

Abstract
The most abundantly produced virion protein in human cytomegalovirus (HCMV) is the immunodominant phosphoprotein 65 (pp65), which is frequently included in CMV vaccines. Although it is nonessential for in vitro CMV growth, pp65 displays immunomodulatory functions that support a potential role in primary and/or persistent infection. To determine the contribution of pp65 to CMV infection and immunity, we generated a rhesus CMV lacking both pp65 orthologs (RhCMVΔpp65ab). While deletion of pp65ab slightly reduced growth in vitro and increased defective particle formation, the protein composition of secreted virions was largely unchanged. Interestingly, pp65 was not required for primary and persistent infection in animals. Immune responses induced by RhCMVΔpp65ab did not prevent reinfection with rhesus CMV; however, reinfection with RhCMVΔUS2-11, which lacks viral-encoded MHC-I antigen presentation inhibitors, was prevented. Unexpectedly, induction of pp65b-specific T cells alone did not protect against RhCMVΔUS2-11 challenge, suggesting that T cells targeting multiple CMV antigens are required for protection. However, pp65-specific immunity was crucial for controlling viral dissemination during primary infection, as indicated by the marked increase of RhCMVΔpp65ab genome copies in CMV-naive, but not CMV-immune, animals. Our data provide rationale for inclusion of pp65 into CMV vaccines but also demonstrate that pp65-induced T cell responses alone do not recapitulate the protective effect of natural infection.
Introduction
Human cytomegalovirus (HCMV) persistently infects most of humanity (1). While the vast majority of these infections are asymptomatic and not associated with any pathologic consequence, HCMV can cause serious disease in the setting of immune deficiency or immaturity, including late-stage AIDS, iatrogenic immune suppression (particularly, organ and stem cell transplantation), and fetal infection (where infection can cause hearing loss and mental retardation) (2, 3). In maternal-to-fetal transmission and, to a certain extent, with transplantation, the most serious disease appears to arise in the setting of primary HCMV infection (3–5). Therefore, vaccination has been proposed as potential intervention to ameliorate these poor outcomes (6). Although it was initially thought that an effective HCMV vaccine might prevent acquisition of HCMV altogether, accumulating data indicate that even the potent natural immunity elicited by persistent HCMV infection of healthy subjects is, at best, only partially protective against superinfection (7). Thus, conceptually, the most realistic goal of an HCMV vaccine would be to establish a similar level of immunity as present in typical HCMV+ individuals, in HCMV– females prior to pregnancy, or all HCMV– subjects prior to transplantation with HCMV+ cells or tissue, so as to prevent the potentially severe consequences of primary infection in these subjects. Indeed, due to the importance of HCMV in causing congenital disease and complications in transplant recipients, vaccine development efforts have been given high priority by the Institute of Medicine of the National Academy of Sciences (8).
While initial approaches to CMV vaccines focused on the development of an attenuated strain of HCMV (Towne) (9, 10), more recently, the focus has shifted toward the development of subunit vaccines (11), either single antigen vaccines (12) or cocktails of antibody-inducing and T cell–inducing subunits (13). A frequently used T cell–inducing subunit in the development of CMV vaccines is the phosphoprotein 65 (pp65), which is consistently a major target for the T cell response in infected individuals (14–17). HCMV pp65 is part of the viral tegument and the most abundant virion protein (18). Multiple functions in modulation of innate and intrinsic immunity (19–21) as well as adaptive immune responses (22–24) have been assigned to HCMV pp65. Moreover, pp65 has been shown to modulate the activity of serine/threonine kinases (25–27), Polo-like kinase 1 (28), and the viral UL97 serine/threonine kinase (29). Nevertheless, pp65 is dispensable for viral replication in HCMV-infected fibroblasts (30), but pp65-deleted HCMV showed decreased virus production in monocyte-derived macrophages (31).
The role of pp65 for the establishment and maintenance of persistent infection by HCMV is unknown, due to the strict species specificity of HCMV that does not infect immunocompetent experimental animals. Thus, animal CMVs are generally used as models to study CMV and CMV vaccines (32–34). Since host restriction resulted in coevolution of CMVs with their respective hosts, infection of rhesus macaques (RMs) with rhesus CMV (RhCMV) represents an animal model that closely resembles infection of humans with HCMV (35). We therefore used this model to study the role of RhCMV pp65 in infection and immunity. RhCMV encodes 2 ORFs, Rh111 and Rh112, with comparable homology to HCMV pp65 (pp65a ~34%, pp65b ~40%) and 40% identity to each other (36, 37). The 2 proteins combined comprise approximately 11% of the entire viral proteome in RhCMV virions (38), which is similar to HCMV, in which the single pp65 protein makes up 15% of the virion proteins (18). To examine the function of pp65 in vitro and in vivo, we deleted both pp65 homologs from the genome of RhCMV. We characterized the impact of pp65 deletion on viral growth in vitro and on the composition of the virion proteome. We then determined the role of pp65 for the ability of RhCMV to establish primary or secondary persistent infection in RhCMV+ or RhCMV– animals, respectively. By challenging with recombinant RhCMV lacking the immunoevasins US2, 3, 6, and 11, a virus incapable of superinfecting, we further evaluated whether pp65-specific T cells are required for the protective effect of preexisting CMV infection or sufficient to recapitulate T cell–mediated protection induced by natural infection. Our observations demonstrate a unique physiologic role for pp65 in CMV biology and, moreover, have implications for the use of pp65 as a subunit vaccine.
Results
RhCMVΔpp65ab shows delayed growth kinetics at low MOI.
To study the function of pp65 in RMs, we deleted the pp65a- and pp65b-encoding genes Rh111 and Rh112 in the RhCMV strain 68-1–derived BAC (39) to generate Δpp65ab. Upon reconstitution of recombinant virus in telomerized rhesus fibroblasts (TRFs), we verified that genes Rh111 and Rh112 were absent, whereas the neighboring genes Rh110 (UL82 [pp71] homolog) and Rh114 (UL84 homolog) were still expressed (Figure 1A).
Figure 1. Characterization of RhCMVΔpp65ab in vitro.

(A) RT-PCR results for cDNA of infected TRFs showing the expression of pp65a (Rh111) and pp65b (Rh112) and their neighboring ORFs Rh110 (pp71) and Rh114. IE1 and GAPDH were included as controls. (B) Single-step (MOI of 3) and (C) multistep (MOI of 0.01) growth curves of RhCMV WT and RhCMVΔpp65ab on TRFs. *P < 0.05, Student’s t test; **P < 0.01, Student’s t test.
To determine whether pp65 deletion affected in vitro growth properties of RhCMV, we compared the growth kinetics of Δpp65ab with those of BAC-derived RhCMV 68-1 (herein referred to as WT control). TRFs were infected with Δpp65ab or WT either at a high MOI of 3 to generate a single-step growth curve or at a low MOI of 0.01 to measure multistep growth. Supernatants collected at high MOI contained similar titers of the 2 viruses, with a peak titer reached on and after 4 days postinfection (dpi) (Figure 1B). However, when multiple rounds of infection were measured, Δpp65ab displayed a modest, but significant, delay in viral growth, ultimately reaching the same peak titer as WT (Figure 1C).
Characterization of Δpp65ab virions.
Since previous reports suggested that pp65 in HCMV affected viral assembly and thus the incorporation of other viral proteins (31), we studied the structure and protein composition of Δpp65ab virions. Using mass spectrometry, we demonstrated recently a remarkable similarity between the predominant viral proteins found in both RhCMV and HCMV, with respect to protein ratios and protein abundance (38). To similarly determine the proteome of Δpp65ab virions, we concentrated viral particles from the supernatant of infected TRFs followed by purification over a discontinuous Nycodenz gradient (see Methods). Compared with WT, we observed an increased appearance of particles that sedimented with higher density in virion preparation of Δpp65ab (Figure 2A). Electron microscopy of this high-density band revealed abnormal structures consistent with capsidless (defective) viral particles. However, the lower density virion band contained an essentially pure preparation of particles with the same general structure as WT RhCMV, including an icosahedral capsid containing the viral DNA as the core of the virion surrounded by a tegument layer and enveloped by a lipid membrane (Figure 2B). NuPAGE and Western blot analysis of gradient-purified WT and Δpp65ab mutant virions demonstrated the absence of pp65a and pp65b in the deletion mutant (Figure 2, C and D). Comparison of the dimensions of the WT versus Δpp65ab virions revealed an overall reduced diameter of Δpp65ab virions (173.4 nm) compared with that of WT virions (222.5 nm) (Figure 2E). This reduction in particle size was primarily due to a significant reduction of the viral tegument layer (38.4 nm [Δpp65ab] compared with 61.2 nm [WT]), consistent with the fact that pp65a and pp65b constitute a major portion of the viral tegument in WT (approximately 24.4%; ref. 38). In addition, the capsid appeared to be diminished in size, although to a lesser degree (89.7 nm [Δpp65ab] compared with 100.2 nm [WT]). Thus, both phenotypically normal, but smaller, virions and defective particles were recovered from the supernatant of cells infected with Δpp65ab.
Figure 2. Intact and defective viral particles are secreted from fibroblasts infected with Δpp65ab.

(A) Image of a Nycodenz gradient loaded with RhCMVΔpp65ab, and electron microscope images of virions (top image) and defective particles (bottom image) contained in the visible bands of the gradient. (B) Electron microscope image of purified RhCMVΔpp65ab virions showing the purity of the sample. (C) Purified RhCMV WT and Δpp65ab virions were lysed, and 10 μg protein was electrophoretically separated using NuPAGE MOPS gradient gels and visualized by Coomassie blue staining. (D) Western blots of 5 μg gradient-purified RhCMV 68-1 WT and viral mutant Δpp65ab stained for RhCMV pp65a, pp65b, or a RhCMV-specific antibody. (E) Various electron microscopy images of purified WT and Δpp65ab virions were taken, and the diameters of virions, capsids, and the tegument were determined in multiple images and magnifications (WT, n = 39; Δpp65ab, n = 45). The mean diameters with their respective SDs are shown, and Student’s t tests were performed to determine the P values. Scale bars: 100 nm.
To further characterize the proteome of the virions contained in the upper band by mass spectrometry, the recovered material was digested with trypsin and analyzed by 1D LC-MS/MS. The resulting mass spectra were initially searched against stop-to-stop translated sequences of the RhCMV 68-1 BAC genome for ORFs ≥30 amino acids. Since all proteins identified by this method corresponded to proteins contained in our recently published reannotation of the RhCMV 68-1 BAC genome (38), we also used a protein library based on the revised annotation for further analysis of protein abundance. The virion preparation was analyzed by LC-MS/MS upon elution from 0% to 100% acetonitrile gradient over 100 minutes, and 5 technical repeats were performed. 68.5 mol% of all identified proteins and 69.9 mol% of all identified peptides corresponded to viral proteins, whereas 31.5 mol% of the proteins and 30.1% of the peptides were derived from the host (Supplemental Figure 1B; supplemental material available online with this article; doi: 10.1172/JCI67420DS1). This result is similar to that previously obtained for WT (64.8 mol% viral proteins, 63.4% viral peptides and 35.2 mol% host proteins, 36.6% host peptides), suggesting a similar level of sample purity. As expected, peptides corresponding to pp65a or pp65b were not detected in the Δpp65ab virion preparation (Table 1). However, a total of 50 different viral proteins could be identified for Δpp65ab, which is comparable to that for WT virions, for which 53 different viral proteins were identified. Every capsid protein found in WT virions was found in the deletion mutant, and besides the deleted pp65 proteins, this also holds true for the tegument proteins (Table 1). Similarly, all major glycoproteins were present in Δpp65ab in equal abundance compared to WT. In fact, most proteins that differed in their abundance between Δpp65ab virions compared with WT were low-abundance proteins, suggesting that these proteins might not be consistently part of the virions or that they were missed in our analysis due to low abundance. If an abundance threshold of 0.25 mol% is applied, 8 proteins differ between Δpp65ab and WT: Rh17 (RL11 family), Rh131 (UL96), Rh211 (US26), and Rh214 (US28) are decreased in the Δpp65ab mutant compared with the WT, whereas Rh05 (RL11 family), Rh13.1 (RL13), Rh173 (RL11 family), and Rh218 (US28) were increased in the Δpp65ab mutant compared with the WT (Figure 3). Of those, Rh211 is the only protein with a substantial presence with 0.85 mol% in WT virions that is completely absent in mutant virions. Rh211 is the homolog of HCMV US26, whose function is unknown. Thus, our proteomics analysis revealed that only 8 proteins with a higher abundance than 0.25 mol% showed marked changes of more than 2-fold between the WT and the pp65ab deletion mutant, with most of these being low-abundance proteins.
Table 1.
Comparison of viral proteins contained in WT and Δpp65ab virions
Figure 3. Δpp65ab establishes primary and secondary infections and protects against superinfection with ΔUS2-11.

(i) Two RhCMV seronegative male RMs (filled circles, Rh22037; open circles, Rh23016) were infected s.c. with 107 PFUs of Δpp65ab at day 1. CD4+ (blue) and CD8+ (red) T cell responses were monitored in peripheral blood (PBMCs) by intracellular cytokine staining at the indicated days using overlapping peptides of pp65ab and IE1/2. (ii) On day 659, the 2 animals were inoculated s.c. with 107 PFUs of ΔUS2-11gag (green dotted line), and the T cell response to SIVgag was measured in addition. Note the absence of a T cell response to SIVgag or pp65 and a lack of boosting of responses to IE1. (iii) On day 876, the 2 RMs were inoculated with 107 PFUs of WTgag (black dotted line), and the T cell response was monitored by intracellular cytokine staining. Note the appearance of de novo responses to SIVgag and pp65 and a boosting of the T cell response to IE1. (iv) On day 1,107, the 2 RMs were inoculated with 107 PFUs of Δpp65ab-rtn (blue dotted line). Using overlapping 15-mer peptides, a de novo response to SIVretanef was detectable, indicating superinfection. Also note a boosting of the IE1 response but not of pp65- or SIVgag-specific responses. The corresponding T cell responses obtained from BAL fluid are shown in Supplemental Figure 2.
In general, there was surprisingly little impact of pp65ab deletion on the presence of other proteins in the virions. There was no substantial difference among nonstructural proteins (transcription/replication machinery or uncharacterized category), whereas the quantities of most structural proteins (capsid, envelope, glycoproteins) were, in fact, slightly elevated in Δpp65ab compared with those in WT (Table 2). Moreover, we did not observe a decrease in specific, non-pp65 tegument proteins, but rather, we saw a decrease in the abundance of all non-pp65 tegument proteins in Δpp65ab virions. This is in contrast to a previous report for HCMV, describing selective lack of specific tegument proteins in pp65-deleted virions (31). Thus, it seems that RhCMV virions assembled normally but with an overall reduced tegument. Indeed, when virion protein abundance is adjusted for the absence of pp65ab by normalizing to a total of 89% (11% of the WT virion is made up by pp65a and pp65b combined), protein quantities are very similar to those of WT (Tables 1 and 2). Despite the lack of major tegument proteins that normally represent 11% of the viral particle mass, there was little change in virion composition.
Table 2.
Relative abundance of the 5 functionally different groups of viral proteins in RhCMV WT and Δpp65ab
In contrast to the limited impact of pp65ab deletion on virion proteins, a number of host proteins were substantially different between WT and Δpp65ab (Supplemental Figure 1). 279 host proteins were identified in WT, whereas 240 host proteins were identified in Δpp65ab. Only 172 host proteins were identified in both viral samples, with the remaining proteins being unique to each sample. The role of host cell proteins in CMV virions is unknown, but it seems likely that these proteins reflect the source or host cell membrane used for envelopment. The differential presence of host cell proteins could thus indicate that envelopment of pp65ab-deleted viruses differs somewhat from that of WT virus. This would be consistent with the increased production of defective particles described above. The differential incorporation of host cell proteins likely reflects changes in viral assembly pathways but is less likely of consequence for virion function.
Infection of RMs with Δpp65ab.
To determine whether pp65ab-deleted viruses would be infectious, we inoculated 2 seronegative male RMs with 107 PFUs of Δpp65ab and monitored the CMV-specific T cell response using overlapping peptides to the RhCMV proteins IE1/2 and, as control, to pp65ab for about 22 months. We also monitored viral shedding by coculture of urine samples with rhesus fibroblasts. In previous experiments, we showed that infection of RhCMV-negative RMs results in the appearance of peak T cell responses within the first 2 weeks of infection, followed by a contraction and stabilization of the T cell response at a level that remains more or less constant for the duration of the life of the animal (40, 41). The maintenance of such a long-lived effector memory T cell response reflects the establishment of persistent infection. Similarly, both animals infected with Δpp65ab responded vigorously to IE1/2, with a peak CD4+ and CD8+ T cell response in PBMCs and bronchoalveolar lavage (BAL) fluid at 2 weeks, followed by a slow decline and stabilization of the response that lasted for the entire time (Figure 3, i, and Supplemental Figure 2A). Importantly, T cell responses to pp65ab were not observed, which is consistent with the IE1/2 responses being induced by the pp65ab-deleted virus. The stable T cell response to IE1/2 suggested that the pp65-deleted virus established persistent infection. Persistence was further confirmed by coculture of urine samples with TRFs, in which IE1 was detected in urine cocultures of Δpp65ab-infected animals but pp65 was not detected, confirming that there was no contamination with WT virus (Figure 4). Thus, these data suggest that RhCMV is able to establish and maintain a persistent infection despite the absence of pp65ab.
Figure 4. RhCMVΔpp65ab is persistently secreted from infected animals.

(A) The time line depicts the time points of inoculation with different RhCMV constructs and the days when cocultures were started from urine. Time points marked with asterisks indicate additional days in which cocultures were positive for Δpp65ab, but the data are not shown. PID, postinoculation day. (B) Immunoblot for the indicated antigens in lysates from representative viral cocultures with urine collected on the indicated dpi. The presence of RhCMV-IE1, RhCMV-pp65b, SIVgag, and SIVretanef in cell lysates was detected by immunoblot using antibodies specific for the respective antigens (IE, pp65) or for epitope tags fused to SIVgag or SIVretanef. Note that, initially, secreted RhCMV expressed IE, but not pp65, whereas superinfection with WTgag and Δpp65retanef is indicated by the appearance of pp65-containing virus expressing the respective antigens. As positive control (Con), coculture lysates from a RM inoculated with WTgag and WTretanef is included.
Although pp65 is one of the major targets of the CMV-specific T cell response in both humans and monkeys (14–17, 37), the contribution of pp65-specific T cells to control of CMV replication is not known. Indeed, the experimental determination of the efficacy of RhCMV-specific T cell responses is complicated by the fact that RhCMV readily superinfects RhCMV+ RMs, overcoming preexisting T cell responses due to the presence of viral proteins that inhibit MHC-I antigen presentation (42). However, RhCMV lacking the genes encoding for homologs of the HCMV US2, 3, 6, and 11 immunoevasins is unable to superinfect CMV+ RMs but is capable of establishing persistent infection in CMV-naive animals or upon depletion of CD8+ T cells from CMV-immune animals (42). Thus, the ability to protect against superinfection with ΔUS2-11 RhCMV is a convenient surrogate marker for the quality of T cell responses, i.e., a T cell response that is as efficient as that induced by natural infection. To test whether the T cell responses induced by Δpp65ab to antigens other than pp65 would be sufficient to prevent superinfection with immunoevasins-deleted virus, we inoculated the 2 Δpp65ab-infected RMs with ΔVIHCEΔUS2-11gag, a previously described recombinant virus that expresses the SIVgag as immunological marker. In addition to US2-11, this virus lacks the RhCMV-specific viral inhibitor of heavy chain expression (VIHCE), which is not required for superinfection (42). As observed for RMs naturally infected with RhCMV, ΔVIHCEΔUS2-11 was unable to superinfect Δpp65ab-infected animals, as evident from the absence of an immune response to SIVgag as well as a lack of a boosting response to IE or a de novo response to pp65 (Figure 3, ii, and Supplemental Figure 2B). In fact, T cell responses to IE1/2 remained stable for the entire duration of this experiment (200 days). Thus, the T cell responses generated by Δpp65ab were as efficacious as T cell responses induced by WT in protecting against immunoevasins-deleted virus challenge, indicating that a pp65-specific T cell response is not required for an effective anti-RhCMV immune response and that T cells specific for other codominant or subdominant antigens are sufficient for protection.
To determine whether animals infected with Δpp65ab are resistant to superinfection by WT RhCMV, we inoculated both RMs with WT-gag, a previously described virus that carries SIVgag inserted into the ORF Rh211 between hypothetical ORFs 213 and 214 (41). Upon inoculation of 107 PFUS WT-gag, both animals displayed clear signs of superinfection, as evident by the development of de novo responses to SIVgag and pp65ab and by boosting of the preexisting T cell response to IE1/2 (Figure 3, iii, and Supplemental Figure 2C). Moreover, cocultures of urine samples from these animals contained SIVgag-expressing virus (Figure 4). These data thus demonstrate that the immune responses induced by Δpp65ab, like those elicited by WT RhCMV, are unable to protect against superinfection with WT RhCMV.
Given the role of HCMV pp65 as modulator of several immune response pathways (including protecting against IE-specific T cells [23] and NK cells [22]), it was possible that pp65 itself contributed to the ability of WT to overcome preexisting immune responses. In fact, our previous finding that evasion of T cell responses plays a central role in overcoming preexisting immune responses does not rule out that evasion of other immune response components, e.g., B cells and NK cells, might also contribute to superinfection (42). To examine whether RhCMV lacking pp65ab would be able to superinfect CMV-positive animals, we inserted an expression cassette for SIVretanef (a fusion protein of rev, int, tat, and nef; refs. 41, 43) into the RhCMV genome by replacing the pp65-encoding genes Rh111 and Rh112. After confirming pp65 deletion, in vitro growth properties, and expression of SIVretanef (data not shown), we inoculated the 2 RMs previously infected with Δpp65ab and WT-gag with Δpp65ab-retanef and monitored the immune response to SIVretanef. As shown in Figure 3, iv (Supplemental Figure 2D), both animals showed clear signs of superinfection, as evident from the development of a de novo T cell response to SIVretanef and a boosting of the preexisting IE1/2 response. Note that the T cell responses to pp65ab and SIVgag were not boosted, confirming the lack of pp65ab and SIVgag. We thus conclude that pp65ab is dispensable for the establishment of both primary and secondary persistent infections.
Vaccine-induced pp65-specific T cells do not recapitulate the protective effect of T cells induced by natural infection.
In the RM model, it was previously demonstrated that vaccination with subunit vaccines consisting of pp65b (with or without IE1) as T cell–inducing components and gB as neutralizing antibody-inducing component reduced RhCMV viremia and shedding (44–46). However, our data also suggest that T cell responses to antigens other than pp65 play an important role in the protective effect of RhCMV infection against ΔUS2-11 challenge. We were therefore wondering whether induction of a T cell response to pp65 alone would be sufficient to recapitulate the protective effect of preexisting infections against challenge with RhCMV lacking the US2-11 immunoevasins (42). Therefore, we used a previously described heterologous prime-boost regimen to induce pp65b-specific T cell responses (45, 46). Three animals were vaccinated with DNA encoding pp65b, followed by 2 boosts with pp65b-expressing modified vaccinia Ankara (MVA). For control, we vaccinated 3 animals with antigen-free plasmid and MVA. As shown in Figure 5A (Supplemental Figure 3), all 3 pp65b-vaccinated animals developed a robust CD4+ and CD8+ T cell response to pp65b after this prime-boost vaccination regimen that, in the 6 weeks following the final boost, was similar in magnitude and phenotype to pp65-specific T cell responses that develop in the context of RhCMV infection (Figure 3, iii and iv, and Supplemental Figure 4). As expected, pp65b-specific T cells were not observed in the control MVA-vaccinated group. Six weeks after the final MVA/pp65 versus control MVA boost, all animals were challenged with RhCMV lacking US2-11 and expressing SIVgag (ΔUS2-11gag). Similar to ΔVIHCEΔUS2-11gag, this virus is unable to overcome preexisting T cell immunity, despite the presence of the RhCMV-specific MHC-I inhibitor VIHCE (42). All 3 control-vaccinated animals developed the expected T cell response to pp65 as well as SIVgag consistent with infection. However, the pp65-vaccinated animals also developed T cell responses to SIVgag with similar kinetics and magnitude compared with those in the control group. We further observed a boost of the T cell response to pp65b consistent with infection by ΔUS2-11gag. These data indicate that pp65-specific T cells alone are unable to provide the level of protection against CMV afforded by natural infection or experimental infection with whole virus.
Figure 5. T cells induced by heterologous prime-boost vaccination with pp65b do not protect against superinfection with ΔUS2-11.

(A) Three CMV-negative RMs were vaccinated with 1 mg pND/pp65b and boosted with 5 × 108 PFUs MVApp65b at 6 and 12 weeks after the initial vaccination (black). As controls, 3 CMV-negative RMs were vaccinated with the parental pND plasmid not expressing any antigen and boosted with WT MVA at 6 and 12 weeks after the initial vaccination (green). At 18 weeks after the initial DNA vaccination, both groups of animals were challenged with 107 PFUs ΔUS2-11gag. The top row shows the specific T cell responses to pp65, whereas the bottom row shows specific T cell responses to SIVgag. T cells were isolated from peripheral blood (PBMCs). The corresponding T cell responses obtained from BAL fluid are shown in Supplemental Figure 3. The production of anti-RhCMV antibodies in pp65-vaccinated animals (Rm23557, Rm27814, Rm27838) was compared to that in control-vaccinated animals (Rm23672, Rm25052, Rm27821) prior to and upon challenge with RhCMV-ΔUS2-11. At the indicated time points, RhCMV-specific end point antibody (IgG, IgA, IgM) titers were measured in plasma from each animal by ELISA using lysates from fibroblasts infected with either (B) RhCMV-Δpp65 or (C) WT-RhCMV as the capture antigen. (D) Viral proteins recognized by the antibodies were detected by Western blotting. Lysates of cells infected with WT-RhCMV or RhCMV-Δpp65 were separated by SDS-PAGE and immunoblotted with antisera from the pp65-vaccinated animal (Rm27838) or a control-vaccinated animal (Rm23672). Asterisks denote the pp65 proteins. The results from these 2 animals are representative of the responses observed in the other animals of each group.
Conceivably, pp65 vaccination could affect the viral loads of ΔUS2-11gag, as has been reported for pp65/gB-vaccinated animals challenged with RhCMV (44). Since primary infection with RhCMV 68-1 (the parental strain of ΔUS2-11gag) does not result in robust viremia (see below), direct measurements of viral loads were unlikely to be informative. Instead, we used the development of anti-CMV antibodies as a surrogate for CMV antigen load, because it was shown previously in the murine model that reduced viral spread correlates with reduced antibody responses but does not affect T cell responses (47). However, when lysates of Δpp65-infected cells were used as antigen, a very modest antibody response was observed in all animals challenged with ΔUS2-11gag, and there was no difference in the kinetics or magnitude specificity of this response between pp65-vaccinated and control-vaccinated animals (Figure 5B), and we did not observe a difference in the specific antigens recognized by immunoblot (Figure 5D). Remarkably, when pp65-containing CMV lysate was used as antigen for our ELISA or immunoblot, we observed an extraordinary increase in the titers of pp65-specific antibodies induced by DNA/MVA vaccination. As shown in Figure 5B, pp65-specific antibodies were above background levels upon boosting with MVApp65. Moreover, subsequent challenge with ΔUS2-11gag increased these pp65-specific antibodies by several orders of magnitude. pp65 antigen was recognized in sera from pp65-vaccinated animals by immunoblot, and this response was strongly increased upon challenge. In contrast, control-vaccinated animals did not recognize the corresponding 65-kDa band even after ΔUS2-11 challenge (Figure 5D). These data indicate that pp65 vaccination did not affect ΔUS2-11 viral load to a level that would affect the induction of CMV-specific antibodies, although a modest reduction, as reported previously, cannot be ruled out. In addition, these observations suggest that antibody responses to pp65, and potentially to other antigens as well, are substantially boosted upon infection with ΔUS2-11 virus.
pp65 limits dissemination of RhCMV in vivo.
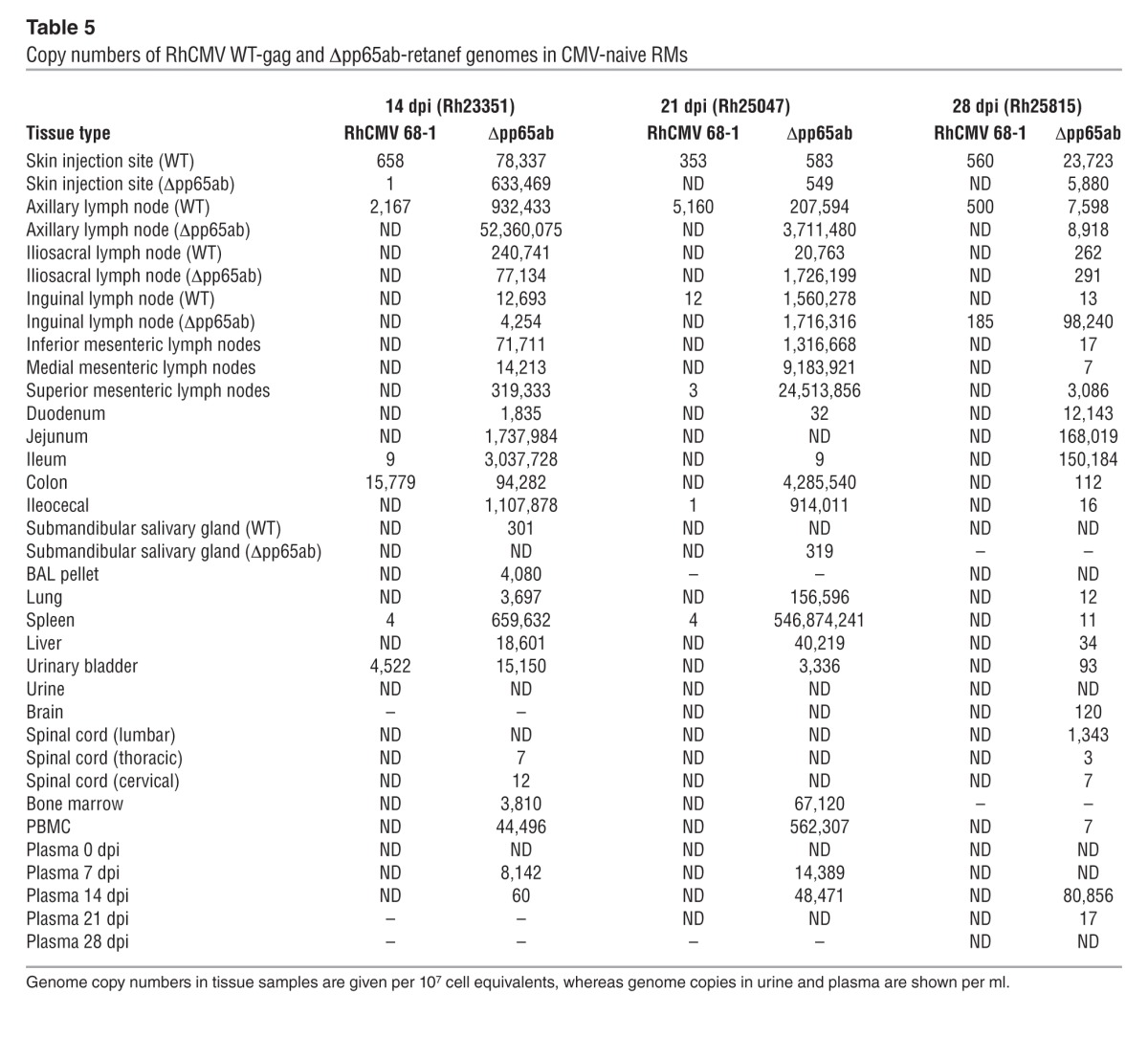
Taken together, these data suggest that neither pp65 itself nor the T cell response to pp65 have a major impact on the overall course of either primary or secondary RhCMV infection. However, it remained possible that the extent of RhCMV replication is affected by the presence or absence of pp65. If lack of pp65 delays RhCMV replication kinetics in vivo, as it does in vitro, infections with pp65-deleted RhCMV would be expected to proceed more slowly and/or manifest reduced spread or peak viral production relative to WT virus. On the other hand, if pp65-induced immune responses have superior efficacy, infections with pp65-deleted RhCMV would manifest greater viral replication and spread than WT RhCMV. To address these possibilities, we performed experiments in which genetically marked WT and Δpp65ab RhCMV constructs (using SIVgag and SIVretanef as the identifying markers, respectively) were simultaneously, but separately (right arm vs. left arm), inoculated into either RhCMV seropositive (n = 2) or RhCMV seronegative RMs (n = 3). These RMs were sacrificed and taken to necropsy at 14, 21, or 28 days after inoculation; DNA was isolated from the sites of inoculation and distant tissues; and the extent and magnitude of viral spread was determined by an ultrasensitive, nested quantitative PCR analysis using primers that specifically amplify fragments of the SIVgag versus SIVretanef inserts (48). In the setting of superinfection, little tissue-associated viral DNA was detected for either the WT or Δpp65ab constructs, with the former only identified in one of the inoculation sites in the RM analyzed at day 14 and the latter identified at very low level in inoculation sites and scattered distant tissues in both the RMs analyzed at day 14 and day 28 after inoculation (Table 3). Although, in these 2 RMs, the extent of spread by the Δpp65ab RhCMV was greater than that of the coadministered WT RhCMV, the level of tissue-associated virus observed in the Δpp65ab RhCMV superinfection was still within the range of that found in WT RhCMV superinfection in other RMs (Table 4), and thus, deletion of pp65 does not seem to significantly affect viral dissemination during superinfection. In striking contrast, the extent of Δpp65ab RhCMV replication in RhCMV-naive RMs was 3 to 4 logs higher than the simultaneously administered WT RhCMV (Table 5). Indeed, the degree of Δpp65ab RhCMV replication during primary infection was astonishing, reaching almost 108 DNA copies in the LN draining the injection site at day 14 after inoculation. Analyses of viral loads in blood indicate that the difference in WT versus Δpp65ab RhCMV replication by can be observed by day 7 after inoculation (Table 5). The levels of Δpp65ab RhCMV in blood and tissue declined dramatically at later time points, indicating that this virus is eventually brought under immune control. Taken together, these data unequivocally indicate that expression of pp65 strongly limits primary viral dissemination over several orders of magnitude and suggest that, while RhCMV-specific T cells can control RhCMV in the absence of pp65, a rapid immune response to pp65 is necessary to limit viral spread during the early days of primary infection.
Table 3.
Copy numbers of RhCMV WT-gag and Δpp65ab-retanef genomes in CMV+ RMs
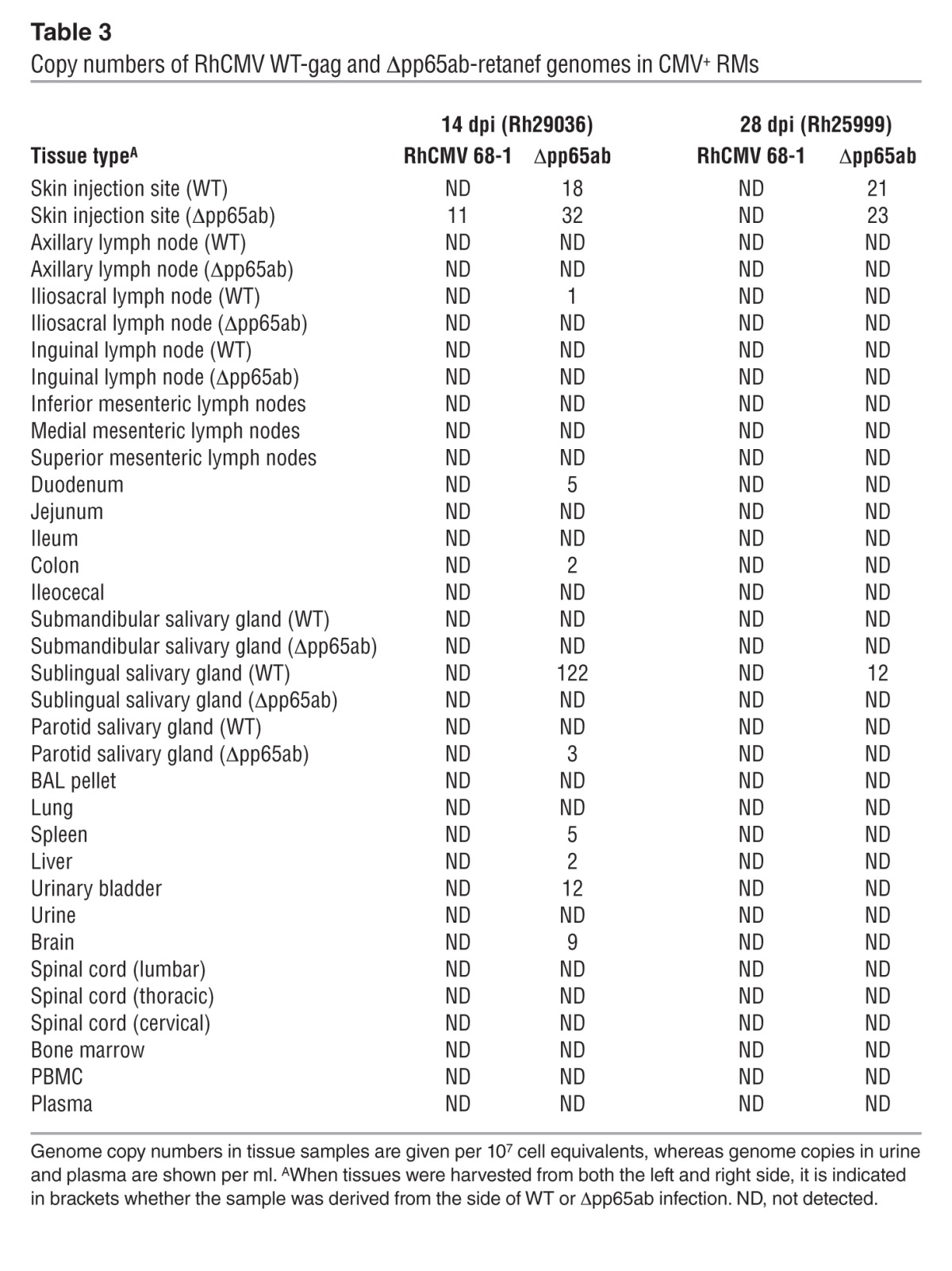
Table 4.
Genome copies of RhCMV 68-1 WT-gag in CMV+ RMs
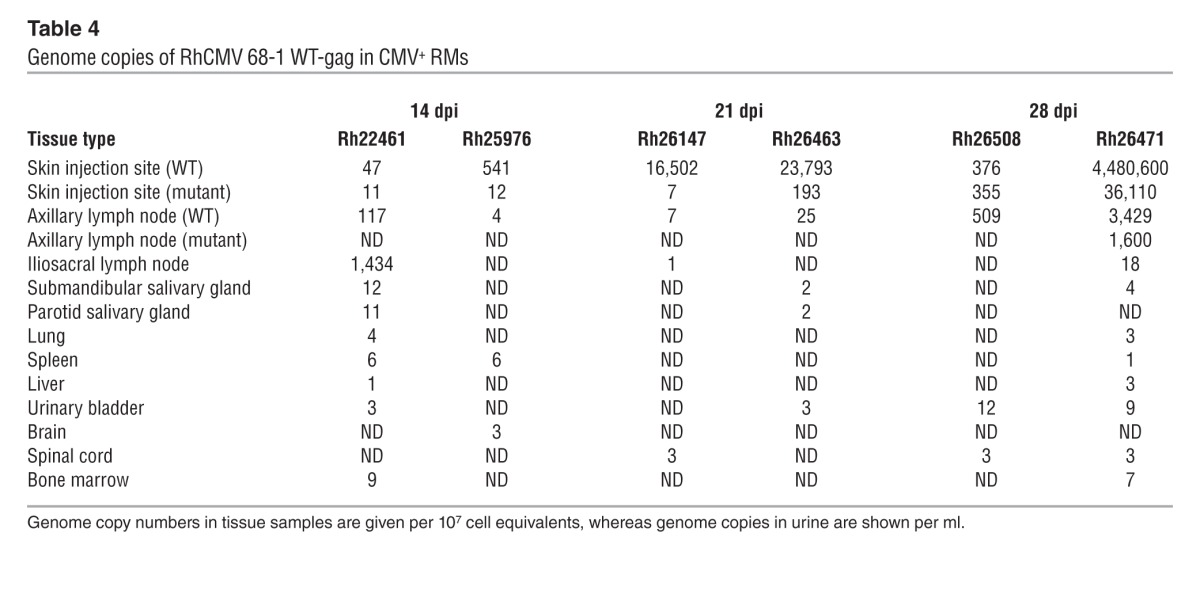
Table 5.
Copy numbers of RhCMV WT-gag and Δpp65ab-retanef genomes in CMV-naive RMs
Discussion
The goal of this study was to assess the role of one of the major structural components of the CMV virion in infection and immunity. Similar to HCMV, a large percentage (approximately 11%) of the protein mass of RhCMV virions consists of the 2 pp65 homologs (38). Nevertheless, in both HCMV and RhCMV, pp65 is not essential for growth in vitro although increased production of defective particles occurs during infection with RhCMVΔpp65ab. This could be reflective of assembly defects due to the lack of pp65ab. For HCMV, it has been reported that pp65 is required for the incorporation of other virion proteins, most notably UL25, UL69, and UL97 (31). However, we did not observe a major skewing in the protein composition of the viral tegument as would have been expected if pp65 selectively controls the incorporation of other viral proteins. Instead, the tegument composition seemed normal but without pp65 present. Conceivably, this could be due to a difference in virion assembly between HCMV and RhCMV. However, the overall virion proteome of RhCMV is highly similar to that of HCMV (38), and UL25, UL69, UL97 are highly conserved in RhCMV. Therefore, it seems more likely that effects of pp65 on incorporation of other tegument proteins are nonselective. The fact that virions are assembled, carrying the same ratios of viral proteins as WT, while lacking pp65, suggests that although pp65 might facilitate virus assembly, once the virus is assembled, the lack of pp65 does not affect the overall viral structure, except for a reduction on overall virion size due to a reduced tegument protein layer.
In addition to viral assembly, pp65 has an immediate function upon release of the tegument into cells during membrane fusion. Similar to other tegument proteins (e.g., pp71 and UL35), pp65 is thought to contribute to setting the stage for optimal viral replication by counteracting intrinsic and innate antiviral host response mechanisms (49). HCMV lacking pp65 showed increased induction of IFN-stimulated genes (ISGs) (19, 20). We reported previously that RhCMV particles inhibited ISG expression (50) and pp65 was a possible candidate for this inhibition. However, Δpp65ab did not induce ISGs, suggesting that inhibitory mechanisms mediated by other RhCMV proteins perform this function (data not shown). It has also been reported that HCMV pp65 binds to and induces the major immediate early promoter (MIEP) in conjunction with the cellular protein IFI16 (21). We have not investigated in detail the impact of pp65 on IE expression in RhCMV. However, in this study, we did observe a delay in virus production in multistep growth curves (Figure 1C), consistent with this effect. Thus, RhCMV pp65 proteins appear to facilitate optimal expression of viral genes in the early stages of cellular infection but are not required for productive infection.
The role of HCMV pp65 for the establishment and maintenance of infection in vivo is unknown due to the strict species specificity of HCMV. The rationale for studying RhCMV pp65 in the RM model was therefore the close evolutionary relationships of both the host to human and of the virus to HCMV. Given the multiple functions assigned to HCMV pp65, it was completely unexpected that deletion of both homologs in RhCMV did not only not affect the ability of RhCMV to establish and maintain a long-term infection in the rhesus host but, in fact, strongly increased the ability of RhCMV to replicate and disseminate during primary infection. In contrast, lack of the pp65 homologous genes M83/M84 and GP83 in murine CMV (MCMV) and guinea pig CMV, respectively, reduced peak viremia during primary infection (51–53). In contrast to primary infection, differences in the replication and dissemination of Δpp65ab versus WT RhCMV were minimal, if not absent, in the setting of superinfection of CMV+ RMs, suggesting that once established, adaptive immune responses to antigens other than pp65 can effectively control the infection. These data suggest that pp65 likely acts as an “immunological brake” during the initial stages of primary infection to limit viral replication and dissemination. We therefore hypothesize that the main function of pp65 is not that of immune evasion, but immune induction, i.e., eliciting a rapid immune response that controls viremia. The most likely candidate mechanism for this effect is the pp65-specific effector T cell response, which, due to the abundance and immunogenicity of pp65 proteins, might appear earlier in primary infection than the response to other CMV proteins. Alternatively or additionally, pp65 might induce innate immune responses that limit viral replication in primary infection. This immune induction function of pp65 is reminiscent of the NK cell–stimulating protein m157 of MCMV, whose deletion or mutation increases viral replication and titers in mice carrying the NK cell receptor Ly49H for which m157 is a ligand (54, 55).
The parental strain used to generate Δpp65ab, RhCMV 68-1, shows reduced secretion from infected animals, most likely due to the lack of genes in the ULb′-homology region required for tissue tropism (56). Since RhCMV 68-1 does not generate robust plasma viremia in infected animals, the appearance of RhCMV-Δpp65ab in plasma samples became particularly striking. Thus, it is conceivable that the increased dissemination of Δpp65ab might be less pronounced in viruses carrying an intact ULb′ region. However, in preliminary observations, we did not observe increased dissemination of RhCMV 68-1.2, a virus that is repaired for tissue tropism (57). Thus, it is likely that pp65 deletion will have a similar effect on a repaired or low-passage viral background, although this still needs to be verified experimentally.
T cells from HCMV-infected individuals recognize a broad spectrum of viral ORFs that are highly variable between individuals (15). Although no ORFs are recognized by all seropositive people, pp65 is one of the most consistently recognized CMV proteins by both CD4+ and CD8+ T cells (15), a level of immunogenicity that has led vaccine developers to include pp65 in HCMV vaccines (11, 58–62). However, relatively little is known about the protective effect of pp65-specific T cells in humans, since vaccine trials generally involve a cocktail of proteins and efficacy cannot be directly correlated to pp65 alone (13). The most direct evidence for a protective effect of pp65-specific T cells comes from adoptive T cell transfer experiments that used pp65-derived peptides to expand HCMV-specific T cells (63–68). In these studies, transfer of pp65-specific T cells accelerated the restoration of antiviral immunity posttransplantation, without graft versus host side effects associated with nonspecific T cell transfer. Our finding that pp65-specific immunity seems to curtail viral dissemination in the early stages of infection would support the inclusion of pp65 in subunit vaccines, provided it is indeed the pp65-specific T cells that are responsible for this effect.
On the other hand, our data also indicate that pp65-specific T cell responses are not sufficient to recapitulate the level of protective immunity generated by actual viral infection. To examine the protective effect of pp65-specific T cells or T cells specific to other CMV antigens we developed a novel challenge strategy. Our approach relies on our previous observation that viral genes encoding the RhCMV homologs of HCMV immunoevasins US2, 3, 6, and 11 are essential for RhCMV to superinfect RhCMV-positive animals (42, 69). The ability to establish secondary persistent infections is also a common occurrence in HCMV, resulting in frequent coinfection with different strains of HCMV (7). Since depletion of CD8+ T cells restores the ability of US2-11–deleted RhCMV to infect seropositive animals, infection with ΔUS2-11 viruses can be used to monitor the quality of a vaccine-induced T cell response. Moreover, the clear protection observed by natural infection allows these studies to take place in a very small group of animals, since the outcome of superinfection is binary. The results shown in Figure 3 are typical: both animals inoculated with Δpp65ab were clearly protected against superinfection with US2-11–deleted RhCMV but not with WT. (In fact, we observed superinfection in more than 200 animals inoculated with recombinant RhCMV.) Evidently, the T cell responses to antigens other than pp65 are protective in this challenge model.
Using the US2-11 challenge approach we were able to examine whether pp65-specific T cells elicited by heterologous prime-boost vaccination were sufficient to recapitulate the protective effect of T cells elicited by preexisting infection. We used a DNA-prime/MVA-boost protocol employed previously to vaccinate animals with a combination of pp65, IE1, and gB (44). In this previous work, it was shown that this vaccination regimen, while unable to protect against superinfection with RhCMV, reduced local and systemic viremia as well as viral shedding. Moreover, reduction in shedding correlated with the magnitude of pp65-specific T cell responses (44). In our hands, the heterologous prime-boost vaccination induced a robust CD4+ and CD8+ T cell response to pp65b that in the blood was similar in magnitude to pp65-specific T cell responses elicited by RhCMV infection. Although prime-boost vaccination would not be expected to maintain the effector-memory–biased T cells responses elicited by RhCMV infection over the long term (41, 48, 69), at the time of challenge (6 weeks after the final MVA boost), the responses generated by the prime-boost vaccine still manifested a predominant effector memory phenotype. Despite this, these vaccine-generated pp65-specific T cell responses were insufficient to protect against infection by ΔUS2-11gag virus, as shown by the induction of Gag-specific T cell responses and CMV-specific antibody responses. This suggests that T cells induced by pp65 alone do not reproduce the protective effect of T cells induced by ongoing persistent infections. Since the T cell response to pp65 was substantial in all 3 animals, it seems unlikely that a different vaccination strategy would have induced a better protection. Rather, it seems more likely that additional antigens might be required to recapitulate the protective effect of natural infection. Thus, our results caution against the use of pp65 as the only T cell stimulatory subunit in a CMV vaccine.
The ΔUS2-11 challenge used in this study provides an excellent tool to evaluate the T cell component of subunit vaccines. Conceivably, a similar approach could be used in human clinical trials to specifically evaluate the T cell immunity generated by a given vaccine. Recently, challenge with the Towne strain was used to evaluate the efficacy of subunit vaccines by monitoring an anamnestic HCMV-specific immune response (61). Conceivably, a US2-11–deleted Towne strain would not generate an anamnestic response, similar to our observation that IE1-specific responses were not boosted when RhCMVΔUS2-11 failed to superinfect (Figure 3). In this case, a second challenge with WT-Towne could be used to monitor protection and T cell boosting as described previously (61). In contrast, a boosting of the pp65-specific T cell and antibody responses, as observed in pp65-vaccinated animals (Figure 5), would be a clear indication of infection by US2-11–deleted Towne and lack of protection by the T cell component of a given vaccine. Furthermore, our observation that vaccine-induced antibody responses against pp65 were strongly boosted by inoculation with ΔUS2-11 virus suggests that a safe, US2-11–deleted HCMV could be used to boost antibody levels induced by a given vaccine. Since high levels of antibodies are a desired feature of many vaccine regimens, inclusion of CMV as a new tool to enhance antibodies should be considered.
In summary, our work revealed a novel and surprising function of pp65, suggesting that this viral protein acts as an immune inducer that generates an immune response that stringently restricts viral replication during primary infection but that has little impact on long-term maintenance, immunogenicity, or viral shedding. A likely explanation for this finding is that the highly abundant and immunogenic protein pp65 induces a rapid T cell response that limits viral dissemination. Since an intact immune system is required for this “immunological brake” mechanism to function, the lack of pp65-mediated control likely contributes to the high level of dissemination observed in immunocompromised individuals, such as transplant recipients, or in fetuses with immature immune systems. In RMs, RhCMV can cause severe sequelae, including spontaneous abortions, when injected into the developing fetus (70, 71). Since the immune-dominance of pp65 is conserved in HCMV, it is likely that this “antivirulence” function is conserved as well. HCMV and RhCMV thus seem to use the adaptive immune response to limit their dissemination during primary infection. It is not immediately obvious why CMV would choose such a self-imposed restriction. However, since the establishment and maintenance of persistent infection, as well as persistent shedding from the infected host, is not affected by the presence or absence of pp65, it seems that the ultimate “goal” of CMV, to establish a benign infection that easily spreads through the human population, is unaffected by pp65. The pp65-mediated immunological control of CMV dissemination might thus serve to soften the impact of primary infection on the host to ensure a healthy host that is able to maintain and transmit the virus for a long time.
Methods
Cells and reagents.
TRFs (72) were maintained in DMEM with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin and were grown at 37°C in humidified air with 5% CO2.
Viruses and construction of recombinant mutants.
BAC-derived RhCMV 68-1 (39) was reconstituted by electroporating BAC DNA into TRFs (250 V, 950 μF), and cytopathic effect was observed after 7 to 10 days. Recombinant RhCMV mutants were created by homologous recombination (73, 74) in E. coli strain EL250, which contains heat-inducible λ-recombination (rec) genes and an arabinose-inducible FLP recombinase (75). Bacterial cultures were grown in LB at 30°C until an OD of 0.35 at 600 nm was reached, and the rec genes were induced through heat induction by shaking the culture at 42°C in a water bath for 15 minutes. The bacteria were subsequently chilled on ice for 10 minutes and made electrocompetent by washing them 4 times with cold, deionized water. Electrocompetent EL250 were always made and used fresh to increase the recombination efficiency.
To construct the pp65a and pp65b (RhCMV-Δpp65ab) double-deletion virus, recombination primers containing 50 bp homology to regions flanking the pp65 ORFs (forward mutagenesis primer 5′-GAAATAAGTGTGCGGTCTCGGGGGATTGGGGTTTTTATATAGGTATGGGT-3′ and reverse mutagenesis primer 5′-ATGAGCCAAGTTGCGCAGCTCAGTCGGCGGTGTCGCCAAAGTCAGACAAC-3′) were used to amplify a kanamycin (Kan) resistance cassette from plasmid pCP015 (76). The pCP015 forward primer binding site (5′gtaaaacgacggccagt) and reverse primer binding site (5′gaaacagctatgaccatg) were added to the 3′ end of the mutagenesis primers.
Competent EL250 bacteria containing WT RhCMV BAC were then electroporated with the PCR product for recombination using a MicroPulser (Bio-Rad) and selected for Kan and chloramphenicol (Cm) resistance at 30°C on LB agar. To induce the FLP recombinase excising the Kanr cassette, clones were grown in LB with Cm until they reached an OD of 0.5 at 600 nm and incubated with 1 mg/ml arabinose for 1 hour. The bacteria were streaked out on an LB plate with Cm selection using an inoculation loop and incubated overnight at 30°C. After colonies were visible, clones were replica plated on LB agar with Kan and Cm and LB agar with Cm only, and colonies were selected that had lost Kanr and characterized by restriction digest, Southern blot, and partial sequencing. Virus was reconstituted by electroporation of TRFs with 5 to 10 μg of BAC DNA.
To construct the second pp65ab double-deletion mutant containing the SIV ref/tat/nef (retanef) fusion protein, which was driven by the EF1α promoter and inserted in place of the pp65ab proteins, recombination primers containing the same 50 bp homology to regions flanking the pp65 ORFs as before were used to amplify a Kan resistance cassette from plasmid pCP015rtndx (41). The pCP015rtndx forward primer binding site (5′gtaaaacgacggccagt) and reverse primer binding site (5′gtatgttgtgtggaattgtgag) were added to the 3′ end of the mutagenesis primers. All subsequent steps to generate the final mutant virus were the same as described above. The lack of expected genes in the recombinant viruses and lack of WT contamination was confirmed by Western blot analysis of purified viral stocks (Figure 2D) and RT-PCR of cDNA from virally infected cells (Figure 1A). The antibodies used in this study to confirm the presence or absence of RhCMV viral proteins were generated at the VGTI Monoclonal Antibody Core (mouse α-RhCMV pp65a clone 3H3.1.2, mouse α-RhCMV pp65b clone 19C12.2, and mouse α-RhCMV clone 6H7.3).
RhCMV particle purification procedures.
RhCMVΔpp65ab virions were purified over a discontinuous Nycodenz gradient, as described before for HCMV AD169 (18) and RhCMV 68-1 BAC-derived WT (38). The virus was isolated from the culture medium of infected TRFs when the cells displayed maximal cytopathic effect. The cellular supernatants were first clarified by centrifugation at 7,500 g for 15 minutes. The clarified medium was layered over a sorbitol cushion (20% D-sorbitol, 50 mM Tris [pH 7.4], 1 mM MgCl2), and virus was pelleted by centrifugation at 64,000 g for 1 hour at 4°C in a Beckman SW28 rotor. The virus pellet was resuspended in TNE buffer (50 mM Tris [pH 7.4], 100 mM NaCl, and 10 mM EDTA). The virus particles were further purified by layering them over a discontinuous 5% to 50% Nycodenz gradient (Sigma-Aldrich) in TNE buffer and centrifuging at 111,000 g for 2 hours at 4°C in a Beckman SW41 Ti rotor. The virion bands in the gradient were isolated with a syringe through the side of the centrifuge tube, and the particles were pelleted in a Beckman TLA-45 rotor in a Beckman Optima TL 100 Ultracentrifuge at 40,000 g for 1 hour and washed twice with TNE buffer. The pellet was resuspended in TNE buffer, and electron microscopy was performed to confirm the purity of the sample. In order to assess the protein content of the purified RhCMVΔpp65ab virions, especially in comparison to a parental RhCMV WT sample, a denatured protein preparation was separated on a NuPAGE morpholine propanesulfonic acid (MOPS) gradient gel (Invitrogen) and visualized by Coomassie brilliant blue staining (Figure 2C).
Quantitative proteomic analysis.
The quantitative proteomic analysis was performed as previously described in detail (38). Briefly, RhCMV particles were denatured in 8 M urea, 100 mM NH4HCO3, and 5 mM DTT, and the cysteine residues were alkylated with 10 mM iodoacetamide. Then, the samples were 4-fold diluted with 25 mM NH4HCO3 and 1 mM CaCl2 and digested overnight with a 1:20 (mass/mass) trypsin-to-protein ratio. The digested peptides were desalted with C18 cartridges and dried in a vacuum centrifuge before being separated in capillary columns (75 μm × 65 cm capillary [Polymicro] packed with 3-μm C18 particles [Phenomenex]) connected to a custom-made 4-column liquid chromatography LC system (77) or a longer capillary column (75 μm × 100 cm) connected to a nanoAcquity system (Waters). Eluting peptides were analyzed directly in a linear ion-trap orbitrap mass spectrometer (LTQ Orbitrap XL, Thermo Scientific).
Collected MS/MS spectra were searched against forward and reverse sequences of the RhCMV ORFs (275 sequences), Macaca mulatta Ensembl database (21,905 sequences, downloaded from http://www.ensembl.org on November 15, 2010), and 186 common contaminants (downloaded from http://www.ncbi.nlm.nih.gov/protein on August, 2006). Identified peptides were first filtered with a mass spectrum–generating function (MS-GF) (78) probability of ≤ 1 × 10–8 and, to ensure a low false discovery rate, each protein was required to have at least one peptide with MS-GF ≤ 1 × 10–10. Protein abundances were estimated by exponentially modified protein abundance index as previously described (79). Data are available at Pacific Northwest National Laboratory ( http://omics.pnl.gov) and in the PeptideAtlas online database ( http://www.peptideatlas.org; dataset identifier: PASS00367).
RMs.
A total of 9 male and 4 female purpose-bred juvenile RMs (M. mulatta) of Indian genetic background were used in this study. All RMs were specific-pathogen free (SPF), as defined by being free of cercopithecine herpesvirus 1, D-type simian retrovirus, simian T lymphotrophic virus type 1, SIV, rhesus rhadinovirus, Mycobacterium tuberculosis, and RhCMV infection at the start of the study. The Δpp65ab-deleted RhCMV vector was tested in vitro by administering the deletant virus s.c. at a dose of 1 × 107 PFUs to 2 SPF (defined above) RMs. To test whether the immune responses generated by the primary infection with Δpp65ab were sufficient to prevent superinfection, the same 2 RMs were inoculated with the same 1 × 107 PFUs dose with ΔVIHCEΔUS2-11gag, followed by superinfection with RhCMVgag and later with Δpp65ab(retanef).
To determine whether vaccine-induced pp65-specifc T cells could prevent CMV infection, 3 SPF RMs were vaccinated intramuscularly with 1.0 mg pND/pp65b DNA followed by an intramuscular boost using 5 × 108 PFUs pp65b-MVA at week 6 and 12 after DNA vaccination. For a control in these experiments, 3 additional SPF RMs were vaccinated following the same vaccine strategy using pND (empty) and MVA (empty), respectively (45, 46). The plasmids pND and pND/pp65b were provided by Peter A. Barry, UCD, Davis, California, USA (45); the empty MVA as well as the pp65b-expressing recombinant MVA were provided by Don J. Diamond, City of Hope, Duarte, California, USA. As a first step, the gene encoding RhCMV 68-1 pp65-2 was amplified from previously described plasmid expression vectors and engineered into the pZWIIA MVA transfer vector using established protocols (46). rMVA expressing RhCMV pp65-2 (Rhpp65-MVA) was generated on BHK-21 cells via homologous recombination. The protein expression levels for RhCMV pp65-2 in infected BHK-21 cells were confirmed by Western blot using polyclonal antibodies to RhCMV pp65-2 by chemiluminescence detection (ECL, Amersham Pharmacia Biotech). The plasmid DNA of pND and pND/pp65-2 was isolated using the EndoFree Plasmid Mega Kit (Qiagen) to avoid any endotoxin contamination of the DNA. Vaccinated animals were challenged s.c. 18 weeks after the initial DNA vaccination with 107 PFUs RhCMV ΔUS2-11gag.
BAL fluid, peripheral blood, and urine samples were collected at specified time points (see Figures 3–5) throughout the entire experiment. Isolated CD4+ and CD8+ T cells from BAL fluid and peripheral blood were stimulated with antigen-specific peptides to examine the immune response induced by the initial vaccination and the subsequent viral challenge.
Nested real-time PCR.
To determine the viral copy numbers of RhCMV 68-1 GAG and RhCMVΔpp65ab retanef, 3 naive and 2 CMV+ RMs were infected s.c. with 107 PFUs of each virus in the opposite arm on the same day. Blood samples were taken once a week to monitor CD4+ and CD8+ T cell responses and to determine the presence of cell-free virus in PBMCs. At the indicated time points after infection, the macaques were necropsied and tissues were harvested. DNA was isolated from the samples by ONPRC’s Molecular Virology Support Core (MVSC). Tissue samples (<100 mg) were prepared using FastPrep (MP Biomedicals) in 1 ml TriReagent (Molecular Research Center Inc.). 100 μl bromochloropropane (MRC Inc.) was added to each homogenized tissue sample to enhance phase separation. 0.5 ml DNA back-extraction buffer (4 M guanidine thiocyanate, 50 mM sodium citrate, and 1 M Tris) was added to the organic phase and interphase materials, which was then mixed by vortexing. The samples were centrifuged at 14,000 g for 15 minutes, and the aqueous phase was transferred to a clean microfuge tube containing 240 μg glycogen and 0.4 ml isopropanol and centrifuged for 15 minutes at 14,000 g. The DNA precipitate was washed twice with 70% ethanol and resuspended in 100 to 500 μl ddH2O. Nested real-time PCR was performed with primer and probe sets for the inserted SIV proteins GAG (first round: for-GAAACCATGCCGAAGACCTCTC and rev-CTCGTTGATGATGTCACGGATG; second round: for-CAACTACGTCCACCTGCCACTGTC, rev-TCCAACGCAGTTCAGCATCTGG, and probe-CCGAGAACCCTGAACGCTTGGGTCAAGC-FAM) and SIVretanef (first round: for-CGGAAGCAGAACATGGACGAC and rev-CCCCTTCTCCTTGATGAAGTGC; second round: for-CGACGAGGAGGACGACGACTTA, rev-CCAACTTGTACGACATCGTCCG, and probe-TCTCAGTGCGGCCGAAGGTCCC-FAM). For each DNA sample, 10 individual replicates (5 μg each) were amplified by first-round PCR synthesis (12 cycles of 95°C for 30 seconds and 60°C for 1 minute) using Platinum Taq in 50 μl reactions. Then, 5 μl of each replicate was analyzed by nested quantitative PCR (45 cycles of 95°C for 15 seconds and 60°C for 1 minute) using Fast Advanced Master Mix (ABI Life Technologies) in an ABI StepOnePlus Real-Time PCR machine. The results for all 10 replicates were analyzed by Poisson distribution and expressed as copies per cell equivalents (80).
Viral detection in urine by coculture.
We centrifuged filter-sterilized (0.4 mm) urine at 16,000 g for 1 hour at 4°C to concentrate virus for coculture on rhesus fibroblasts. Cell lysates were prepared after we observed extensive cytopathic effects or after 42 days of coculture if cytopathic effects were minimal or not observed. The prepared cell lysates were assessed for the presence of RhCMV on the basis of expression of RhCMV- or SIV-specific antigens by Western immunoblotting.
Immunologic assays.
RhCMV- and SIV-specific CD4+ and CD8+ T cell responses were measured in mononuclear cell preparations from blood and BAL fluid by flow cytometric intracellular cytokine analysis, as previously described (41). Briefly, sequential 15-mer peptides (overlapping by 11 amino acids) comprising the SIVMAC239Gag, or Rev/Nef/Tat and RhCMV 68.1pp65ab, or IE1/2 were used in the presence of costimulatory CD28 and CD49d monoclonal antibodies (BD Biosciences). Cells were incubated with antigen and costimulatory molecules alone for 1 hour, followed by addition of Brefeldin A (Sigma-Aldrich) for an additional 8 hours. Costimulation without antigen served as a background control. Cells were then stained with fluorochrome-conjugated monoclonal antibodies, flow cytometric data were collected on a LSR II (BD Biosciences), and data were analyzed using the FlowJo software program (version 8.8.7; Tree Star). Responses frequencies (CD69+/TNF+ and/or CD69+/IFN+) were first determined in the overall CD4+ and CD8+ population and then memory corrected (with memory T cell subset populations delineated on the basis of CD28 and CD95 expression).
Measuring RhCMV-specific antibody responses by ELISA.
Antibody levels to RhCMV were measured in circulating plasma of RMs by standard ELISA using plates coated with lysates of fibroblasts infected with either WT-RhCMV or RhCMV-Δpp65 at 10 μg total protein per well. Nonspecific binding sites were blocked with 2% milk powder resuspended in PBS. Serial 2-fold dilutions of plasma were incubated for 1.5 hours prior to washing 3 times with ELISA wash buffer (PBS with 0.1% Tween-20; 200 μl per well). Primary antibody binding was detected and quantified with HRP-conjugated anti-rhesus IgG/IgA/IgM secondary antibody and addition of o-phenylenediamine chromogen substrate. A log-log transformation was performed on the linear portion of the curve and end point titers were calculated using 0.1 OD units as the cutoff point. Each plate contained a positive control sample to normalize ELISA titers between assays and a negative control sample to ensure assay specificity and to subtract background. Graphical data was generated using Prism GraphPad software.
Measuring RhCMV-specific antibody responses by SDS-PAGE and Western blotting.
Rhesus fibroblasts infected with WT-RhCMV or RhCMV-Δpp65 were solubilized in 2× Laemmeli’s sample buffer, and 200 μg total protein per lane were loaded onto NuPAGE 4%–12% Bis-Tris gradient gels (Invitrogen, Life Technologies) and run in MOPS buffer. Proteins were transferred to Immobilon-P blotting membrane, and nonspecific binding sites were blocked in 2% milk powder in Tris-buffered saline with 0.02% Tween-20 for 60 minutes. Primary rhesus monkey polyclonal antisera was added at 1:700 dilution in blocking buffer for 60 minutes, and membranes were washed 3 times for 10 minutes each in Tris-buffered saline plus 0.2% Tween (TBST). Secondary goat anti-rhesus-HRP conjugate was added at 1:5,000 in blocking buffer for 30 minutes, and membranes were washed 3 times for 10 minutes each in TBST. Membranes were developed using ECL Advance Lumigen-TNA (GE Healthcare) for 1 minute and exposed to Biomax Light Film (Kodak) at various exposure times.
Ethics statement.
All RMs were handled in accordance with good animal practice, as defined by relevant national and/or local animal welfare bodies. The use of nonhuman primates was approved by the ONPRC Institutional Animal Care and Use Committee (IACUC no. 0691). The ONPRC is fully accredited by the Assessment and Accreditation of Laboratory Animal Care International. For blood and BAL collection, monkeys were anesthetized with ketamine by intramuscular injection. Monkeys were humanely euthanized by the veterinary staff at ONPRC in accordance with end point policies. Euthanasia was conducted under anesthesia with ketamine, followed by overdose with sodium pentobarbital. This method is consistent with the recommendation of the American Veterinary Medical Association.
Statistics.
All P values in this study were determined using a 1-tailed unpaired Student’s t test. The cutoff for statistical significance was defined as P < 0.05 unless otherwise specified. All error bars shown in all presented figures represent mean ± SD.
Study approval.
All animals in the presented study were used with approval of the ONPRC Animal Care and Use Committee, under the standards of the US NIH Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Acknowledgments
This work was funded by NIH grants R01 AI070890, RO1 AI070890-03S1, and RO1 AI059457 to K. Früh and R01 AI095113-02 to L.J. Picker. Further funding came from the Bill & Melinda Gates Foundation through Global Health Proposal no. 103312 to L.J. Picker. E.E. Marshall was supported as a trainee on an institutional training grant from the NIH (T32 AI078903). This project was also supported by the National Center for Research Resources and the Office of Research Infrastructure Programs of the NIH through grant P51OD011092. Portions of this research were supported by grants from the National Center for Research Resources (5P41RR018522-10) and the National Institute of General Medical Sciences (8 P41 GM103493-10) from the NIH (to R.D. Smith). The proteomics experimental work described here was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Department of Energy (DOE) and located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the DOE under contract DEAC05-76RL0 1830. We are grateful to Don Diamond, City of Hope, for providing recombinant MVA and to Peter Barry, UCD, for plasmid vaccines. We would like to acknowledge the help of Dina Alzhanova for growing the MVA constructs, the MVSC for their assistance in isolating DNA from animal tissues, and Michael Denton, Laura Springgay, Sam Purvine, Ron Moore, and Tom Fillmore for their technical assistance. We also would like to thank Teresa Sawyer and the electron microscopy facility at Oregon State University for taking the electron microscope images and Jay Nelson for reagents and help with interpreting the data presented in this publication.
Footnotes
Conflict of interest: Oregon Health and Science University and Klaus Früh, Scott G. Hansen, and Louis J. Picker have a significant financial interest in TomegaVax Inc., a company that may have a commercial interest in the results of this research and in this technology. This potential individual and institutional conflict of interest has been reviewed and managed by Oregon Health and Science University.
Citation for this article: J Clin Invest. 2014;124(5):1928–1944. doi:10.1172/JCI67420.
Ernesto S. Nakayasu’s present address is: Bindley Bioscience Center, Purdue University, West Lafayette, Indiana, USA.
Kasinath Viswanathan’s present address is: Advinus Therapeutics Ltd., Hinjewadi, Pune, India.
References
- 1.Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010;20(4):202–213. doi: 10.1002/rmv.655. [DOI] [PubMed] [Google Scholar]
- 2.Boppana SB, Fowler KB, Britt WJ, Stagno S, Pass RF. Symptomatic congenital cytomegalovirus infection in infants born to mothers with preexisting immunity to cytomegalovirus. Pediatrics. 1999;104(1 pt 1):55–60. doi: 10.1542/peds.104.1.55. [DOI] [PubMed] [Google Scholar]
- 3.Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326(10):663–667. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- 4.Fowler KB, Stagno S, Pass RF. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA. 2003;289(8):1008–1011. doi: 10.1001/jama.289.8.1008. [DOI] [PubMed] [Google Scholar]
- 5.Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am. 2011;25(1):151–169. doi: 10.1016/j.hoc.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis. 2004;39(2):233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 7.Ross SA, Arora N, Novak Z, Fowler KB, Britt WJ, Boppana SB. Cytomegalovirus reinfections in healthy seroimmune women. J Infect Dis. 2010;201(3):386–389. doi: 10.1086/649903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stratton KR, Durch JS, Lawrence RS. Vaccines For The 21St Century. Washington, DC, USA: National Academy Press; 2000. [PubMed] [Google Scholar]
- 9.Plotkin SA, Starr SE, Friedman HM, Gonczol E, Weibel RE. Protective effects of Towne cytomegalovirus vaccine against low-passage cytomegalovirus administered as a challenge. J Infect Dis. 1989;159(5):860–865. doi: 10.1093/infdis/159.5.860. [DOI] [PubMed] [Google Scholar]
- 10.Plotkin SA, et al. Towne-vaccine-induced prevention of cytomegalovirus disease after renal transplants. Lancet. 1984;1(8376):528–530. doi: 10.1016/s0140-6736(84)90930-9. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein DI. Vaccines for cytomegalovirus. Infect Disord Drug Targets. 2011;11(5):514–525. doi: 10.2174/187152611797636695. [DOI] [PubMed] [Google Scholar]
- 12.Pass RF, et al. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med. 2009;360(12):1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kharfan-Dabaja MA, et al. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis. 2012;12(4):290–299. doi: 10.1016/S1473-3099(11)70344-9. [DOI] [PubMed] [Google Scholar]
- 14.Kern F, et al. Cytomegalovirus (CMV) phosphoprotein 65 makes a large contribution to shaping the T cell repertoire in CMV-exposed individuals. J Infect Dis. 2002;185(12):1709–1716. doi: 10.1086/340637. [DOI] [PubMed] [Google Scholar]
- 15.Sylwester AW, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLaughlin-Taylor E, et al. Identification of the major late human cytomegalovirus matrix protein pp65 as a target antigen for CD8+ virus-specific cytotoxic T lymphocytes. J Med Virol. 1994;43(1):103–110. doi: 10.1002/jmv.1890430119. [DOI] [PubMed] [Google Scholar]
- 17.Wills MR, et al. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. 1996;70(11):7569–7579. doi: 10.1128/jvi.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varnum SM, et al. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol. 2004;78(20):10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browne EP, Shenk T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A. 2003;100(20):11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abate DA, Watanabe S, Mocarski ES. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol. 2004;78(20):10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cristea IM, et al. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol. 2010;84(15):7803–7814. doi: 10.1128/JVI.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnon TI, et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol. 2005;6(5):515–523. doi: 10.1038/ni1190. [DOI] [PubMed] [Google Scholar]
- 23.Gilbert MJ, Riddell SR, Plachter B, Greenberg PD. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate-early gene product. Nature. 1996;383(6602):720–722. doi: 10.1038/383720a0. [DOI] [PubMed] [Google Scholar]
- 24.Odeberg J, Plachter B, Branden L, Soderberg-Naucler C. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR alpha-chain. Blood. 2003;101(12):4870–4877. doi: 10.1182/blood-2002-05-1504. [DOI] [PubMed] [Google Scholar]
- 25.Britt WJ, Auger D. Human cytomegalovirus virion-associated protein with kinase activity. J Virol. 1986;59(1):185–188. doi: 10.1128/jvi.59.1.185-188.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somogyi T, Michelson S, Masse MJ. Genomic location of a human cytomegalovirus protein with protein kinase activity (PK68). Virology. 1990;174(1):276–285. doi: 10.1016/0042-6822(90)90075-3. [DOI] [PubMed] [Google Scholar]
- 27.Yao ZQ, et al. Site-directed mutation in a conserved kinase domain of human cytomegalovirus-pp65 with preservation of cytotoxic T lymphocyte targeting. Vaccine. 2001;19(13–14):1628–1635. doi: 10.1016/S0264-410X(00)00423-0. [DOI] [PubMed] [Google Scholar]
- 28.Gallina A, et al. Polo-like kinase 1 as a target for human cytomegalovirus pp65 lower matrix protein. J Virol. 1999;73(2):1468–1478. doi: 10.1128/jvi.73.2.1468-1478.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamil JP, Coen DM. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J Virol. 2007;81(19):10659–10668. doi: 10.1128/JVI.00497-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmolke S, Kern HF, Drescher P, Jahn G, Plachter B. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J Virol. 1995;69(10):5959–5968. doi: 10.1128/jvi.69.10.5959-5968.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chevillotte M, et al. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J Virol. 2009;83(6):2480–2490. doi: 10.1128/JVI.01818-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brune W, Hengel H, Koszinowski UH. A mouse model for cytomegalovirus infection. Curr Protoc Immunol. 2001;Chapter 19:Unit 19.7. doi: 10.1002/0471142735.im1907s43. [DOI] [PubMed] [Google Scholar]
- 33.Schleiss MR. Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV). J Clin Virol. 2002;25(suppl 2):S37–S49. doi: 10.1016/S1386-6532(02)00100-2. [DOI] [PubMed] [Google Scholar]
- 34.Powers C, Fruh K. Rhesus CMV: an emerging animal model for human CMV. Med Microbiol Immunol. 2008;197(2):109–115. doi: 10.1007/s00430-007-0073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue Y, Barry PA. Rhesus cytomegalovirus a nonhuman primate model for the study of human cytomegalovirus. Adv Virus Res. 2008;72:207–226. doi: 10.1016/S0065-3527(08)00405-3. [DOI] [PubMed] [Google Scholar]
- 36.Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW. Complete sequence and genomic analysis of rhesus cytomegalovirus. J Virol. 2003;77(12):6620–6636. doi: 10.1128/JVI.77.12.6620-6636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue Y, Kaur A, Zhou SS, Barry PA. Characterization and immunological analysis of the rhesus cytomegalovirus homologue (Rh112) of the human cytomegalovirus UL83 lower matrix phosphoprotein (pp65). J Gen Virol. 2006;87(pt 4):777–787. doi: 10.1099/vir.0.81516-0. [DOI] [PubMed] [Google Scholar]
- 38.Malouli D, et al. Reevaluation of the coding potential and proteomic analysis of the BAC-derived rhesus cytomegalovirus strain 68-1. J Virol. 2012;86(17):8959–8973. doi: 10.1128/JVI.01132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang WL, Barry PA. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. J Virol. 2003;77(9):5073–5083. doi: 10.1128/JVI.77.9.5073-5083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price DA, et al. Induction and evolution of cytomegalovirus-specific CD4+ T cell clonotypes in rhesus macaques. J Immunol. 2008;180(1):269–280. doi: 10.4049/jimmunol.180.1.269. [DOI] [PubMed] [Google Scholar]
- 41.Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15(3):293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansen SG, et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science. 2010;328(5974):102–106. doi: 10.1126/science.1185350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hel Z, et al. A novel chimeric Rev, Tat, and Nef (Retanef) antigen as a component of an SIV/HIV vaccine. Vaccine. 2002;20(25–26):3171–3186. doi: 10.1016/S0264-410X(02)00258-X. [DOI] [PubMed] [Google Scholar]
- 44.Abel K, et al. Vaccine-induced control of viral shedding following rhesus cytomegalovirus challenge in rhesus macaques. J Virol. 2011;85(6):2878–2890. doi: 10.1128/JVI.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue Y, et al. Immunogenicity and protective efficacy of DNA vaccines expressing rhesus cytomegalovirus glycoprotein B, phosphoprotein 65-2, and viral interleukin-10 in rhesus macaques. J Virol. 2007;81(3):1095–1109. doi: 10.1128/JVI.01708-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue Y, et al. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med Microbiol Immunol. 2008;197(2):117–123. doi: 10.1007/s00430-008-0074-5. [DOI] [PubMed] [Google Scholar]
- 47.Mohr CA, et al. A spread-deficient cytomegalovirus for assessment of first-target cells in vaccination. J Virol. 2010;84(15):7730–7742. doi: 10.1128/JVI.02696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen SG, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473(7348):523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalejta RF. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev. 2008;72(2):249–265. doi: 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeFilippis V, Früh K. Rhesus cytomegalovirus particles prevent activation of interferon regulatory factor 3. J Virol. 2005;79(10):6419–6431. doi: 10.1128/JVI.79.10.6419-6431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morello CS, Cranmer LD, Spector DH. In vivo replication, latency, and immunogenicity of murine cytomegalovirus mutants with deletions in the M83 and M84 genes, the putative homologs of human cytomegalovirus pp65 (UL83). J Virol. 1999;73(9):7678–7693. doi: 10.1128/jvi.73.9.7678-7693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cranmer LD, Clark CL, Morello CS, Farrell HE, Rawlinson WD, Spector DH. Identification, analysis, and evolutionary relationships of the putative murine cytomegalovirus homologs of the human cytomegalovirus UL82 (pp71) and UL83 (pp65) matrix phosphoproteins. J Virol. 1996;70(11):7929–7939. doi: 10.1128/jvi.70.11.7929-7939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGregor A, Liu F, Schleiss MR. Molecular, biological, and in vivo characterization of the guinea pig cytomegalovirus (CMV) homologs of the human CMV matrix proteins pp71 (UL82) and pp65 (UL83). J Virol. 2004;78(18):9872–9889. doi: 10.1128/JVI.78.18.9872-9889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voigt V, et al. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci U S A. 2003;100(23):13483–13488. doi: 10.1073/pnas.2233572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bubic I, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78(14):7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oxford KL, et al. Protein coding content of the UL)b’ region of wild-type rhesus cytomegalovirus. Virology. 2008;373(1):181–188. doi: 10.1016/j.virol.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lilja AE, Shenk T. Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc Natl Acad Sci U S A. 2008;105(50):19950–19955. doi: 10.1073/pnas.0811063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berencsi K, et al. A canarypox vector-expressing cytomegalovirus (CMV) phosphoprotein 65 induces long-lasting cytotoxic T cell responses in human CMV-seronegative subjects. J Infect Dis. 2001;183(8):1171–1179. doi: 10.1086/319680. [DOI] [PubMed] [Google Scholar]
- 59.Selinsky C, et al. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum Vaccin. 2005;1(1):16–23. doi: 10.4161/hv.1.1.1335. [DOI] [PubMed] [Google Scholar]
- 60.Bernstein DI, et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine. 2009;28(2):484–493. doi: 10.1016/j.vaccine.2009.09.135. [DOI] [PubMed] [Google Scholar]
- 61.Jacobson MA, et al. A CMV DNA vaccine primes for memory immune responses to live-attenuated CMV (Towne strain). Vaccine. 2009;27(10):1540–1548. doi: 10.1016/j.vaccine.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 62.La Rosa C, et al. Clinical evaluation of safety and immunogenicity of PADRE-cytomegalovirus (CMV) and tetanus-CMV fusion peptide vaccines with or without PF03512676 adjuvant. J Infect Dis. 2012;205(8):1294–1304. doi: 10.1093/infdis/jis107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feuchtinger T, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. 2010;116(20):4360–4367. doi: 10.1182/blood-2010-01-262089. [DOI] [PubMed] [Google Scholar]
- 64.Peggs KS, et al. Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis. 2011;52(1):49–57. doi: 10.1093/cid/ciq042. [DOI] [PubMed] [Google Scholar]
- 65.Mackinnon S, Thomson K, Verfuerth S, Peggs K, Lowdell M. Adoptive cellular therapy for cytomegalovirus infection following allogeneic stem cell transplantation using virus-specific T cells. Blood Cells Mol Dis. 2008;40(1):63–67. doi: 10.1016/j.bcmd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 66.Micklethwaite K, et al. Ex vivo expansion and prophylactic infusion of CMV-pp65 peptide-specific cytotoxic T-lymphocytes following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(6):707–714. doi: 10.1016/j.bbmt.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Peggs KS, Mackinnon S. Clinical trials with CMV-specific T cells. Cytotherapy. 2002;4(1):21–28. doi: 10.1080/146532402317251491. [DOI] [PubMed] [Google Scholar]
- 68.Schub A, Schuster IG, Hammerschmidt W, Moosmann A. CMV-specific TCR-transgenic T cells for immunotherapy. J Immunol. 2009;183(10):6819–6830. doi: 10.4049/jimmunol.0902233. [DOI] [PubMed] [Google Scholar]
- 69.Hansen SG, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science. 2013;340(6135):1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barry PA, et al. Nonhuman primate models of intrauterine cytomegalovirus infection. ILAR J. 2006;47(1):49–64. doi: 10.1093/ilar.47.1.49. [DOI] [PubMed] [Google Scholar]
- 71.Tarantal AF, Salamat MS, Britt WJ, Luciw PA, Hendrickx AG, Barry PA. Neuropathogenesis induced by rhesus cytomegalovirus in fetal rhesus monkeys (Macaca mulatta). J Infect Dis. 1998;177(2):446–450. doi: 10.1086/514206. [DOI] [PubMed] [Google Scholar]
- 72.Chang WL, Kirchoff V, Pari GS, Barry PA. Replication of rhesus cytomegalovirus in life-expanded rhesus fibroblasts expressing human telomerase. J Virol Methods. 2002;104(2):135–146. doi: 10.1016/S0166-0934(02)00060-5. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20(2):123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 74.Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27(6):1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee EC, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73(1):56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 76.Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158(1):9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 77.Livesay EA, et al. Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal Chem. 2008;80(1):294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J Proteome Res. 2008;7(8):3354–3363. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishihama Y, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4(9):1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 80.Cline AN, Bess JW, Piatak M, Piatak M, Jr, Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34(5–6):303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.