promoters contain the PPAR response element25. Thus the PPARγ agonist RGZ may potentially regulate the expression of SOD and CAT. The increase in anti-oxidative capacity by RGZ may contribute to its protection of SH-SY5Y cells against ROS caused by AGEs.
There is now a body of evidence that has demonstrated the efficacy of PPARγ agonists in protecting neuronal cells from apoptosis26, 27. However, how RGZ protects against AGEs-induced apoptosis in neuronal cells is poorly understood. The present study demonstrated that AGEs caused a significant suppression of Bcl2 expression levels which were restored by treatment with RGZ (Figure 4A and 4D). Restoration of Bcl2 expression was abrogated by the PPARγ pharmacological specific inhibitor GW9662, indicating that the anti-apoptotic effect of RGZ is dependent on PPARγ. In addition, the present results indicate that RGZ prevents the increase of Bax and Caspase3 in AGEs-induced SH-SY5Y cells (Figure 4B, 4C, 4E, and 4F), which effects were also blocked by PPARγ antagonist GW9662. Therefore, these findings, taken together, support the notion that RGZ-mediated cytoprotection is due to its inhibition of the oxidative stress and mitochondrial apoptotic pathway in a PPARγ-dependent manner.
PPARγ involvement in ameliorating AD-related pathology has been the focus of studies that have been directed at dissecting the mechanisms through which PPARγ regulates Aβ metabolism28, 29, 30. Sastre and colleagues30 reported that PPARγ agonists modulate the processing of APP through regulation of β-secretase, whereas Camacho and co-workers28 demonstrated that PPARγ did not affect expression or activity of any of the secretases involved in the generation of Aβ, but instead induces a fast, cell-bound clearing mechanism responsible for the removal of the Aβ peptide from the medium. In the present study, RGZ treatment resulted in a dramatic reduction of Aβ secretion, which was accompanied by a reduction of APP at a post-transcriptional level (Figure 5A, 5C, and 6A). Moreover, RGZ did not affect either mRNA or protein levels of BACE1 in this process (Figure 5B and 5D), indicating that RGZ decreases Aβ secretion by promoting its degradation. IDE is a protease that has been demonstrated to play a key role in Aβ degradation31. However, little is known about the cellular regulation of IDE in this process. The present study demonstrates that the PPARγ agonist RGZ regulates the transcription and translation of IDE in neuronal cells (Figure 6B and 6C). Given that RGZ does not affect the expression of BACE1 involved in the generation of Aβ, this indicates that RGZ may regulate Aβ degradation rather than generation in neuronal cells.
In summary, RGZ protects SH-SY5Y cells against AGEs-induced cytotoxicity. Its anti-oxidative and anti-apoptotic as well as anti-inflammatory properties, such as reducing Aβ secretion and enhancing Aβ degradation, render this effective molecule potentially protective against AGEs-induced cytotoxicity. This report may offer a useful strategy for the treatment of progressive neurodegenerative diseases such as AD.
Author contribution
Prof Yi-na ZHANG designed the research; Dr Chun-jiang YU wrote and revised the manuscript; Dr Li WANG performed the experiments; Dr Wei LIU assisted with the experiments; and Dr Lu-yang CHENG assisted with the data statistical analysis.
Acknowledgments
We thank Medjaden Bioscience Limited for assisting in the preparation of this manuscript. This research was supported by Educational Commission Foundation of Heilongjiang Province of China (No 11531080).
References
- Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, et al. AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation Neurobiol Aging 2010. doi: 10.1016/j.neurobiolaging.2010.05.026 [DOI] [PubMed]
- McCall AL. The impact of diabetes on the CNS. Diabetes. 1992;41:557–70. doi: 10.2337/diab.41.5.557. [DOI] [PubMed] [Google Scholar]
- Wang SH, Sun ZL, Guo YJ, Yuan Y, Li L. PPARγ-mediated advanced glycation end products regulation of neural stem cells. Mol Cell Endocrinol. 2009;307:176–84. doi: 10.1016/j.mce.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Munch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, et al. Alzheimer's disease — synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105:439–61. doi: 10.1007/s007020050069. [DOI] [PubMed] [Google Scholar]
- Mruthinti S, Sood A, Humphrey CL, Swamy-Mruthinti S, Buccafusco JJ. The induction of surface beta-amyloid binding proteins and enhanced cytotoxicity in cultured PC-12 and IMR-32 cells by advanced glycation end products. Neuroscience. 2006;142:463–73. doi: 10.1016/j.neuroscience.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Woltjer RL, Maezawa I, Ou JJ, Montine KS, Montine TJ. Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J Alzheimers Dis. 2003;5:467–76. doi: 10.3233/jad-2003-5607. [DOI] [PubMed] [Google Scholar]
- Loske C, Neumann A, Cunningham AM, Nichol K, Schinzel R, Riederer P, et al. Cytotoxicity of advanced glycation endproducts is mediated by oxidative stress. J Neural Transm. 1998;105:1005–15. doi: 10.1007/s007020050108. [DOI] [PubMed] [Google Scholar]
- Gasic-Milenkovic J, Loske C, Deuther-Conrad W, Munch G. Protein “AGEing” — cytotoxicity of a glycated protein increases with its degree of AGE-modification. Z Gerontol Geriatr. 2001;34:457–60. doi: 10.1007/s003910170018. [DOI] [PubMed] [Google Scholar]
- Deuther-Conrad W, Loske C, Schinzel R, Dringen R, Riederer P, Munch G. Advanced glycation endproducts change glutathione redox status in SH-SY5Y human neuroblastoma cells by a hydrogen peroxide dependent mechanism. Neurosci Lett. 2001;312:29–32. doi: 10.1016/s0304-3940(01)02174-7. [DOI] [PubMed] [Google Scholar]
- Wei Y, Chen L, Chen J, Ge L, He RQ. Rapid glycation with D-ribose induces globular amyloid-like aggregations of BSA with high cytotoxicity to SH-SY5Y cells. BMC Cell Biol. 2009;10:10. doi: 10.1186/1471-2121-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla B, Loske C, Garcia De Arriba S, Schinzel R, Huber J, Munch G. Differential effects of “Advanced glycation endproducts” and beta-amyloid peptide on glucose utilization and ATP levels in the neuronal cell line SH-SY5Y. J Neural Transm. 2004;111:427–39. doi: 10.1007/s00702-003-0038-2. [DOI] [PubMed] [Google Scholar]
- de Arriba SG, Stuchbury G, Yarin J, Burnell J, Loske C, Munch G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells — protection by carbonyl scavengers. Neurobiol Aging. 2007;28:1044–50. doi: 10.1016/j.neurobiolaging.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–18. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, et al. Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am J Pathol. 1998;153:1149–55. doi: 10.1016/S0002-9440(10)65659-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest. 1991;87:432–8. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Vlassara H, Kirstein M, Yamada Y, Striker GE, Striker LJ. Receptor-specific increase in extracellular matrix production in mouse mesangial cells by advanced glycosylation end products is mediated via platelet-derived growth factor. Proc Natl Acad Sci U S A. 1992;89:2873–7. doi: 10.1073/pnas.89.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese G, Pricci F, Romeo G, Pugliese F, Mene P, Giannini S, et al. Upregulation of mesangial growth factor and extracellular matrix synthesis by advanced glycation end products via a receptor-mediated mechanism. Diabetes. 1997;46:1881–7. doi: 10.2337/diab.46.11.1881. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Kikuchi S, Sasaki N, Suzuki T, Watai T, Iwaki M, et al. Involvement of advanced glycation end-products (AGEs) in Alzheimer's disease. Curr Alzheimer Res. 2004;1:39–46. doi: 10.2174/1567205043480582. [DOI] [PubMed] [Google Scholar]
- Sato T, Shimogaito N, Wu X, Kikuchi S, Yamagishi S, Takeuchi M. Toxic advanced glycation end products (TAGE) theory in Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2006;21:197–208. doi: 10.1177/1533317506289277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M. Advanced glycation end products inhibit de novo protein synthesis and induce TGF-beta overxepression in proximal tubular cells. Kidney Int. 2003;63:464–73. doi: 10.1046/j.1523-1755.2003.00752.x. [DOI] [PubMed] [Google Scholar]
- Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int. 2009;55:496–504. doi: 10.1016/j.neuint.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci. 2007;253:53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics. 2008;5:481–9. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistrosch F, Passauer J, Fischer S, Fuecker K, Hanefeld M, Gross P. In type 2 diabetes, rosiglitazone therapy for insulin resistance ameliorates endothelial dysfunction independent of glucose control. Diabetes Care. 2004;27:484–90. doi: 10.2337/diacare.27.2.484. [DOI] [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, et al. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–15. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. J Biol Chem. 2002;277:31781–8. doi: 10.1074/jbc.M204279200. [DOI] [PubMed] [Google Scholar]
- Camacho IE, Serneels L, Spittaels K, Merchiers P, Dominguez D, De Strooper B. Peroxisome-proliferator-activated receptor gamma induces a clearance mechanism for the amyloid-beta peptide. J Neurosci. 2004;24:10908–17. doi: 10.1523/JNEUROSCI.3987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Abramo C, Massone S, Zingg JM, Pizzuti A, Marambaud P, Dalla Piccola B, et al. Role of peroxisome proliferator-activated receptor gamma in amyloid precursor protein processing and amyloid beta-mediated cell death. Biochem J. 2005;391:693–8. doi: 10.1042/BJ20050560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003;162:313–9. doi: 10.1016/s0002-9440(10)63822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]