

Abstract
Asthma is one of the most common chronic airway inflammatory diseases. The clinical hallmarks of asthma include elevated serum levels of immunoglobulin E (IgE), eosinophilic inflammation and airway hyper-responsiveness (AHR). Arsenic trioxide (As2O3) is considered a carcinogen; however, it has also been used to treat diseases, such as syphilis, in traditional Chinese and Western medicine. Today, As2O3 is used as one of the standard therapies for acute promyelocytic leukemia (APL). Previous studies have indicated that As2O3 can induce apoptosis in eosinophils. However, the effect of As2O3 on asthma has not been investigated. We used ovalbumin (OVA)-immunized mice as a model for asthma and treated mice with As2O3 at doses of 2.5 and 5 mg/kg. The mice were then monitored for OVA-specific IgE production, airway inflammatory cell infiltration and AHR. We found that administration of As2O3 in OVA-immunized mice abrogated airway eosinophil recruitment by downregulating eotaxin expression but did not alter serum IgE or IL-5 levels in bronchoalveolar lavage fluid (BALF). Furthermore, the development of AHR and cellular infiltration into the airway were reduced by treating mice with As2O3. In vitro data suggested that low concentrations of As2O3 could induce only a small degree of apoptosis in primary pulmonary cells but could significantly inhibit the secretion of eotaxin by these cells. These results indicate that the administration of As2O3 to OVA-immunized mice can suppress lung allergic inflammatory responses. As2O3 might therefore have therapeutic potential in treating allergic airway inflammatory diseases.
Keywords: arsenic trioxide, asthma, eosinophils
Background
Asthma is a chronic inflammatory disease caused by allergic airway inflammation, which is characterized by increased mucus secretion, airway remodeling and airway hyper-responsiveness (AHR).1 Many cells, particularly mast cells, eosinophils and T lymphocytes, have been suggested to play a critical role in the pathogenesis of asthma. In addition, other parameters, such as high serum immunoglobulin E (IgE) levels and cellular infiltration of the airways by eosinophils and T lymphocytes expressing T helper 2 (Th2) cytokines, are also pathological features of asthma. IgE binds to mast cells, providing for the recognition of specific antigens. Th2 cells account for the accumulation of eosinophils, which release reactive oxygen species and toxic granular proteins when activated. The pathological symptoms of asthma appear to be correlated with the degree of airway inflammation and the level of local Th2 cytokine production.2, 3
Arsenic has been considered a poison for a long time. Since the 1820s, many studies have shown arsenic to be a potent environmental carcinogen in human malignancies, especially in skin and lung cancers.4, 5, 6 However, in traditional Chinese medicine, arsenous compounds, including arsenous acid and arsenic trioxide (As2O3), are often used to treat tooth marrow disease, psoriasis and rheumatosis.7 Arsenic has been used as both a therapeutic agent and a poison in ancient Greece and Rome. In Western medicine, arsenic has been used more recently in the treatment of syphilis and trypanosomiasis, which affect the central nervous system.8, 9 In the 1970s, As2O3 was introduced as a treatment for acute promyelocytic leukemia (APL) and showed striking effectiveness. Today, As2O3 has become one of the standard therapies for APL and is thought to be the major factor in achieving complete remission of APL.
Previous studies have found that As2O3 promotes apoptosis of pulmonary eosinophils in a guinea pig model of asthma.10 As2O3 also reduces eosinophil recruitment. In current studies, the ovalbumin (OVA)-induced mouse model of asthma was used to investigate the possible therapeutic effects of As2O3. Our data demonstrated that As2O3 treatment could block eosinophil infiltration into the airway by downregulating eotaxin levels and decreasing AHR. In addition, we found that low concentrations of As2O3 could significantly inhibit the secretion of eotaxin and regulated upon activation, normal T cell expressed and secreted (RANTES) by primary lung epithelial cells without damaging the cells. Therefore, we believe that As2O3 has therapeutic potential in the treatment of asthma.
Methods
Reagents
As2O3 (1 mg/ml) was purchased from TTY Biopharm Co., Ltd (Taoyuan, Taiwan) and stored at 4 °C before use.
Animals
Female BALB/c mice aged 6–8 weeks were obtained from and maintained in the Animal Center of the College of Medicine, National Taiwan University. Animal study protocols were approved by the Animal Research Committee, College of Medicine, National Taiwan University.
Immunization and As2O3 treatment
We modified a previously established murine model of airway inflammation as described.11 For systemic immunization, mice were sensitized by intraperitoneal injection of 50 µg of OVA (Sigma, St Louis, MO, USA) mixed with 4 mg of alum on day 0; they were then given 25 µg of OVA mixed with 4 mg of alum on days 14, 21 and 28. On days 42, 43 and 44, mice were challenged with 100 µg/mouse (in a total volume of 40 µl) of OVA by intranasal administration. Four days prior to the OVA challenge, As2O3 was administered intraperitoneally for 7 days. For the positive control group, the sensitized mice were treated with phosphate-buffered saline (PBS) intraperitoneally prior to OVA challenge. The naive group received OVA challenge but were not sensitized.
Measurement of OVA-specific antibodies
Blood was collected from the retro-orbital venous plexus at days 0 and 47 and centrifuged to separate the sera for antibody assays. The amount of OVA-specific IgE was determined by ELISA. Briefly, 96-well plates were coated with OVA at 10 µg/well. After the plates were blocked, 100 µl/well of diluted sera was added, and the plates were incubated at room temperature for 2 h or at 4 °C overnight. After incubation, the plates were washed five times with PBS with Tween 20 buffer, the secondary antibody (biotinylated rat anti-mouse IgE; AbD Serotec, Kidlington, UK) was added and plates were incubated for 1 h at room temperature. After plates were washed, avidin-horseradish peroxidase (Pierce Chemical, Rockford, IL, USA) was added, and samples were incubated at room temperature for 30 min. The avidin-horseradish peroxidase was removed by washing with PBS with Tween 20 buffer, and the bound enzyme substrate was detected by adding tetramethylbenzidine reagent (KPL, Gaithersburg, MD, USA). After incubation at room temperature for a short time, the reaction was stopped by adding 50 µl/well of 2 N H2SO4. Optical density was measured at 450 nm (550 nm was used as a reference filter) in a microplate autoreader (Anthos Reader 2010; Anthos Labtec Instruments GmbH, Salzburg, Austria).
Measurement of AHR
On day 45, the airway response to aerosolized methacholine (Sigma) was measured in unrestrained, conscious mice as previously described.12 The mice were placed in the main chamber of a whole body plethysmograph (Buxco Electronics, Inc., Sharon, CT, USA) and challenged with aerosolized 0.9% normal saline accompanied by increasing doses of methacholine (6.25–50 mg/ml). Each nebulization lasted for 3 min, and after each nebulization, recordings were taken and averaged for the 3 min. The Penh (enhanced pause 5 pause 3 (peak expiratory box flow/peak inspiratory box flow)) values were determined, and the data were expressed as Penh values.
Bronchoalveolar lavage fluid (BALF) assessment
To measure airway inflammation, we examined the accumulation of inflammatory cells in the BALF.13 After assessment of pulmonary function, the mice were killed, and the trachea was cannulated and immediately lavaged three times with 1 ml of Hank's balanced salt solution without calcium and magnesium. The lavage fluid was kept on ice and then centrifuged (400g) at 4 °C for 10 min. After being washed, cell pellets were resuspended in 1 ml of Hank's balanced salt solution, and the total number of cells in the BALF was counted with a standard hemocytometer. A differential count was performed on a smear prepared with a cytocentrifuge and stained with Liu's stain solution. A minimum of 200 cells were counted and classified as macrophages, lymphocytes, neutrophils and eosinophils based on standard morphological criteria.
BALF cytokine measurement
The levels of eotaxin and IL-5 in BALF supernatants were evaluated using an appropriate ELISA Kit (R&D, Minneapolis, MN, USA). These eotaxin and IL-5 assays have a threshold of detection of 500 pg/ml and 2 ng/ml, respectively. The cytokine levels were calculated by linear regression analysis based on the values obtained from a standard curve.
Histopathological analysis
To evaluate the effects of As2O3 treatment on allergen-induced pulmonary inflammation, each group of animals was killed for histopathological examination. After lavage, the lungs were immediately removed and fixed in 10% buffered formalin. Pulmonary tissues were subsequently sliced, embedded in paraffin and cut into 5-µμ thick sections. Sections were stained with hematoxylin–eosin and examined by light microscopy for histological changes.
Primary mouse lung cell culture
Three-week-old BALB/c female mice were killed by cervical dislocation to avoid the influence of ether or pentobarbital on lung tissue. The lungs were removed and washed with 1× PBS buffer until all blood was removed. All connective tissues and blood vessels were removed, and the lung tissue was subsequently cut into small pieces. The resulting single cell suspension was centrifuged, and the cell precipitate was collected. Lung cells were cultured with alpha-minimum essential medium (Life Technologies, Grand Island, NY, USA) complete medium including 10% fetal bovine serum, 4 mM L-glutamine, 25 mM N-2-hydro-xyethylpiperazine-N′-2-ethanesulfonic acids (pH 7.2), 5×10−5 M 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin and 0.25 mg/ml amphotericin. After 10–14 days, the primary cell population was able to reach 80% confluence.14 After confluence, cells were seeded at 2×105 cells/well in 24-well plates and cultured at 37 °C with 5% CO2 until the cells again attained 80% confluence. For eotaxin detection, cells were treated with 3000 U/ml of recombinant IL-4 (R&D) and As2O3. For RANTES detection, cells were treated with 20 ng/ml of tumor necrosis factor-α (PeproTech, Rocky Hill, NJ, USA) and As2O3. After 48 h of incubation, the supernatant from each well was collected and frozen at −20 °C prior to analysis.
Detection of apoptosis in vitro
Primary lung epithelial cells were seeded at 2×105 cells/well in 12-well plates. The cells were cultured at 37 °C with 5% CO2 incubation until the cells attained 80% confluence and subsequently treated with 0, 0.05, 0.1, 0.5, 1 and 5 µΜ of As2O3. After 24 h, the cells were collected and analyzed for apoptotic cells by flow cytometry using 7-amino-actinomycin D and annexin V staining (Apoptosis Detection Kit, PharMingen; Becton Dickinson & Co., San Jose, CA, USA).
Statistical analysis
All data are expressed as mean±SEM. For in vivo experiments, each group consisted of 3–6 mice. Single pairs of the groups were compared by Student's t-test. Differences were considered statistically significant when P<0.05.
Results
As2O3 treatment did not affect the serum IgE level
After OVA sensitization, mice were treated with As2O3 on days 38–44. On days 42–44, mice were challenged by intranasal administration of OVA. Figure 1 shows that there was no significant difference in serum IgE levels between OVA sensitized mice treated either with or without As2O3.
Figure 1.
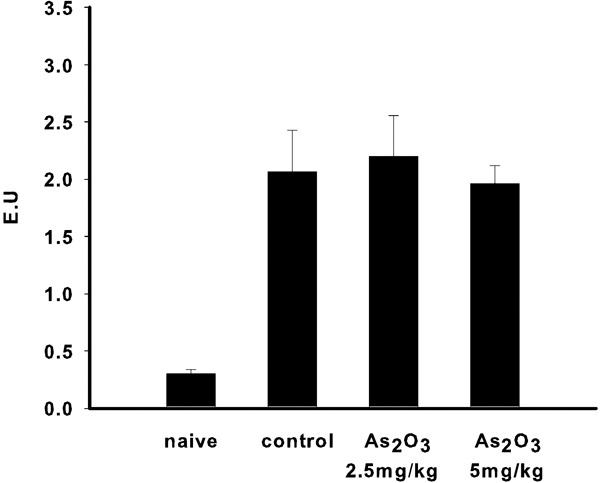
OVA-specific serum IgE levels. After treatment with As2O3, there was no significant difference in serum IgE between the control and As2O3 treatment groups. Each group included four to five mice. Values are expressed as mean±SEM. As2O3, arsenic trioxide; IgE, immunoglobulin E; OVA, ovalbumin.
The effect of As2O3 on the AHR of mice
We next explored whether As2O3 treatment could protect mice from AHR. Penh, which is a measurement of airway resistance, was used as a readout to determine the degree of AHR. After the final intranasal OVA challenge, mice were subjected to a methacholine stimulation test. As shown in Figure 2, mice treated with As2O3 had a lower Penh ratio compared with mice without As2O3 treatment. These data indicate that As2O3 treatment decreased AHR in mice.
Figure 2.
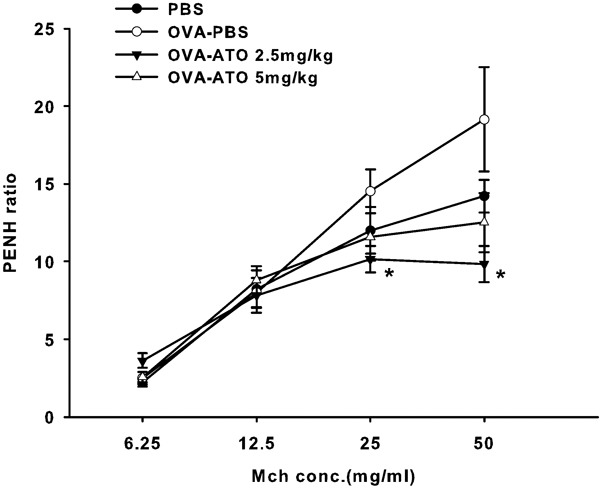
Airway hyperresponsiveness in mice treated with different concentrations of As2O3. The mice treated with 2.5 mg/kg of As2O3 had a significant decrease in airway hyperresponsiveness. Animals administered 5 mg/kg of As2O3 had a lower Penh value than the control group. Each group included four to five mice. Values are expressed as mean±SEM. The difference is statistically significant compared with the control group (*P<0.05). As2O3, arsenic trioxide; ATO, arsenic trioxide; PBS, phosphate-buffered saline; Penh, enhanced pause; OVA, ovalbumin.
The effect of As2O3 on cellular composition in the BALF
To evaluate whether As2O3 could modulate the recruitment of inflammatory cells in the airway, differential cell counts were determined by Liu's stain. The data show that mice treated with As2O3 had fewer eosinophils and increased macrophages in their BALF (Figure 3). In contrast, there were a higher number of eosinophils in mice sensitized with OVA but not given As2O3 treatment. Thus, mice treated with As2O3 had significantly decreased eosinophilia in their lungs.
Figure 3.
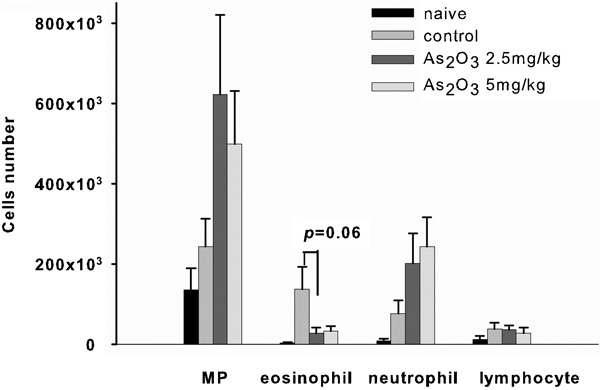
Cellular composition in the BALF. Mice treated with As2O3 had decreased numbers of eosinophils but increased numbers of inflammatory cells compared with the control group. Each group included four to five mice. Values are expressed as mean±SEM. The difference is statistically significant compared with the control group (*P<0.05). As2O3, arsenic trioxide; BALF, bronchoalveolar lavage fluid; MP, macrophages.
IL-5 and eotaxin levels in the BALF
IL-5 is a key cytokine that promotes eosinophil differentiation, maturation, recruitment and activation at sites of inflammation.15 We thus determined the level of IL-5 in BALF by ELISA. Figure 4a shows no significant difference in IL-5 protein levels in mice either treated with As2O3 or left untreated. To further examine how As2O3 might decrease the number of eosinophils, the level of eotaxin was also determined. The data show that mice treated with 2.5 mg/kg of As2O3 had a decreased concentration of eotaxin in BALF (Figure 4b).
Figure 4.
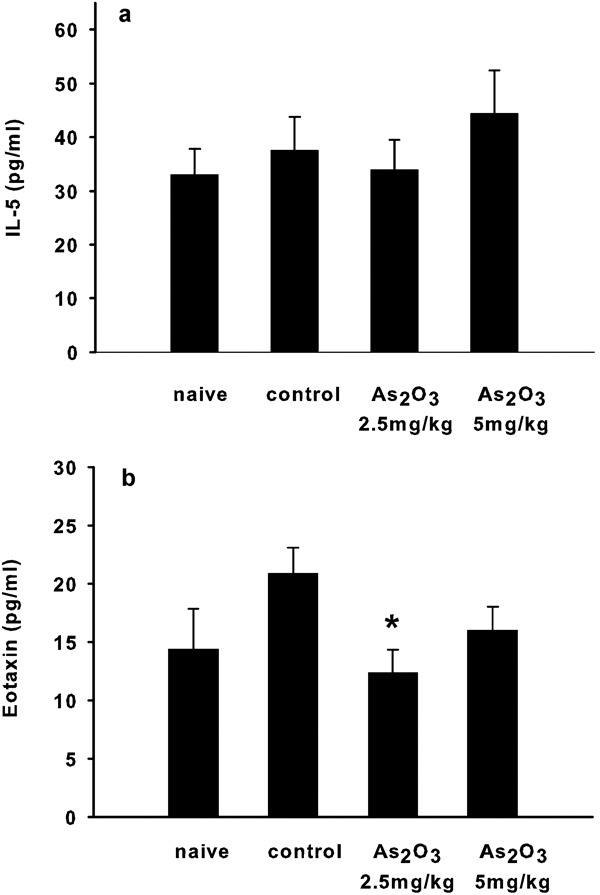
Cytokine levels in the BALF in each group of mice. (a) The IL-5 level detected in the BALF was not different between the groups of mice. (b) There was a significant decrease in the concentration of eotaxin in the BALF of mice treated with 2.5 mg/kg of As2O3. Each group included four to five mice. Values are expressed as mean±SEM. The difference is statistically significant compared with the control group (*P<0.05). As2O3, arsenic trioxide; BALF, bronchoalveolar lavage fluid.
Recruitment of inflammatory cells in the airway
To determine the effect of As2O3 treatment on the recruitment of inflammatory cells, lungs of mice were prepared for histopathological staining. In the untreated group, the cellular infiltration consisted mainly of mononuclear cells predominantly in the peribronchial and perivascular areas (Figure 5b). In contrast, As2O3 treatment resulted in reduced cellular infiltration (Figure 5c and d); indeed, cellular infiltration levels were as low as that observed in the naive (unsensitized) group (Figure 5a).
Figure 5.
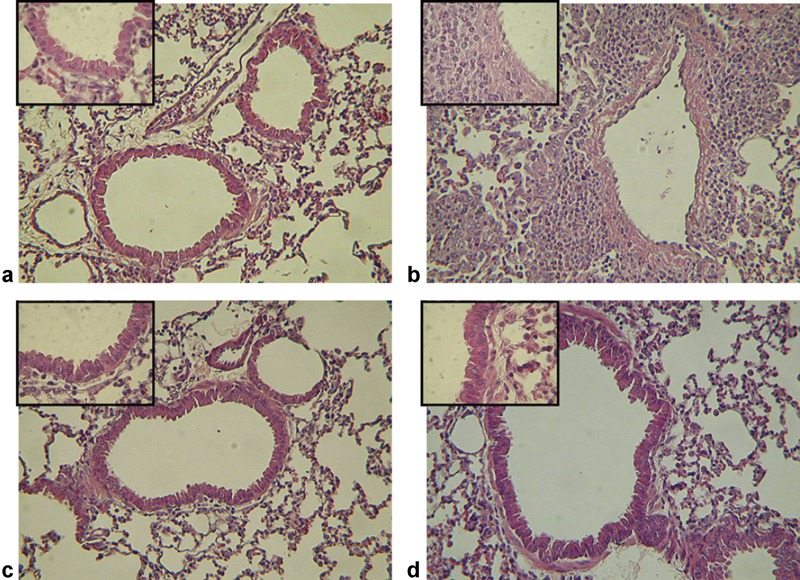
Histological analysis of pulmonary sections from immunized mice with or without As2O3 treatment. (a) Unsensitized mice show healthy pulmonary tissue. (b) Mice sensitized and challenged with OVA show cells infiltrating the airways. After As2O3 treatment (2.5 mg/kg (c) and 5 mg/kg (d)), cellular infiltration was reduced. (a–d) Sections are stained with H&E. Original magnification in a–d, ×100; upper left figures: ×400. As2O3, arsenic trioxide; H&E, hematoxylin–eosin; OVA, ovalbumin.
The effect of As2O3 on lung epithelial cells
To investigate the effect of As2O3 on pulmonary cell secretion of eotaxin, we isolated and cultured primary pulmonary cells from BALB/c mice. The optimal concentration of As2O3 in the assay was determined by assessing apoptosis. The data shown in Figure 6a indicate that there was slightly increased cell apoptosis after 24 and 72 h of treatment with 0.05 µΜ of As2O3 (14.29±0.2%, data not shown) compared with cells without treatment (10.21±0.49%). Furthermore, As2O3 induced pulmonary cell apoptosis in a dose-dependent manner. We thus chose the lowest concentration (0.05 µΜ) for further analysis, because this concentration did not affect cell viability. The secretion of eotaxin by primary pulmonary cells was stimulated by IL-4 but significantly inhibited when the cells were concurrently treated with As2O3 (Figure 6b, 297.8±49.2 pg/ml and 94.7±24.4 pg/ml, respectively). In addition, the RANTES level was also decreased in the As2O3-treated group (Figure 6c, 366.8±30.4 pg/ml and 242.4±7.9 pg/ml, respectively).
Figure 6.
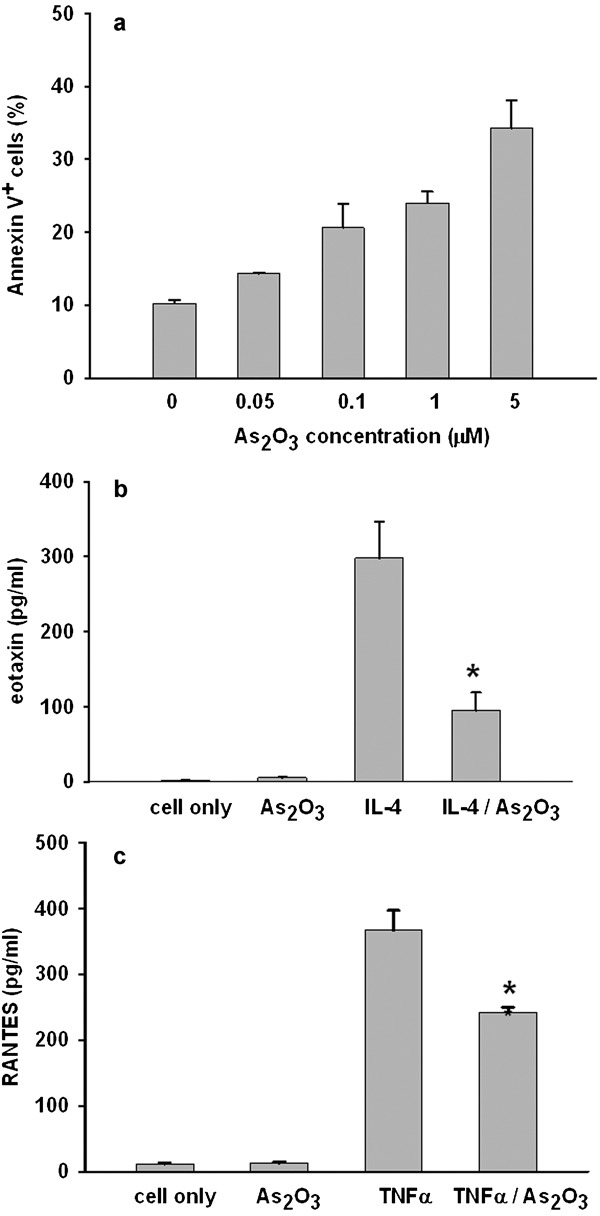
Twenty-four hours of treatment with As2O3 induces apoptosis in primary pulmonary cells in a dose-dependent manner (a). As2O3 of 0.05 µΜ inhibits the secretion of eotaxin by pulmonary cells stimulated with IL-4 (3000 U/ml) (b) and the secretion of RANTES by pulmonary cells stimulated with TNF-α (20 ng/ml) (c). Values are expressed as mean±SEM. The difference is statistically significant compared with the control group (*P<0.05). As2O3, arsenic trioxide; RANTES, regulated upon activation, normal T cell expressed and secreted; TNF, tumor necrosis factor.
Discussion
Although arsenic is often associated with environmental contamination, As2O3 is currently used in the clinic to treat cancer. In traditional Chinese medicine, arsenic has been used as a powerful therapeutic for various illnesses, on the principle that ‘using a toxic substance against another toxic substance' could treat the illness. Therapeutic As2O3 has been found to induce the differentiation and apoptosis of all-trans retinoic acid-resistant APL cells in vitro and in vivo.16, 17, 18 It has also been suggested that arsenic compounds have proinflammatory properties when the concentration of arsenite is approximately 5 µΜ.19 In contrast, another group found that As2O3 exerts anti-inflammatory effects through augmentation of IκB and suppression of NF-κB activation. In the current study, we investigated the function of As2O3 in alleviating AHR in a murine model of asthma.
In mice, IgE-mediated activation of mast cells enhanced pulmonary responsiveness to cholinergic stimulation.20 Itami et al. demonstrated that IgE was not required for the induction of the late phase of immediate hypersensitivity reactions; however, IgE enhanced pulmonary inflammation and hyperresponsiveness.21, 22 We found that As2O3 did not alter IgE levels in the serum, indicating that the inhibition of AHR by As2O3 was not associated with decreased levels of IgE. As2O3 might therefore target the lung directly.
Eosinophil accumulation in peripheral blood and tissue is a hallmark feature of several important medical diseases, including atopic disorders (allergic rhinitis and asthma), parasitic infections and various systemic diseases (e.g., eosinophilic pneumonia).23 IL-5 is an eosinophil-specific cytokine that can regulate eosinophil growth, differentiation and survival, and can stimulate the release of eosinophils and eosinophil precursors from the bone marrow into peripheral circulation.24 IL-5 was originally discovered in guinea pigs and found to be responsible for allergen-induced eosinophil accumulation in the lungs.25 Eotaxin is an eosinophil-selective chemoattractant that has been identified as a potent activator of eosinophils, inducing eosinophils to generate superoxide and release granule proteins. Early studies suggested that eosinophil recruitment in allergic reactions was regulated by Th2 lymphocytes and that eotaxin production was T cell-dependent.23 Many cell types in the lung, including airway epithelial cells, airway smooth muscle cells, vascular endothelial cells, macrophages and eosinophils, appear to be capable of synthesizing eotaxin.20, 24, 26 In our studies, we found no difference in the level of IL-5 in the BALF of mice with or without As2O3 treatment. However, As2O3 treatment decreased the level of eotaxin. These findings suggest that eosinophils can leave the bone marrow and enter the circulation in OVA-induced airway hyper-responsive mice. Furthermore, As2O3 directly inhibited the secretion of eotaxin by lung epithelial cells, resulting in decreased numbers of eosinophils recruited into the airway (Figure 4b). Activated eosinophils release highly toxic granule proteins and free radicals, which can result in connective tissue matrix remodeling, smooth muscle contraction, increases in vascular permeability and leukocyte activation; these processes promote AHR. Our data showed that 2.5 mg/kg of As2O3 decreased the Penh value in OVA-sensitized mice. We speculate that As2O3 inhibits AHR by downregulating eotaxin production by cells in pulmonary tissue, thus decreasing the accumulation of eosinophils. However, we cannot exclude the possibility that As2O3 might inhibit eosinophils from leaving the bone marrow and entering the circulation.
The pathology of asthma is thought to be mediated by CD4+ T cells producing type-2 cytokines, IL-4 and IL-5, which are elevated in bronchial biopsies, bronchoalveolar lavage and peripheral blood of allergic patients.21, 27, 28 These cytokines promote the accumulation and activation of eosinophils and induce IgE synthesis by B cells. We found no significant difference in either IL-5 or IgE levels between the groups, implying that the therapeutic effect of As2O3 is not due to blockade of eosinophils or mast cell activation. In other words, As2O3 alleviates the severity of asthma at the late phase of the reaction rather than at the acute stage. Therefore, As2O3 might be potentially beneficial for asthmatic patients.
In a study by Zhou et al., eosinophils treated with As2O3 were found to be defective in their ability to chemoattract to eotaxin and RANTES. Herein, we treated primary pulmonary cells with much lower concentrations of As2O3 (0.05 µΜ), which would not induce pulmonary cell apoptosis, and found that As2O3 inhibited both eotaxin and RANTES secretion. Other studies have shown that As2O3 can abolish eosinophilia by downregulating chemoattractants released by pulmonary cells.29 Although we did not investigate the effect of As2O3 on dendritic cells or on the levels of other cytokines (except IL-5 and eotaxin) in BALF, the possibility that As2O3 might promote dendritic cell maturation and stimulate cells to produce Th1 cytokines cannot be ruled out.
In conclusion, we have shown that As2O3 has a direct inhibitory effect on the production of eotaxin by pulmonary cells without affecting IgE levels in the serum or IL-5 levels in BALF. Decreased eotaxin secretion due to treatment with As2O3 results in the ablation of eosinophilia in the lung and the alleviation of AHR in an OVA-induced asthmatic murine model. As2O3 may therefore have therapeutic potential in the treatment of asthma.
References
- Arm JP, Lee TH. The pathobiology of bronchial asthma. Adv Immunol. 1992;51:323–382. doi: 10.1016/s0065-2776(08)60491-5. [DOI] [PubMed] [Google Scholar]
- Azzawi M, Bradley B, Jeffery PK, Frew AJ, Wardlaw AJ, Knowles G, et al. Identification of activated T lymphocytes and eosinophils in bronchial biopsies in stable atopic asthma. Am Rev Respir Dis. 1990;142:1407–1413. doi: 10.1164/ajrccm/142.6_Pt_1.1407. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- Nielsen FH, Uthus EO.ArsenicIn: Frieden F (ed.)Biochemistry of the Essential Ultratrace Elements. New York: Plenum; 1981319 [Google Scholar]
- Bishop C, Kipling MD.Arsenic and cancer J Soc Occup Med978283–5. [DOI] [PubMed] [Google Scholar]
- Lerda D. Sister-chromatic exchange (SCE) among individuals chronically exposed to arsenic in drinking water. Mutat Res. 1994;312:111. doi: 10.1016/0165-1161(94)90015-9. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Liu TP, Yang Y.(eds). Manual of Clinical Drugs. Shanghai: Shanghai Science and Technology Press; 1995830 [Google Scholar]
- Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Perdrup A. Penicillin versus arsenical-bismuth treatment in early syphilis. Acta Derm Venereol Suppl (Stockh) 1950;31 (Suppl. 24:44–48. [PubMed] [Google Scholar]
- Zhou LF, Yin KS. Effect of arsenic trioxide on apoptosis of pulmonary eosinophils in asthmatic guinea-pigs. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2002;22:292–294. [PubMed] [Google Scholar]
- Chuang YH, Fu CL, Lo YC, Chiang BL. Adenovirus expressing Fas ligand gene decreases airway hyperresponsiveness and eosinophilia in a murine model of asthma. Gene Ther. 2004;11:1497–1505. doi: 10.1038/sj.gt.3302325. [DOI] [PubMed] [Google Scholar]
- Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, et al. Non-invasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- Lee YL, Fu CL, Ye YL, Chiang BL. Administration of interleukin-12 prevents mite Der p1 allergen-IgE antibody production and airway eosinophil infiltration in an animal model of airway inflammation. Scand J Immunol. 1999;49:229–236. doi: 10.1046/j.1365-3083.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Ye YL, Huang WC, Lee YL, Chiang BL. Interleukin-12 inhibits eotaxin secretion of cultured primary lung cells and alleviates airway inflammation in vivo. Cytokine. 2002;19:76–84. doi: 10.1006/cyto.2002.1950. [DOI] [PubMed] [Google Scholar]
- Kay AB. The role of eosinophils in the pathogenesis of asthma. Trends Mol Med. 2005;11:148–152. doi: 10.1016/j.molmed.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Chen GQ. Use of arsenic trioxide in the treatment of acute promyelocytic leukemia (APL): I. arsenic trioxide exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- Shao W. Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst. 1998;90:124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- Soigner SL. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Eng J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- Trouba KJ, Germolec DR. Micromolar concentrations of sodium arsenite induce cyclooxygenase-2 expression and stimulate p42/44 mitogen-activated protein kinase phosphorylation in normal human epidermal keratinocytes. Toxicol Sci. 2004;9:248–257. doi: 10.1093/toxsci/kfh132. [DOI] [PubMed] [Google Scholar]
- MacLean JA, Wenbey R, Luster AD. T cell-dependent regulation of eotaxin in antigen-induced pulmonary eosinophilia. J Exp Med. 1996;184:1461–1469. doi: 10.1084/jem.184.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami DM, Latinne D, Bazin H, Garcia ML, Perini A, Martins MA, et al. Immunoglobin E is not required for but enhances airway inflammation and hyperresponsiveness. Allergy. 2003;58:1117–1124. doi: 10.1034/j.1398-9995.2003.00206.x. [DOI] [PubMed] [Google Scholar]
- Ying S, Robinson DS, Meng Q, Rottman J, Kennedy R, Ringler DJ, et al. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. Association with airway hyperresponsiveness and predominant colocalization of eotaxin mRNA to bronchial epithelial and endothelial cells. Eur J Immunol. 1997;27:3507–3516. doi: 10.1002/eji.1830271252. [DOI] [PubMed] [Google Scholar]
- Rothenberg ME. Eosinophilia. N Eng J Med. 1998;338:1592–1600. doi: 10.1056/NEJM199805283382206. [DOI] [PubMed] [Google Scholar]
- Palframan RT, Collins PD, Williams TJ, Rankin SM. Eotaxin induces a rapid release of eosinophils and their progenitors from the bone marrow. Blood. 1998;91:2240–2248. [PubMed] [Google Scholar]
- Humbles AA, Conroy DM, Marleau S, Rankin SM, Palframan RT, Proudfoot AE, et al. Kinetics of eotaxin generation and its relationship to eosinophil accumulation in allergic airways disease: analysis in a guinea pig model in vivo. J Exp Med. 1997;186:601–612. doi: 10.1084/jem.186.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose PT, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, et al. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J Exp Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkhioued B, Renzi PM, Younes A, Garcia-Zepeda EA, Allakhverdi Z, Ghaffar O, et al. Increased expression of eotaxin in bronchoalveolar lavage and airways of asthmatics contributes to the chemotaxis of eosinophils to the site of inflammation. J Immunol. 1997;159:4593–4601. [PubMed] [Google Scholar]
- Martin TR, Takeishi T, Katz HR, Austen KF, Drazen JM, Galli SJ. Mast cell activation enhances airway responsiveness to methacholine in the mouse. J Clin Invest. 1993;91:1176–1182. doi: 10.1172/JCI116277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LF, Zhu Y, Cui XF, Xie WP, Hu AH, Yin KS. Arsenic trioxide, a potent inhibitor of NF-κB, abrogates allergen-induced airway hyperresponsiveness and inflammation. Resperatory Res. 2006;7:146–158. doi: 10.1186/1465-9921-7-146. [DOI] [PMC free article] [PubMed] [Google Scholar]