

Abstract
Aim:
ΦC31 integrase mediates site-specific recombination between two short sequences, attP and attB, in phage and bacterial genomes, which is a promising tool in gene regulation-based therapy since the zinc finger structure is probably the DNA recognizing domain that can further be engineered. The aim of this study was to screen potential pseudo att sites of ΦC31 integrase in the human genome, and evaluate the risks of its application in human gene therapy.
Methods:
TFBS (transcription factor binding sites) were found on the basis of reported pseudo att sites using multiple motif-finding tools, including AlignACE, BioProspector, Consensus, MEME, and Weeder. The human genome with the proposed motif was scanned to find the potential pseudo att sites of ΦC31 integrase.
Results:
The possible recognition motif of ΦC31 integrase was identified, which was composed of two co-occurrence conserved elements that were reverse complement to each other flanking the core sequence TTG. In the human genome, a total of 27924 potential pseudo att sites of ΦC31 integrase were found, which were distributed in each human chromosome with high-risk specificity values in the chromosomes 16, 17, and 19. When the risks of the sites were evaluate more rigorously, 53 hits were discovered, and some of them were just the vital functional genes or regulatory regions, such as ACYP2, AKR1B1, DUSP4, etc.
Conclusion:
The results provide clues for more comprehensive evaluation of the risks of using ΦC31 integrase in human gene therapy and for drug discovery.
Keywords: gene therapy, ΦC31 integrase, pseudo attP, human genome, motif finding, drug discovery
Introduction
In the year 2002 in France, three children who were SCID (Severe Combined Immunodeficiency) patients developed a T-cell leukemia several years after they had undergone treatment using retroviral vectors. It is now believed that the leukemia developed as a result of the activation of a known proto-oncogene, LMO2, adjacent to the “disabled” retroviral vector insertion sites1,2. Because several other failures had been reported earlier, this severe adverse event aroused great interest among scientists studying the non-viral and site-specific Streptomyces phage ΦC31 Int system, which is expected to be a new tool in human gene therapy3,4.
The presence of ΦC31 Int mediates site-specific recombination without host factors between two short sequences, attP and attB, in phage and bacterial genomes5. As a result, the extrinsic gene could be integrated into the host chromosomes at various pseudo attachment (att) sites, flanked by two hybrid att sites, attL and attR6 (Figure 1). All the previously identified pseudo sites share a common TT(C)G core sequence. By using the ΦC31 Int system, Olivares et al achieved an enhanced long-term expression of human a1-antitrypsin (hAAT) and human factor IX (hFIX) in mice7, and therapeutic levels of the protein (4000 ng/mL) were successfully maintained for 8 months. However, one major question about this novel system remains: how many “risk” sites are buried in the human genome? Because the system can still integrate at various locations in the human genome, the evaluation of the safety and efficacy of this method ultimately depends on being able to predict its particular bias. However, validating all these sites in the whole genome by using experimental techniques alone seems impossible. Thus, a genome-wide computation-aided analysis is greatly desired to help identify the Int's recognition motif and the corresponding distribution of those potential sites in the human genome.
Figure 1.

The ΦC31 Int system can also be used for mammalian genome modification, especially in basic research of gene therapy. In the presence of this Int, without host factors, the integration reaction mediates recombination between a short sequence of mammalian genome DNA, the pseudo attachment site-attP, and a short sequence in extrinsic DNA vectors, the attachment site-attB. The extrinsic gene integrates into the mammalian chromosome where it is flanked by two hybrid att sites, attL and attR. Once the integration occurs, the extrinsic genes can be stably integrated into the host genome with high efficiency.
In this study, a classical representation of the conserved motifs, Position Specific Scoring Matrix (PSSM), was applied to calculate an approximation of the specific protein-DNA interaction8,9,10. PSSMs have been widely used in modeling transcription factor binding sites (TFBSs), and various computational tools have been developed and successfully applied to distinguish TFBSs from promoter regions where the true binding sites are embedded. These tools provide techniques for the analysis of the reported sites of the ΦC31 Int because both the transcription factor DNA binding and the Int att sites recognition share the similar character of specific protein–DNA interaction. Understanding the target preference of the ΦC31 Int would help researchers evaluate the risks associated with this new method, prior to its use in human gene therapy.
Material and methods
Previous work has identified some “minimal” recognition sites, including the wild-type attP sites, attB sites and pseudo attachment sites in different genomes, such as bacterial, human and mouse5,6,11,12. Twenty different sites have been retrieved from the literature, and these sites, ranging from 39 bp to 161 bp, share an average length of 83 bp. Considering both strands of the att sites and allowing for some mutations, we performed a sequence comparison between one strand and its reverse complementary strand, for each sequence.
TRANSFAC and Jaspar were used to image the profile of the TFBSs13,14. Let S be a set of N aligned binding sites with length l for a particular protein; let nj(b) be the number of times base b is in position j, and let fj(b) be the frequency of this event. Usually, PSSM assumes independence between positions. Often a Bayesian estimate15 is used to handle the zero frequency case and fj(b) is replaced with,
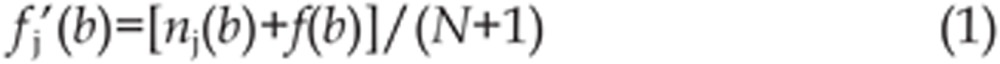 |
where f(b) is the overall background frequency of base b. In optimized MatInspector16, the variant of information content Ci-value used to measure the conservation of position i was calculated using the equation,
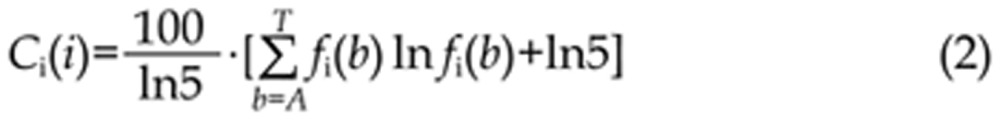 |
To search for a candidate sequence, optimized MatInspector uses this information to scan the candidate sequence s=(s1, ..., sl) and measures the similarity between the candidate and the most conserved nucleotides at each position. The score function is given by,
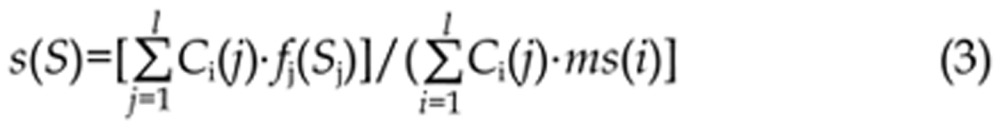 |
where ms(i) is the maximum frequency of bases in position i.
Eq (1) is used to represent the PSSM of a group of reported binding sites, and Eq (3) is used to score a candidate binding site.
Methods for conserved elements discovery
Putative recognition motifs were identified by a suite of motif discovery programs.
AlignACE is based on a Gibbs sampling algorithm and returns a series of motifs that are overrepresented in the input set17;
BioProspector modifies the motif model used in the earlier Gibbs samplers to allow for the modeling of gapped motifs and motifs with palindromic patterns18;
Consensus is based on a greedy algorithm and models motifs using PSSM with a maximum information content19;
MEME discovers one or more motifs in a collection of DNA or protein sequences by using the technique of expectation maximization to fit a two-component finite mixture model to the set of sequences20;
Weeder uses a pattern-driven method that exhaustively enumerates all the oligos up to a maximum length21;
MotifSampler, also based on Gibbs sampling algorithm, improves performance through a high-order background model22.
The different strategies (Greedy method, enumerating method and statistical method) are used in the implementation of these programs. All these methods have been widely and successfully applied to infer potential TFBSs. According to Tompa et al's assessment of these computational techniques, they are favorable for short sequences23.
Similarity of inter-motifs and intra-motifs
We construct a distance metric using a Pearson Correlation Coefficient. A similarity of two columns x=(xA; xC; xG; xT) and y=(yA; yC; yG; yT) in two PSSMs can be measured by Ref24,
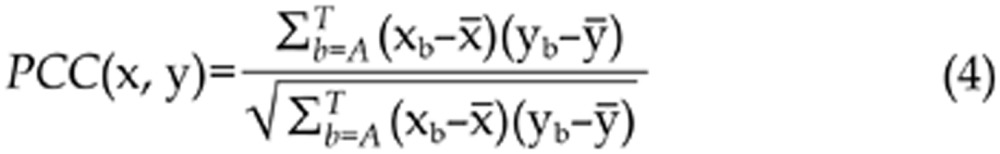 |
where
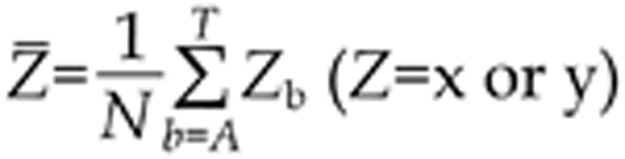 |
.
In practice, the different motifs may not be optimally aligned. We adopt the cores of length k in the measuring procedure as described in Ref24, wherein the optimal continuation of the k-cores are considered to be ideal to the aligned motifs. Alignments to the reverse complement of the motifs are included here. To compare two matrices consisting of multiple columns, the scores of individual columns are summed up.
Structure motif model and specificity of risks in chromosomes
One of the challenges in scanning candidate sites is to control for false positives. A motif co-occurrence strategy can be useful in solving this problem induced by TFBS identification25. Therefore, the triplet structure motif in Figure 2 can be represented by the co-occurrence model T=M1NaMcoreNbM2(Na and Nb are any a- and b-length bases between the two adjacent elements, respectively), where M1 and M2 are two conserved complementary motifs and Mcore is TTG or AAC and their reverse complements GTT or CAA. The model can also be represented by M1NgM2 (g=a+b+3 and TTG is included in Ng), which can be conveniently used to screen genome sequences.
Figure 2.

The two conserved pattern are reverse complement to each other. In the middle, the TTG core (or AAC) is the attaching sites. The logos are generated by Weblogo48.
To describe risks specificity in different chromosomes, we adopt the ratio between the predicted sites and the size of chromosome i, rsi(T)=Ni(T)/Li, rather than the number of the predicted sites. This can help parameterize the risks specificity involved in one chromosome, where the Li (Mbp) is the length of chromosome i and Ni(T) is the number of sites in the chromosome.
Results
Conserved elements and their PSSMs
Usually, DNA-binding proteins bind to different DNA sequences that are not necessarily identical but highly conserved. One DNA-binding domain could recognize DNA sequences of 4–10 bp that share a conserved pattern called a motif or profile26,27. A large number of computational tools have been designed to infer the binding elements on a set of promoter sequences of co-regulated genes17,18,19,20,21,22,23. These tools have been successfully applied in identifying binding sites in various organisms28. Therefore, we uses these motif-finding tools to detect the conserved pattern in the att sites of the ΦC31 Int. Six motif-discovering methods, AlignACE, Consensus, MEME, BioProspector, Weeder, MotifSampler (as detailed in Material and methods), were used to analyze the 20 reported sequences. Acquiring the results from multiple tools can improve the accuracy of the final prediction, compared to any of these tools used alone29. Subsequently, we clustered these motifs according to k-means strategy with PCC similarity. The clustering result showed that the consistent consensus is GGGGTKBS (IUPAC nomenclatures for DNA consensus). Consensus only finds an approximate substring GGTGCC of the consensus GGGGTKBS.
Although the consensus' found by the tools are consistent, it is still hard to extract the exact positions from the long sequences because of some noisy signals. However, the occurrences in the sites, except the ones in mouse chromosomes 7, 10, 12, 14, 17, and X (These sites are named “Long-sites” for they are relatively long), can be easily extracted manually. Consequently, the pattern GGGGTKNC and its reverse complement, GNMACCCC, separated by the Core TT(C)G in the middle, were identified. The profile of the palindrome structure embedded with the core TTG (TCG was ignored for further screening in the next section) forms a triplet structure motif T (Figure 2), which we used further to discover the occurrences of the structure motif in the Long-sites. We found that the Long-sites contain the triplet structure motif, but the occurrences of the structure motif in the Long-sites are more difficult to discriminate from the background signal noise, as compared to the short sites. Because the PSSM-scoring method is sensitive to the occurrences used for constructing the profile, we excluded the occurrences in the Long-sites to ensure the accuracy of the screened results.
Potential sites in the human genome
To identify the potential recognition sites in the human genome, we have screened the genome sequences with the triplet structure T shown in Figure 2. We focused on sites with a distance between 3 and 33 bp from the consensus GGGGTKBS and its reverse complement GNMACCCC because the lengths of the known sites used to construct the profile are no more than 46 bp. Thus, there is a spacer of at most 30 bp split by the 3-bp long core TT(C)G between the 8-nucleotide conserved motifs GGGGTKNC and its reverse complement GNMACCCC. In our analysis, we have used variant information content, which is a common PSSM scoring strategy (detailed in Materials and methods), to score each arm of a candidate for further understanding the occurrence of the structural motif. We then merged the occurrences of both arms into one site if 1) the distance between the two arms was between 3 and 33 bp and 2) TTG (or AAC) existed between the two arms.
One problem with the PSSM scoring strategy is setting up a good cutoff scoring value. The assumption that the score of the candidate follows a normal distribution appears to be valid, and a vast majority of the known sites fall within two standard deviations (μ±2δ) of the mean of the previous elements' scoring value25,30. The problem is that many of the false-positive signals cannot be discriminated from the result when μ-2δ is set as the threshold of the score value.
We used P-values to calculate the significance of a candidate. In general, the lower the P-value, the more significant the site. The relationship between thresholds of the PSSM score values and the number of sites, and between thresholds of P-values and the number of sites, are shown in Figure 3. We can see that as the thresholds increase, the number of sites decrease dramatically. Therefore, there is a trade-off between the accuracy of the result and the completeness of the list of potential sites.
Figure 3.

The number of sites is sensitive to the threshold value of scoring function. And the inter box show the highest P-value of the sites which scoring under the cressponding scoring value.
We set the significance for the threshold value to 0.0005 for both arms of the structural motif. Therefore, each arm of a candidate that needs identification must have a P-value lower than or equal to 0.0005. We identified a total of 27 924 sites, when overlap was not allowed. Details of the sites are listed in additional file 2. Because the degrees of risk vary in different chromosomes, we measured the specificity of the risk in each chromosome i by rsi(T) (Materials and methods). The results are shown in Figure 4.
Figure 4.

The risk specificity value are used to measure the ratio of recognizing the potential sites. From the result, the expected risk specificity under null hypothesis and the actural risk specificity correlate well. And it also can be found that the chromosome 19, 17, 20, 16 have relatively higher risks. Similar result has also been found in the HIV integration sites preference31.
Table 1 lists the distribution of the sites in each human chromosome. The risk in different chromosomes is also of interest, and rsi (T) and rsi' (T) were measured (Figure 4). From the results, we can see that human chromosomes 16, 17, and 19 have high-risk specificity values; these chromosomes have relatively high gene density, and they are very active in gene transcription (Figure 4). Surprisingly, this result correlates well with Schröder et al's findings that the HIV integrase also has the most integration sites in chromosome 19, and a considerably high number of integration sites in chromosomes 16 and 1731. The expected risks are measured by the probability of M1, M2, and Mcore under the null hypothesis and correlate well with the actual risk specificity. To evaluate the risks of the sites more rigorously, we reset the significance for each arm element to 2×10-5; 53 hits were discovered. These 53 were then used to Blast-search the human genome. Among these sites, 14 sites were located within or near the coding region of important functional genes, such as ACYP2, AKR1B1, DUSP4, etc (Table 2). In Table 2, we also list some representative potential risk sites that are quite similar to the wild-type attP sites in length, although they have lower significance value for each arm; these include STK11, LENG4, CYP2B6, RYR1, and ICAM1. Mitchell et al compared retroviral vectors derived from three viruses, including two common gene therapy vectors and reported 3127 sites where the retroviruses typically integrated into the human genome32. Different vectors show different target preferences, and many of them are notably prone to target active genes. Previously, “disabled” retroviral systems have been shown to trigger several lethal and rare hereditary diseases. The site-specific ΦC31 Int system is also likely to recognize some important genes or regulatory regions, such as the pseudo attP sites in the human chromosomes, though little evidence of risks has been found in previous studies. The further use of “molecular monitoring” to screen all the potential pseudo att sites to find its exact risks, is not a very feasible option. The data that are currently available, including previous experimental results, may constitute a very small part of the big picture, and they cannot confirm that the Int system is safer than the previously used systems. Although the ΦC31 Int system is highly efficient, the occurrence of unexpected pathological changes cannot be ruled out.
Table 1. Targets of the ΦC31 Int in human chromosomes.
| Chromosome ID | rs'(T) (×10-6) | Number of potential sites | Chromosome length (bp) |
|---|---|---|---|
| Chr1 | 5.29 | 2255 | 245 203 898 |
| Chr2 | 4.96 | 2092 | 243 315 028 |
| Chr3 | 4.23 | 1475 | 199 411 731 |
| Chr4 | 3.71 | 1205 | 191 610 523 |
| Chr5 | 3.97 | 1262 | 180 967 295 |
| Chr6 | 4.25 | 1227 | 170 740 541 |
| Chr7 | 6.05 | 1555 | 158 431 299 |
| Chr8 | 5 | 1237 | 145 908 738 |
| Chr9 | 4.96 | 1216 | 134 505 819 |
| Chr10 | 5.51 | 1380 | 135 480 874 |
| Chr11 | 6.4 | 1470 | 134 978 784 |
| Chr12 | 5.77 | 1284 | 133 464 434 |
| Chr13 | 3.05 | 600 | 114 151 656 |
| Chr14 | 4.62 | 854 | 105 311 216 |
| Chr15 | 5.37 | 871 | 100 114 055 |
| Chr16 | 8.49 | 1294 | 89 995 999 |
| Chr17 | 10.2 | 1403 | 81 691 216 |
| Chr18 | 4.1 | 575 | 77 753 510 |
| Chr19 | 14.4 | 1429 | 63 790 860 |
| Chr20 | 7.27 | 836 | 63 644 868 |
| Chr21 | 4.26 | 360 | 46 976 537 |
| Chr22 | 8.79 | 755 | 49 476 972 |
| ChrX | 4.33 | 1111 | 152 634 166 |
| ChrY | 1.86 | 178 | 50 961 097 |
Table 2. Some potential risk sites found in the human genome.
| Gene | Hits | Brief discription |
|---|---|---|
| Acyp2 | GGGGTTCCCCCTTGTTCTTGGGTCGGGATGCAGTCCAGGAACCCC | Acylphosphatase 2, muscle type |
| Akr1b1 | GGGGTGCCCAGATTTTTCTCCCGAGTTCCAGACCCAGGGCACCCC | Aldo-ketoreductase family 1, member B1 |
| Dusp4 | GGGGTTCCTTATCCTTCCCACCCGCCCTCAAACCCAGGAACCCC | Dual specificity phosphatase 4 |
| Ptpn5 | GGGGTGCCCCCATGCGGCAAGGTCCGAGATGTGCCGGCACCCC | Protein tyrosine phosphatase, non-receptor |
| Fli1 | GGGGTTCCTTTTACAGAGACAATTGTTGGGTCAAGAAGGAACCCC | Friend leukemia virus integration 1 |
| Gpt2 | GGGGTTCCCTTGAACATGCGTAGGCTGGAACCCC | Glutamic pyruvate transaminase (alanine aminotransferase) |
| Slc7a5 | GGGGTGCCTTGGGGGCAGTGCATTGGAGGAACCCC | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 5 |
| Dhdh | GGGGTTCCAGGTAGAGGTTGAAAGGACCAAGGAACCCC | Dihydrodiol dehydrogenase (dimeric) |
| Phf2 | CCCCAAGGAGCCCAGGACTTGGCCTTGGGG | PHD finger protein 2 |
| Igf1r | CCCCAAGGAAAGCATATCATAAACAAGTTTTCCCTTGGGG | Insulin-like growth factor 1 receptor |
| Wwp2 | CCCCAAGGGTACAGAACAGTGTCACCTTTGCCGTGGGG | WW domain containing E3 ubiquitin protein ligase 2 |
| Flj39501 | CCCCAAGGTCACATTAATTGAGCGATCCGTGGGG | Cytochrome P450, family 2, subfamily E, polypeptide 2 homolog |
| Plcb1 | CCCCAAGGCTCCATGTCTGAACATCATCACCTTGGGG | Phospholipase C, beta 1 (phosphoinositide -specific) |
| Flj10945 | CCCCAAGGGCACACGTAGAAGCAGAGTTCCTTGGGG | Hypothetical protein FLJ10945 |
| Stk11* | CCCCAGGGAGGCGGGGCTTTTGTGCAGAAATGTAGGGTTGGGG | This gene encodes a tumor suppressor |
| Leng4 | CCCCAGTTGAGAAGCACTTGTCTAAACACTGGGG | This gene is malignant cell expression-enhanced gene/ |
| Tumor progression -enhanced gene | ||
| Cyp2b6 | CCCCACTATTTATTTTTGTAGAGATGTGTTTGGGG | Cytochrome P450, family 2, subfamily |
| Ryr1 | CCCCAAGGTCCGGGTTGGGGACCTTGTGCTGGGG | Ryanodine receptor 1 (skeletal); Multi-process involved |
| Icam1 | CCCCAGCCGAGAATTTCTCTTTGCGTCCTTCCTACTTTGGGG | Intercellular adhesion molecule 1 (CD54); Multi-process involved |
* Hit in STK11 has a relatively lower score but it has a similar motif to the wide type attPsites. So does the last four sites, LENG4, CYP2B6, RYR1, and ICAM1.
Discussion
Our work aims to screen for potential target sites of the ΦC31 in the human genome. By combining the PSSM-based score function and the co-occurrence model, we can significantly reduce the number of false-positive signals, and the multiple computational techniques that were combined in this study can more accurately identify conserved motifs than any of the techniques used alone.
There were several questions that we wanted to answer. Which motif would this integrase prefer to recognize in the more evolved human genomes? Why are the pseudo-att sites so different from the wild type att sites derived from its original host, even though they are all recognized by the same integrase? Previous studies remind us that the answers may be found in the basic principles of the protein-DNA interaction33. In other words, certain DNA-binding domains formed by combinations of α helices, β-sheets and loops would strongly select and bind to specific sites in the genome, and protein and DNA can adapt to different conformations with sufficient flexibility. This flexibility affects target site preference and also the diversity of sites that the proteins can bind to. For example, proteins that form dimers tend to bind the palindromic sites34, and the specificity of binding sites of the C2H2 zinc finger protein relies on the critical basic regions of the protein35. Moreover, Benos et al 's experiments with the EGR family proteins, and Mandel-Gutfreund et al's quantitative modeling method, have shown the specificity of the Arg-G recognition pair; if placed appropriately, the arginine (Arg) usually specifically recognizes guanine (G) in most cases, independent of the protein family36,37.
Generally, most recombinases and integrases are composed of three distinct domains: the DNA recognition domain, the catalytic domain and sometimes a dimerization domain. The DNA-binding regions result in site-specific enzymes, such as λ Int, γδ resolvase and Cre recombinase; these enzymes display a similar three-dimensional organization to other Int family members34. By assembling α helices, β-sheets and loops, they recognize “minimal” DNA substrates, in which typically 4–10-bp long inverted repeats are separated by a spacer that is 6 bp or longer, and each of the repeats binds to a monomer of the recombinase33,34. The inverted repeats are also called “core-type” binding sequences. The structure usually has a U-shaped cavity in which the DNA is bound38, and the DNA is usually severely deformed as a result of the binding process.
However, no crystal structure of the large serine integrases, including ΦC31, has been resolved till date, and the precise nature of the molecular events during strand exchange are not clearly understood. Also the results obtained from protein structure tools, such as 3D-PSSM39, FUGUE40, mGenTHREADER41, SAMT9942, PDB-blast (http://bioinformatics.burnham-inst.org/pdbblast/) are disappointing. Though the ΦC31 Int shares low sequence similarity with other members of the serine Int family, they are predicted to be highly conserved in the C terminus as an HLH structure, which often mediates dimerization between proteins. Specifically, a C4 motif conserved in the Int serine family has been identified to be a zinc ribbon DNA-binding structure by multiple sequence alignment (Figure 5). Another member of the serine Int family, ccrB (cassette chromosome recombinase B, Q8RPD2), has been identified by Pfam43 to have a topoisomerase DNA-binding C4 zinc ribbon domain44,45. This particular domain is mainly found in topoisomerases from prokaryotes. A tyrosine in this domain is involved in the transient breakage of a DNA strand, with subsequent formation of a covalent protein-DNA intermediate. Similarly, a tyrosine residue is highly conserved in the center of the C4 motifs. Local structure predictions for the above proteins show that they are all conserved in the C4 motif (Figure 5) as repetitive three β-sheets, which are highly conserved in the typical zinc ribbon domains.
Figure 5.

Alignment of C4 motifs in the serine Int family aligned by ClustalX with manual editing. All of them share a repetitive three β-sheets followed by a HLH structure. A tyrosine is also conserved in these proteins. Q9T221 is the ΦC31 Int derived from Streptomyces. Uniquely, an arginine-rich basic region lies in the centre of the C4 motif of the ΦC31 Int and a conserved HLH structure follows. The alignment is colored and the 80% consensus sequence of the domain calculated using Chroma tool49. Capital letters represent amino acids. Lower-case letters: b, big; h, hydrophobic; l, aliphatic; p, polar; s, small. A secondary structure of this alignment profile is predicted using Jpred50 and PROF51.
Interestingly, we noticed that there is a G-rich perfect reverse complementary motif on both arms of the core sequence in the wild type attP site derived from the original bacterial host genome. A similar sequence occurs in the human ψA site, which is preferentially targeted compared to the already found att sites in human cells6. This result emphasizes the fact that if there are no perfectly matched wild type att sites in the human genome, then there can be a series of particular biased targets, which might share a low sequence similarity, but can be recognized by certain key amino acids in the specific DNA-binding domain. Thus, a general profile of the recognition sites, most likely a core-type motif instead of a concrete and consecutive oligonucleotide sequence, would actually be preferentially recognized and would even allow for some mutants or variants.
A minimum of 39-bp long attP and 34-bp attB have been proven to be sufficient to enable an efficient integration. Additionally, reduction experiments showed that some nucleotides, such as G (first G in the GGGGT) and C (last C in ACCCC) in the flanked region, are necessary for efficient integration5. The reduction experiments show that deletion of the last guanine in the G-rich region of the 39-bp minimal wild-type attP site leads to a significant drop in the recombination rate from 100% to 71.1%, and a further deletion of both the adjacent guanine and a cytosine in the C-rich region located symmetrically across the core sequence results in an almost complete loss of the recombination activity. Importantly, other parts within the minimal site contribute less to site recognition, and they are poorly conserved in the primary sequences5,46. This implies that there are certain nucleotides that play a very important role in the specific recognition on both arms of the core sequence. Therefore, considering that the implicit mechanism of the integration may be similar to previously reported recombinases/integrases, we used multiple motif-finding methods to give a more accurate description of the hidden conservation, and a “core-type” binding motif was found to be preferred by the ΦC31 Int.
The motif identified in this work also seems to be an echo of our structure prediction. It is interesting that the Arg(R)-rich basic region and a guanine (G)-rich motif are likely to shape a specific complex structure; each region seems to enhance the other's function. Thus, the DNA-binding domain of the ΦC31 Int positions itself into the major groove and specifically interacts with the base edges in the G-rich motif, while the N-terminal catalytic domain envelopes the substrates in a synapse using networks of secondary structures. Then, a site-specific integration occurs via a Holliday intermediate, during which four helical arms flank the crossover point. Of course, the exact nature of the recognition process will be uncovered through structure parsing, and further experiments need to be conducted. Nevertheless, we have provided an alternative method to identify the underlying recognition rules by computational analysis that is both time- and fund-saving. Furthermore, our work provides a genome-wide estimation that more than twenty thousand sites in the human genome are likely to be recognized by the ΦC31 Int, some of which are located in very important human genes.
As listed in Table 2, FLJ39501 and CYP2B6 belong to the Cytochrome P450 family. Drug metabolism by Cytochrome P450s plays an important role in the disposition and in the pharmacological and toxicological effects of drugs, which is an early consideration for ADME (Absorption, Distribution, Metabolism, Elimination). CYP2B6 is the major enzyme responsible for the metabolism of selegiline, a drug used in the treatment of Parkinson's disease. GPT2 (also known as Alanine aminotransferase) is a widely used index of liver integrity or hepatocellular damage in clinics, as well as a key enzyme in intermediary metabolism. Stk11 is a tumor suppressor gene and also the major pathogenic gene of human Peutz-Jeghers syndrome (PJS), a rare hereditary disease in which there is predisposition to benign and malignant tumors of many organ systems.
Because the ΦC31 Int can integrate at various sites in the human genome and because the Int system is highly efficient in genome modification, not only the number of the targets based on its preference but also the exact position of these targets comprise a vital index for risk evaluation. However, the data that is available, both from laboratory experiments and computer-aided analysis, are not sufficient for an accurate conclusion. Additionally, considering the complexity of the protein-DNA interaction and the influence of other factors, including cell cycle and chromosomal states, risk evaluation for this system is far more complex than that had been previously imagined. Further research is necessary to identify the detailed molecular mechanism of how the ΦC31 Int finds its specific DNA targets. However, ΦC31 Int has proven quite promising in human gene therapy, and as we have concluded, if the number of targets can be reduced to 53 well-matched hits in the human genome with the increase of significant value, the Int can be modified to have not just a safer target preference but also better efficiency by directed evolution of this integrase47. Thus, the system is expected to be more powerful in future clinical research.
Author contribution
Zhi-peng HU, Lu-sheng CHEN, Wei WANG, Huan-zhang ZHU, and Jiang ZHONG designed research; Zhi-peng HU, Lu-sheng CHEN, Cai-yan JIA, and Wei WANG performed research; Huan-zhang ZHU and Jiang ZHONG contributed new reagents or analytic tools; Lu-shen CHEN and Cai-yan JIA analyzed data; Zhi-peng HU and Wei WANG wrote the paper.
Acknowledgments
We thank Fei WANG from the Intelligent Information Processing Lab, Department of Computer Science of Fudan University for her kindly offering of related materials. We also thank Rodolf FLEISCHER, also from Department of Computer Science of Fudan University, for his improvement of the manuscript.
References
- Cavazzana-Calvo M, Thrasher A, Mavilio F. The future of gene therapy. Nature. 2004;427:779–81. doi: 10.1038/427779a. [DOI] [PubMed] [Google Scholar]
- Kohn DB, Sadelain M, Glorioso JC. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat Rev Cancer. 2003;3:477–88. doi: 10.1038/nrc1122. [DOI] [PubMed] [Google Scholar]
- Check E. A tragic setback. Nature. 2002;420:116–8. doi: 10.1038/420116a. [DOI] [PubMed] [Google Scholar]
- Khan MS, Khalid AM, Malik KA. Phage phiC31 integrase: a new tool in plastid genome engineering. Trends Plant Sci. 2005;10:1–3. doi: 10.1016/j.tplants.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci U S A. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol Cell Biol. 2001;21:3926–34. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat Biotechnol. 2002;20:1124–8. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Benos PV, Bulyk ML, Stormo GD. Additivity in protein-DNA interactions: how good an approximation is it. Nucleic Acids Res. 2002;30:4442–51. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stormo GD. Quantitative analysis of EGR proteins binding to DNA: assessing additivity in both the binding site and the protein. BMC Bioinformatics. 2005;6:176. doi: 10.1186/1471-2105-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes P, Till R, Bee S, Smith MC. The streptomyces genome contains multiple pseudo-attB sites for the (phi)C31-encoded site-specific recombination system. J Bacteriol. 2002;184:5746–52. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–41. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–4. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–14. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–45. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Liu X, Brutlag DL, Liu JS. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac Symp Biocomput. 2001. pp. 127–38. [PubMed]
- Stormo GD, Hartzell GW. Identifying protein-binding sites from unaligned DNA fragments. Proc Natl Acad Sci U S A. 1989;86:1183–7. doi: 10.1073/pnas.86.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Pavesi G, Mereghetti P, Mauri G, Pesole G. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 2004;32:W199–203. doi: 10.1093/nar/gkh465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs G, Lescot M, Marchal K, Rombauts S, De Moor B, Rouzé P, et al. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001;17:1113–22. doi: 10.1093/bioinformatics/17.12.1113. [DOI] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–44. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Schones DE, Sumazin P, Zhang MQ. Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics. 2005;21:307–13. doi: 10.1093/bioinformatics/bth480. [DOI] [PubMed] [Google Scholar]
- Bulyk ML, McGuire AM, Masuda N, Church GM. A motif co-occurrence approach for genome-wide prediction of transcription-factor-binding sites in Escherichia coli. Genome Res. 2004;14:201–8. doi: 10.1101/gr.1448004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Liu JS. De novo cis-regulatory module elicitation for eukaryotic genomes. Proc Natl Acad Sci U S A. 2005;102:7079–84. doi: 10.1073/pnas.0408743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol. 2002;20:835–9. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ST, Liu JS. BioOptimizer: a Bayesian scoring function approach to motif discovery. Bioinformatics. 2004;20:1557–64. doi: 10.1093/bioinformatics/bth127. [DOI] [PubMed] [Google Scholar]
- Robison K, McGuire AM, Church GM. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J Mol Biol. 1998;284:241–54. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Austin SE, Berman HM, Thornton JM.An overview of the structures of protein-DNA complexes Genome Biol 20001REVIEWS001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voziyanov Y, Pathania S, Jayaram M. A general model for site-specific recombination by the integrase family recombinases. Nucleic Acids Res. 1999;27:930–41. doi: 10.1093/nar/27.4.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Friedman N, Margalit H. Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol. 2005;1:e1. doi: 10.1371/journal.pcbi.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benos PV, Lapedes AS, Stormo GD. Probabilistic code for DNA recognition by proteins of the EGR family. J Mol Biol. 2002;323:701–27. doi: 10.1016/s0022-2836(02)00917-8. [DOI] [PubMed] [Google Scholar]
- Mandel-Gutfreund Y, Margalit H. Quantitative parameters for amino acid-base interaction: implications for prediction of protein-DNA binding sites. Nucleic Acids Res. 1998;26:2306–12. doi: 10.1093/nar/26.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: A structural analysis. J Mol Biol. 1999;287:877–96. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–57. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–56. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–80. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahumada A, Tse-Dinh YC. The Zn(II) binding motifs of E coli DNA topoisomerase I is part of a high-affinity DNA binding domain. Biochem Biophys Res Commun. 1998;251:509–14. doi: 10.1006/bbrc.1998.9500. [DOI] [PubMed] [Google Scholar]
- Tse-Dinh YC, Beran-Steed RK. Escherichia coli DNA topoisomerase I is a zinc metalloprotein with three repetitive zinc-binding domains. J Biol Chem. 1988;263:15857–9. [PubMed] [Google Scholar]
- Groth AC, Calos MP. Phage integrases: biology and applications. J Mol Biol. 2004;335:667–78. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- Sclimenti CR, Thyagarajan B, Calos MP. Directed evolution of a recombinase for improved genomic integration at a native human sequence. Nucleic Acids Res. 2001;29:5044–51. doi: 10.1093/nar/29.24.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstadt L, Ponting CP. CHROMA: consensus-based colouring of multiple alignments for publication. Bioinformatics. 2001;17:845–6. doi: 10.1093/bioinformatics/17.9.845. [DOI] [PubMed] [Google Scholar]
- Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. JPred: a consensus secondary structure prediction server. Bioinformatics. 1998;14:892–3. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- Ouali M, King RD. Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 2000;9:1162–76. doi: 10.1110/ps.9.6.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]