

Abstract
Viral RNAs produced during viral infection are recognized by the cytoplasmic RNA helicases retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5). A central adapter protein downstream of RIG-I and MDA5 is the mitochondrial membrane protein virus-induced signaling adaptor (VISA), which mediates the induction of type I interferons (IFNs) through the activation of transcription factors such as nuclear factor-kappaB (NF-κB) and IFN-regulatory factor-3 (IRF3). Here we found that hepatitis B virus (HBV)-encoded X protein (HBx) acts as an inhibitor of virus-triggered IRF3 activation and IFN-β induction. Reporter and plaque assays indicate that HBx inhibits signaling by components upstream but not downstream of VISA. Immunoprecipitation experiments indicate that HBx interacts with VISA and disrupts the association of VISA with its upstream and downstream components. These findings suggest that HBx acts as a suppressor of virus-triggered induction of type I IFNs, which explains the observation that HBV causes transient and chronic infection in hepatocytes but fails to activate the pattern recognition receptor-mediated IFN induction pathways.
Keywords: HBx, innate immunity, signal transduction, type I interferons, VISA
Introduction
Mammalian cells employ a variety of pattern recognition receptors (PRRs) to detect conserved components of infectious microbes known as pathogen-associated molecular patterns.1 Detection of pathogen-associated molecular patterns by the PRRs of host cells triggers a series of signaling events that lead to the induction of type I interferons (IFNs) consisting of IFN-α and IFN-β family cytokines.2 Type I IFNs further activate the JAK–STAT signal transduction pathway to induce downstream proteins that inhibit viral replication and mediate the clearance of virus-infected cells, thereby driving the cellular antiviral response.3, 4, 5
There are three major classes of PRRs, including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and nucleotide oligomerization domain-like receptors.1 Viral RNAs are recognized by certain TLR family members, including TLR3, -7 and -8, and RLRs. Recently, it has been demonstrated that viral DNA is transcribed into RNA by RNA polymerase III and that this viral RNA is recognized by RIG-I of the RLR family.6 The pathways triggered by RLR-mediated recognition of viral RNAs have begun to emerge. The RLR family members RIG-I and MDA5 contain two CARD modules at their N-terminus and a DexD/H-box helicase domain at their C-terminus. The helicase domains of RIG-I and MDA5 serve as intracellular viral RNA receptors, whereas the CARD modules are responsible for transmitting signals to the downstream CARD-containing adapter protein VISA (also known as MAVS, IPS-1 and Cardif).7, 8, 9, 10 The C-terminus of VISA contains a transmembrane domain that anchors VISA to the outer membrane of the mitochondria, implying an important role of mitochondria in innate antiviral immunity.8 On the outer membrane of the mitochondria, VISA acts as a central adapter for assembling a virus-induced complex that activates distinct signaling pathways leading to IFN-regulatory factor-3 (IRF3) and nuclear factor-kappaB (NF-κB) activation. VISA is constitutively associated with another mitochondrion-associated adapter protein, MITA/STING, which is also essential for virus-triggered signaling.11, 12, 13 VISA is associated with TRAF2 and TRAF6 through its TRAF interaction motifs.7 TRAF2 and TRAF6 facilitate K63-linked polyubiquitination of receptor-interacting protein and NF-κB essential modulator, respectively. These processes lead to the activation of IκB kinase and subsequent NF-κB activation.14 VISA is also associated with TRAF3 and TANK-binding kinase 1 (TBK1), leading to phosphorylation and activation of the transcription factor IRF3. In the nucleus, NF-κB and IRF3, as well as other transcription factors and coactivators, collaborate to facilitate the transcription of type I IFN genes.15, 16, 17
Complicated virus–host interactions occur during the course of a viral infection. The host cells utilize PRRs to recognize viral components that trigger the induction of type I IFNs and other antiviral genes. On the other hand, viruses evolve strategies to evade the host immune system by interrupting IFN-inducing cascades. For example, the NS1 protein of the influenza A virus inhibits the function of RIG-I, thus downregulating type I IFN expression.18, 19 The V proteins of paramyxoviruses interact with MDA-5 to block its activity, which leads to a reduction in IFN-β promoter activation.20 The cysteine protease NS3/4A of hepatitis C virus cleaves VISA and the TLR3 adapter protein TRIF, thereby blocking RIG-I- and TLR3-mediated induction of the type I IFNs.21, 22 The A52 and A46 proteins of vaccinia virus target multiple TIR proteins, including TRIF, to block TLR3- and TLR4-mediated induction of type I IFNs.23, 24 The results of virus–host interactions may determine the outcome of a viral infection: elimination of the viruses or the establishment of successful infection.
Hepatitis B virus (HBV) is the prototype of hepadnaviruses, one of the rare type of DNA viruses known to replicate their DNA by reverse transcription of a viral pre-genome RNA. HBV causes transient and chronic infection of the liver.25 Unlike most viruses, HBV infection does not induce the expression of antiviral genes in chimpanzees; thus, the virus spreads throughout the liver.26 Recently, several groups demonstrated that forced induction of type I IFNs by the overexpression of TLR or RLR signaling components inhibits HBV replication in the following experimental systems: in vivo,27 in mice non-parenchymal cells28 and in HepG2 and Huh7 cells.29 The Wu group also determined that Toll-like receptors mediate IRF3 and IFN-β activation and can be inhibited by a high level of HBV replication in HBV-Met cells.30 These results suggest that HBV has evolved mechanisms to overcome the innate immune response of the host cell.
The HBV-encoded X protein (HBx) is a multifunctional regulator that modulates transcription, signal transduction, cell cycle progression, protein degradation, apoptosis and genetic stability by directly or indirectly interacting with virus and host factors. HBx has been reported to transactivate HBV and other viral promoters. A wide variety of cis-elements, designated as xenobiotic responsive elements, have been documented to be responsive to HBx, including binding sites for AP-1, AP-2, NF-κB, SRF, c/EBP, Ets, ATF1 and CREB.25, 31, 32 Replication of the HBx-deficient HBV replicon in HepG2 cells was reduced by threefold compared with wild-type HBV, suggesting that HBx plays a stimulatory role in HBV replication.33 The information on HBx protein continues to grow, although many of the roles and functions of HBx remain controversial.
As HBx is the only regulatory protein encoded in the HBV genome, we speculate that HBx may play a role in IFN signaling. In this study, we found that HBx negatively regulated virus-triggered activation of IRF3 and induction of IFN-β by interacting with VISA and disrupting the VISA-associated complex. These findings provide new insights into the mechanism by which HBV escapes from the host innate immune system.
Materials and methods
Cell culture and reagents
BHK and 293 cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (ExCell). Tumor-necrosis factor-α and IL-1β (R&D Systems, Minneapolis, MN, USA), mouse monoclonal antibodies against Flag and HA (Sigma, St Louis, MO, USA) and rabbit polyclonal antibodies against IRF3 and MDA5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were purchased from the indicated manufacturers; SeV and VSV were previously described.7, 36, 37
Plasmids
A plasmid carrying a greater-than-unit-length (129%) HBV genome (payw1.2; subtype ayw) and the same plasmid with a stop codon replacing the coding sequence for amino acid 7 of HBx (payw*7) were previously described.34, 35 The vector pEGFP-C1 was a gift provided by Yan Wu. Luciferase reporter plasmids for NF-κB, ISRE and the IFN-β promoters, as well as mammalian expression plasmids for HA- or Flag-tagged RIG-I, RIG-I-N, MDA5, MDA5-N, MDA5-C, VISA, TBK1, MITA and IRF3, were previously described.7, 11, 36, 37 Expression plasmids for human HA-, Flag- or GFP-tagged HBx and RFP-tagged RIG-I, MDA5 and VISA were constructed by standard molecular biology techniques.
Transfection and reporter assays
The 293 cells (∼1×105) were seeded on 24-well dishes and transfected the following day by standard calcium phosphate precipitation. In the same experiment, empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, we added 0.01 µg of pRL-TK Renilla luciferase reporter plasmid to each transfection. Luciferase assays were performed with a dual-specific luciferase assay kit (Promega, Madison, WI, USA). In these assays, firefly luciferase activity was normalized to the activity of Renilla luciferase. All reporter assays were repeated at least three times. Data shown are the mean±SD from one representative experiment.
Coimmunoprecipitation, immunoblot analysis, native polyacrylamide gel electrophoresis (PAGE) and VSV plaque assays7, 36, 37
Coimmunoprecipitation and western blot analysis
For transient transfection and coimmunoprecipitation experiments, 293 cells (∼1×106) plated on 10-cm dishes were transfected with indicated plasmids for 24 h. The transfected cells were lysed in 800 µl of lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton, 1 mM EDTA, 10 µl/ml aprotinin, 10 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). For each immunoprecipitation, a 400 µl aliquot of lysate was incubated with 0.5 µg of the indicated antibody or control IgG and 25 µl of a 1∶1 slurry of GammaBind G Plus Sepharose (Amersham Biosciences, Piscataway, NJ, USA) for 4 h. The Sepharose beads were washed three times with 1 ml of lysis buffer containing 500 mM NaCl. The precipitates were then analyzed by standard immunoblot procedures.
Native PAGE
For endogenous IRF3 dimerization analysis, 293 cells (2×105) plated on six-well plates were transfected with the indicated amount of plasmids. Twenty-four hours after transfection, cells were infected with SeV or left uninfected for 6 h. Native PAGE was performed using Ready Gels J 7.5% as follows: The gel was prerun with 25 mM Tris and 192 mM glycine, pH 8.4, with and without 1% deoxycholate in the cathode and anode chamber, respectively, for 30 min at 40 mA. Samples in the native sample buffer (10 µg protein, 62.5 mM Tris-Cl, pH 6.8, 15% glycerol and 1% deoxycholate) were applied to the gel and electrophoresed for 60 min at 25 mA. Immunoblotting was performed as described above.
VSV plaque assay
The 293 cells (1×105) were transfected with the indicated plasmids for 24 h before VSV infection (multiplicity of infection of 0.1). At 1 h after infection, cells were washed with phosphate-buffered saline three times and then medium was added. The supernatants were harvested 12 h after washing. The supernatants were diluted 1∶5×105 and then used to infect confluent BHK21 cells cultured on 24-well plates. At 1 h postinfection, supernatant was removed and 3% methylcellulose was overlaid. At 3 days postinfection, the methylcellulose overlay was removed, and cells were fixed with 4% formaldehyde for 1 h and stained with 0.2% crystal violet in 20% methanol. Plaques were counted, averaged and multiplied by the dilution factor to determine viral titer as PFU/ml.
RT-PCR
Total RNA was isolated from 293 cells using Trizol reagent (invitrogen) and subjected to RT-PCR analysis to measure the expression of IFN-B1, Rantes, ISG15 and GAPDH according to the manufacturer's instructions. Gene-specific primer sequences were as follows: IFN-β, 5′-CAGCAATTTTCAGTGTCAGAAGCT-3′ and 5′-TCATCCTGTCCTTGAGGCAGTAT-3′ Rantes, 5′-ATGAAGGTCTCCGCGGCACGCCT-3′ and 5′-CTAGCTCATCTCCAAAGAGTTG-3′ ISG15, 5′-ATGGGCTGGGACCTGACGG-3′ and 5′-TTAGCTCCGCCCGCCAGGCT-3′ and GAPDH, 5′-AAAATCAAGTGGGGCGATGCT-3′ and 5′-GGGCAGAGATGATGACCCTTT-3′.
Immunofluorescent confocal microscopy
The transfected 293 cells were incubated with MitoTracker Red (Molecular Probes, Eugene, OR, USA) for 30 min. The cells were then fixed with 4% paraformaldehyde for 10 min and observed with a Leica confocal microscope under a ×100 oil objective.
Results
HBx inhibits virus-induced activation of the IFN-β promoter
Various studies have demonstrated that viruses can evade host immune responses by encoding proteins that impair the function of certain antiviral signaling components. In our preliminary screens of several HIV- and HBV-encoded proteins, we found that the HBV-encoded regulatory protein HBx could inhibit Sendai virus (SeV)-induced activation of the IFN-β promoter (Figure 1a). To determine whether HBx is truly an inhibitor of virus-induced IFN-β induction, it is ideal to compare the ability of wild-type and HBx-less HBV to induce type I IFNs. However, HBV-related research is hindered by the lack of an efficient in vitro infection system and the difficulty of obtaining stock virus. As an alternative, we utilized a payw1.2 plasmid, which encodes all of the HBV proteins and is packaged into HBV virus after transfection into mammalian cells. In tandem, we used the same plasmid with a stop codon for amino acid 7 of HBx, termed payw1.2*734, 35 In reporter assays, we found that transfection of cells with payw1.2 had no effect on the activation of the IFN-β promoter, whereas transfection with payw1.2*7 weakly activated the IFN-β promoter (Figure 1b). Interestingly, transfection of payw1.2*7 markedly potentiated SeV-induced activation of the IFN-β promoter, whereas payw1.2 had little effect (Figure 1b). These results indicate that HBx had an inhibitory effect on virus-induced activation of the IFN-β promoter. Overexpression of HBx also inhibited SeV-induced transcription of endogenous IFNB1 and Rantes genes (Figure 1c). These results suggest that HBx is a suppressor of virus-induced IFN-β expression.
Figure 1.
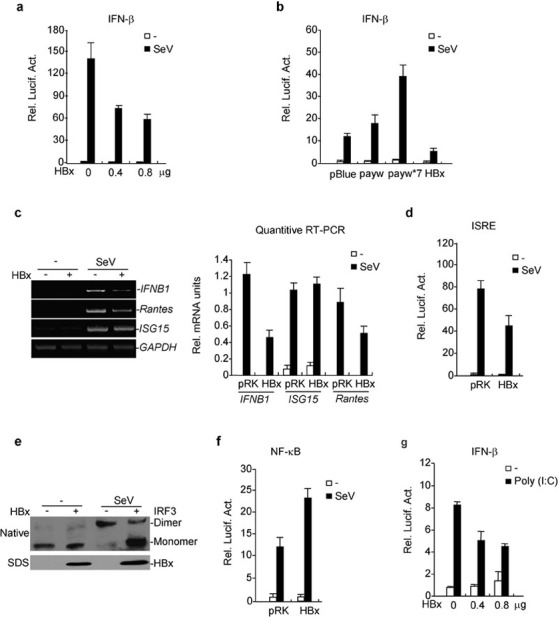
HBx inhibits virus-triggered IRF3 activation and IFN-β induction. (a) HBx inhibits SeV-induced activation of the IFN-β promoter in a dose-dependent manner. The 293 cells (1×105) were transfected with the IFN-β promoter reporter plasmid (0.1 µg) and the indicated amount of expression plasmid for HBx. Twenty-four hours after transfection, cells were left uninfected or infected with SeV for 12 h before luciferase assays were performed. (b) Effects of HBV and mutant replicons on SeV-triggered activation of the IFN-β promoter. Experiments were performed as in (a). (c) HBx inhibits transcription of IFNB1 and RANTES genes. The 293 cells (1×105) were transfected with the indicated plasmids for 24 h. RT-PCR was performed 10 h after SeV infection. The quantitative software Quantity One was used to examine the amount of DNA in agarose gels; the IFN-B1, Rantes and ISG15 values were then normalized to GAPDH values. The specific values are used for graphic display. (d) HBx inhibits SeV-induced ISRE activation. The experiments were performed as in (a). (e) HBx inhibits SeV-induced dimerization of endogenous IRF3. The 293 cells (2×105) were transfected with the indicated plasmids. Twenty-four hours after transfection, cells were infected with SeV or left uninfected for 6 h. Cell lysates were separated by native (upper panel) or SDS (bottom two panels) PAGE and analyzed with the indicated antibodies. (f) HBx does not inhibit SeV-induced NF-κB activation. The experiments were performed as in (a). (g) HBx inhibits poly(I:C)-induced activation of the IFN-β promoter. The 293 cells (1×105) were transfected with the IFN-β reporter and HBx expression plasmids. Twenty-four hours after transfection, cells were transfected with poly(I:C) (0.5 µg/well) or left untransfected for 12 h before luciferase assays were performed. payw: a plasmid carrying a greater-than-length (129%) HBV genome (subtype ayw); payw*7: an HBx-deficient version of the same plasmid of payw. HBV, hepatitis B virus; HBx, HBV-encoded X protein; NF-κB, nuclear factor-kappaB; IFN, interferon; ISRE, IFN-stimulated response element; IRF, IFN-regulatory factor; PAGE, polyacrylamide gel electrophoresis; SeV, Sendai virus.
The induction of IFN-β requires coordinated and cooperative actions of IRF3 and NF-κB. We next determined the effects of HBx on virus-induced IRF3 and NF-κB activation. In reporter assays, HBx inhibited SeV-induced IFN-stimulated response element (ISRE) activation (Figure 1d). HBx also inhibited SeV-induced dimerization of endogenous IRF3 (Figure 1e), which is a hallmark of IRF3 activation. In contrast, HBx potentiated SeV-induced NF-κB activation (Figure 1f). These results suggest that HBx suppresses virus-triggered IFN-β expression through inhibition of the IRF3 activation pathway.
In addition to viral infection, HBx also inhibited cytoplasmic poly(I:C)-induced activation of the IFN-β promoter (Figure 1g), but not tumor-necrosis factor- and IL-1-induced NF-κB activation (data not shown). These results suggest that HBx specifically inhibits virus- and RNA-induced IRF3 activation and type I IFN expression.
HBx targets VISA or acts downstream of VISA
Since HBx inhibits virus-triggered IRF3 activation and IFN-β induction, we next determined at which signaling step HBx targets. In reporter assays, HBx inhibited RIG-I-N-, MDA5-N-, VISA-, and MITA-mediated activation of the IFN-β promoter in a dose-dependent manner. In the same experiments, HBx had no inhibitory effect on TBK1-mediated activation of the IFN-β promoter (Figure 2a–e), and reporter assays with ISRE reporter had similar results (Figure 2f). In plaque assays, HBx reversed MDA5- and VISA-mediated inhibition, but not TBK1-mediated inhibition, of vesicular stomatitis virus (VSV) replication in 293 cells (Figure 2g), suggesting that HBx targets VISA or a signaling component downstream of VISA and upstream of TBK1.
Figure 2.
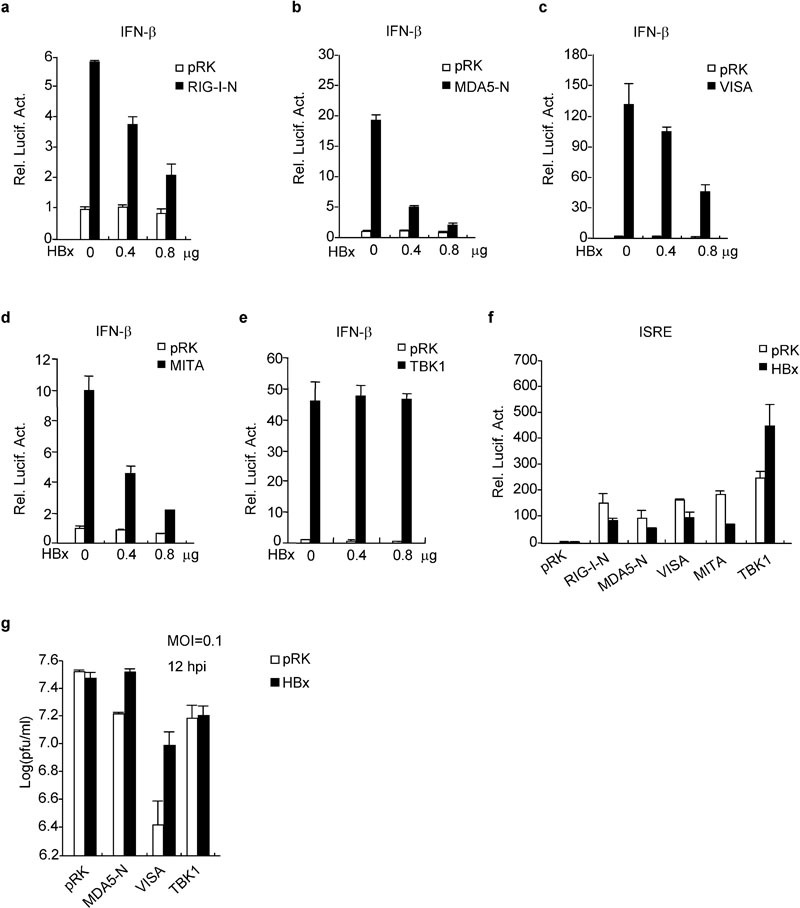
HBx inhibits virus-triggered signaling at the level of VISA. HBx inhibits activation of the IFN-β promoter mediated by RIG-I-N (a), MDA5-N (b), VISA (c) and MITA (d), but not TBK1 (e). The 293 cells (1×105) were transfected with the IFN-β promoter plasmid (0.1 µg) and the indicated expression plasmids. Luciferase assays were performed 24 h after transfection. (f) HBx inhibits virus-triggered ISRE activation at a level upstream of TBK1. Experiments were performed as in (a)–(e), with the exception that an ISRE reporter was used. (g) The role of HBx in the cellular antiviral response. The 293 cells (1×105) were transfected with the indicated expression plasmids. Twenty-four hours after transfection, cells were infected with VSV (MOI=0.1) and supernatants were harvested at 12 hpi. Supernatants were analyzed for VSV production by standard plaque assay. HBx, HBV-encoded X protein; hpi, hours postinfection; IFN, interferon; ISRE, IFN-stimulated response element; MOI, multiplicity of infection; TBK1, TANK-binding kinase 1; VSV, vesicular stomatitis virus.
HBx interacts with VISA
To further determine the mechanism of HBx-mediated inhibition of virus-induced IRF3 activation and IFN-β induction, we performed coimmunoprecipitation experiments. We transfected 293 cells (2×106) with Flag-tagged HBx and HA-tagged RIG-I, MDA5, VISA and TBK1. Twenty-four hours after transfection, cells were lysed and cell lysates were immunoprecipitated with anti-Flag or control IgG. The immunoprecipitates were analyzed by immunoblot with anti-HA (Figure 3a, b and c, top panel). Expression of the transfected proteins was analyzed by immunoblotting with anti-HA and anti-Flag antibodies (Figure 3a, b and c, middle and bottom panels, respectively). As shown in Figure 3a, HBx interacted with VISA and MDA5 but not RIG-I or TBK1. In similar experiments, we found that the N-terminal CARD domain of MDA5 and the C-terminal transmembrane domain-containing region of VISA mediate the observed interactions with HBx (Figure 3b and c). Interestingly, previous studies have demonstrated that the C-terminal region of VISA is important for activating IRF3.8 Confocal microscopy indicated that HBx colocalized with VISA and MDA5 (Figure 3d), consistent with the observation by immunoprecipitation that HBx interacted with VISA and MDA5.
Figure 3.
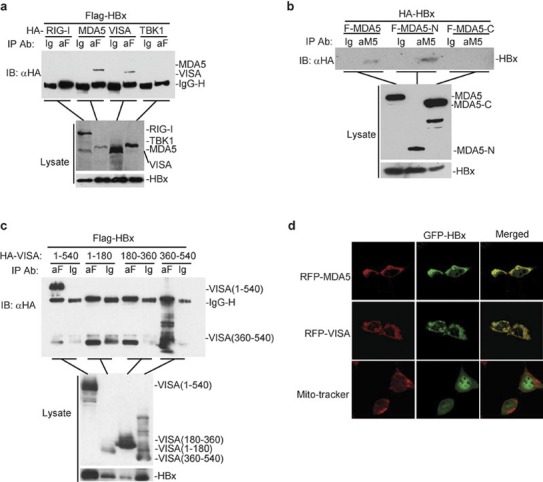
Analysis of the interactions between HBx and MDA5, VISA or TBK1. (a) HBx interacts with MDA5 and VISA but not TBK1. The 293 cells (2×106) were transfected with the indicated plasmids (8 µg each). Twenty-four hours after transfection, cells were lysed and coimmunoprecipitation experiments were performed. (b) HBx binds to the N-terminal CARD domain of MDA5. The experiments were performed as described in (a). (c) HBx interacts with the C-terminal domain (aa 360–540) of VISA. The experiments were performed as in (a). (d) Colocalization of HBx with MDA5 and VISA. The 293 cells were transfected with the indicated plasmids. Transfected cells were stained with MitoTracker Red and observed by confocal microscopy. aa, amino acid; HBx, HBV-encoded X protein; TBK1, TANK-binding kinase 1.
HBx disrupts the VISA-associated complex
Since HBx interacts with VISA, we next determined the effects of HBx on the VISA-associated complex. To do this, we performed competitive coimmunoprecipitation experiments. Eight micrograms each of the indicated plasmids was transfected into 293 cells (2×106). Twenty-four hours after transfection, cells were lysed and cell lysates were immunoprecipitated as above. The results indicated that the interaction between VISA and MDA5 or MITA was obviously inhibited when HBx was co-transfected compared to pRK. Similarly, interactions between VISA and RIG-I or TBK1 were also weakened when cells were co-transfected with HBx, albeit to a lesser degree. Together, these results suggest that an overexpression of HBx disrupts the interactions between VISA and other proteins within the VISA-associated complex, including RIG-I, MDA5, MITA and TBK1 (Figure 4). Thus, HBx suppresses virus-triggered IRF3 activation and IFN induction by disrupting the VISA-associated complex.
Figure 4.
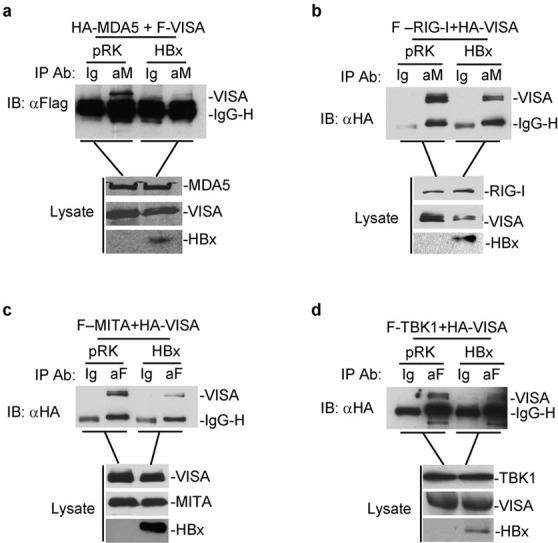
HBx disrupts the VISA-associated complex. Overexpression of HBx disrupts the interaction between VISA and MDA5 (a), RIG-I (b), MITA (c) and TBK1 (d). The 293 cells (2×106) were transfected with the indicated plasmids. Coimmunoprecipitation and immunoblot analysis were performed as in Figure 3a. In (a)–(d), while the expression levels of VISA and VISA-associated proteins were similar, the amount of coimmunoprecipitated VISA was much less after overexpression of HBx. HBx, HBV-encoded X protein; TBK1, TANK-binding kinase 1.
Discussion
The HBx protein has a variety of regulatory functions, such as modulating transcription, signal transduction, cell cycle progression, protein degradation pathways, apoptosis and genetic stability through direct or indirect interactions with virus and host factors. HBx has also been studied as a transactivator of AP-1, AP-2, NF-κB, SRF, c/EBP, Ets, ATF1 and CREB promoters. The exact function of HBx during host–virus interactions remains controversial. Our current study demonstrates that HBx plays an inhibitory role in virus-triggered IRF3 activation and IFN-β induction, revealing a new function for HBx.
HBV is one of the rare type of DNA viruses that replicate their DNA by reverse transcription of a viral pregenome RNA. Several mRNA sequences, designated pre-S (L), S and X mRNA, are produced for viral protein expression during infection and replication. These RNA products can be recognized by cytosolic PRRs, such as RIG-I and MDA5, and trigger IFN signaling. However, HBV frequently results in a chronic infection and finally HCC, which is partly due to the failure of the activation of IFN signaling during the early stages of virus infection. Recent work has demonstrated that an overexpression of the TLR adapters MyD88 or TRIF, or an overexpression of the RIG-I and MDA5 adapters, inhibits HBV replication in HepG2 and Huh7 cells, suggesting that HBV is sensitive to PRR-mediated IFN signaling and the cellular antiviral response.29 Our observation that HBx is an inhibitor of virus-triggered induction of type I IFNs provides a mechanistic explanation for the observation that HBV fails to induce type I IFNs.
Virus-triggered induction of type I IFNs requires activation of both NF-κB and IRF3. Our results indicate that HBx inhibits only the IRF3 activation pathway, which is sufficient to inhibit type I IFN induction. VISA is a central adapter of the virus-triggered signaling pathway. Reporter assays indicate that HBx inhibits virus-triggered signaling through the inhibition of VISA. Further, coimmunoprecipitation experiments indicate that HBx interacts with VISA and markedly reduces the interaction of VISA with its upstream components, RIG-I and MDA5, as well as its downstream components, MITA and TBK1. These results suggest that HBx inhibits virus-triggered IFN signaling by disrupting VISA-associated complexes. Previous studies demonstrate that HBV has evolved mechanisms to overcome TLR-mediated IFN signaling.30 In accordance with these previous studies, the identification of an inhibitory role for HBx in virus-triggered IFN signaling helps to explain how HBV evades the PRR-mediated innate immune response, which results in viral evasion of the immune system and chronic infection of the host.
Acknowledgments
We thank Dr Deying Guo and Dr Yan Wu for providing plasmids and cells. This work was supported by grants from the 973 Program of China (No. 2006CB504301).
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol. 2000;164:4220–4228. doi: 10.4049/jimmunol.164.8.4220. [DOI] [PubMed] [Google Scholar]
- Levy DE, Garcia-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001;12:143–156. doi: 10.1016/s1359-6101(00)00027-7. [DOI] [PubMed] [Google Scholar]
- Levy DE, Marie IJ. RIGging an antiviral defense–it's in the CARDs. Nat Immunol. 2004;5:699–701. doi: 10.1038/ni0704-699. [DOI] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, et al. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, et al. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol. 2007;36:263–269. doi: 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, et al. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007;9:930–938. doi: 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- Childs KS, Andrejeva J, Randall RE, Goodbourn S. Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J Virol. 2009;83:1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, et al. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner RG, Young RA. Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. . J Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A, et al. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol. 2009;83:847–858. doi: 10.1128/JVI.02008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, et al. Hepatitis B virus suppresses Toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology. 2009;49:1132–1140. doi: 10.1002/hep.22751. [DOI] [PubMed] [Google Scholar]
- Murakami S. Hepatitis B virus X protein: a multifunctional viral regulator. J Gastroenterol. 2001;36:651–660. doi: 10.1007/s005350170027. [DOI] [PubMed] [Google Scholar]
- Tang H, Oishi N, Kaneko S, Murakami S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006;97:977–983. doi: 10.1111/j.1349-7006.2006.00299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F, et al. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol. 2005;79:5548–5556. doi: 10.1128/JVI.79.9.5548-5556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melegari M, Scaglioni PP, Wands JR. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol. 1998;72:1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglioni PP, Melegari M, Wands JR. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J Virol. 1997;71:345–353. doi: 10.1128/jvi.71.1.345-353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao F, Li S, Tian Y, Zhang M, Xu LG, Zhang Y, et al. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc Natl Acad Sci USA. 2007;104:11706–11711. doi: 10.1073/pnas.0700544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB. SIKE is an IKK epsilon/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways. EMBO J. 2005;24:4018–4028. doi: 10.1038/sj.emboj.7600863. [DOI] [PMC free article] [PubMed] [Google Scholar]