

Abstract
Adoptive cell-transfer therapy (ACT) has been reported to suppress growing tumors and to overcome tumor escape in animal models. As a candidate ACT effector, γ9δ2T cells can be activated and expanded in vitro and in vivo and display strong antitumor activity against colorectal, lung, prostate, ovarian and renal cell carcinomas. However, it is difficult to obtain a large enough number of γδT cells to meet the need for immunotherapy that can overcome the cancer patients' immune suppressive tumor microenvironment. In previous studies, our lab confirmed that γ9δ2T cells recognized tumor cells via the CDR3δ region of the γδ-T-cell receptor (TCR). We constructed full-length human peripheral blood mononuclear cell (PBMC)-derived γ9 and δ2 chains in which the CDR3 region was replaced by an ovarian epithelial carcinoma (OEC)-derived CDR3. We transferred the CDR3δ-grafted γ9δ2TCR into peripheral blood lymphocytes (PBLs) to develop genetically modified γ9δ2T cells. In vitro studies have shown that these CDR3δ-grafted γ9δ2T cells can produce cytokines after stimulation with tumor cell extracts and exhibit cytotoxicity towards tumor cells, including human OEC and cervical adenocarcinoma. CDR3δ-grafted γ9δ2T cells adoptively transferred into nude mice bearing a human OEC cell line demonstrated significant antitumor effects. These results indicate that CDR3δ-grafted γ9δ2T cells might be candidates for clinical tumor immunotherapy.
Keywords: CDR3δ-grafted γ9δ2TCR, tumor immunotherapy, γδT cells
Introduction
Adoptive cell-transfer therapy (ACT) is a promising immunotherapy strategy that has been reported to suppress growing tumors and to overcome tumor escape in both animal models and human patients1, 2, 3, 4 However, ACT has had limited clinical success in cancer treatment due to the requirement of preexisting tumor-reactive cells5 Genetic modification of autologous lymphocytes with antigen-specific T-cell receptors (TCRs) is a promising potential tool for solving this problem, as these cells have shown antitumor effects both in vitro and in vivo.6, 7, 8, 9 Moreover, clinical studies have shown that transfer of genetically modified T cells resulted in objective regression of metastatic melanoma in a certain percentage of patients10 The challenge of applying genetically modified T cells in cancer immunotherapy is the autoreactivity of those cells, as their TCRs are hybrids consisting of one exogenous and one endogenous chain.11 A novel strategy for generating non-autoreactive T cells with antitumor specificity could greatly improve upon current cancer immunotherapies.
γδT cells, a T-cell subset, directly recognize tumor antigens through the γδTCR, bypassing traditional antigen-presenting cell-mediated antigen recognition12 γδT cells have a broad TCR repertoire, recognizing an array of antigens including nonpeptide phosphoantigens, mitochondrial ATPase and human mutS homolog 2,13, 14, 15 some of which cannot be recognized by αβT cells. γ9δ2T cells belong to the major subset of γδT cells in peripheral blood, and are regarded as a potential candidate for tumor immunotherapy because this subset has been shown to display strong antitumor activity against different type of cancer cells.16, 17, 18, 19, 20 However, the low frequency of γ9δ2T cells in peripheral blood greatly limits their potential clinical applications. γ9δ2T cells account for only 2–5% of the total peripheral blood T-cell repertoire. Expansion of these cells for ACT requires laborious and time-consuming cell culture procedures. Moreover, in the patients with median-stage or late-stage malignant tumors or who are being treated with chemotherapy, neither the quantity nor immunological activity of their seed γ9δ2T cells can meet the treatment demands. As the antitumor effect of γ9δ2T cells mainly depends on the γδTCR, which cannot exchange chains with the αβTCR,21, 22 we hypothesized that large numbers of cytolytic γδT cells could be generated through forced expression of γδTCRs on lymphocytes.
Antigen recognition by γδTCRs depends on the sequence of its peptide, especially that of complementarity determining region 3 (CDR3). Compared with the γ-chain, the sequence of the δ-chain CDR3 region is both longer and more variable.23 This suggests that the CDR3δ may play a vital role in antigen recognition by the γδTCR. Adams et al. analyzed the crystal structures of the G8 γδTCR with a T22 molecule as its antigen and found that the G8 CDR3δ formed a loop which directly contacted T2224 Previously, our lab confirmed that the γ9δ2TCR recognized tumor antigens via the CDR3δ region. We synthesized OT3, a CDR3δ peptide derived from tumor-infiltrating lymphocytes (TILs) in ovarian epithelial carcinoma (OEC). Our data demonstrated specific interactions between OT3 and tumor cells, tumor tissues and tumor cell extracts.25 The OT3 sequence is sufficient for tumor recognition by the TCR, as a CDR3δ-grafted antibody, in which the heavy chain CDR3 region was replaced by the OT3 sequence, showed specific binding to tumor cell lines.19
We have generated genetically modified T cells by forcing the expression of CDR3δ-grafted γ9δ2TCR on peripheral blood lymphocytes (PBLs). PBLs transfected with the γ9δ2 (OT3) vector successfully express the γ9δ2 (OT3) TCR on the cell surface. CDR3δ-grafted γ9δ2T cells showed cytotoxic activity against tumor cells, including human OEC and cervical adeno-carcinoma, and marked cytokine production when stimulated with tumor extracts. Adoptive transfer of CDR3δ-grafted γ9δ2T cells into nude mice bearing a human OEC cell line resulted in significant therapeutic effects. These results implicate CDR3δ-grafted γ9δ2T cells as a promising tumor-repression tool with applications in clinical cancer therapy.
Materials and methods
Cell culture
Human tumor cell lines, including HeLa (a human uterine cervix carcinoma), Daudi (a Human Burkitt's lymphoma) and Raji (a Human Burkitt's lymphoma cell line), were obtained from the Cell Culture Center, Institute of Basic Medicine, Chinese Academy of Medical Sciences, Beijing, China. The human OEC lines SKOV3 and HO8910 were provided by Dr Keng Shen (Department of Gynecology, Peking Union Medical College Hospital, Beijing, China). The RetroPack PT67 cell line was purchased from Clontech Laboratories, Inc, Clontech Laboratories, Mountain View, CA, USA. SKOV3 cells were cultured in McCoy5A medium (Sigma, St Louis, MO, USA) with 10% fetal bovine serum (HyClone). The HO8910, Daudi and Raji cell lines were maintained in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum. HeLa and RetroPack PT67 cells were propagated in Dulbecco's modified Eagle's medium (Sigma) with 10% fetal bovine serum. Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors granting informed consent by density gradient centrifugation on Ficoll-Hypaque (Pharmacia, Piscataway, NJ, USA). PBMCs were stimulated for 3 days with immobilized anti-CD3 antibody (UCHT1; BD Pharmingen, San Diego, CA, USA) and IL-2 (200 IU/ml).
Plasmid construction and viral production
The full-length sequence of the γ9 and δ2 chains with a tumor antigen-specific CDR3 region derived from OEC TILs has been described previously26 Briefly, the full-length γ9 and δ2 sequence was amplified from PBMC cDNA from a healthy donor by PCR using primers directed at the 5'-untranslated region (UTR) and 3'-UTR of each chain. The first half of the full-length γ9 or δ2 sequence was amplified using a forward primer targeting the 5'-UTR and a reverse complement primer directed at the CDR3. The second half of the full-length γ9 or δ2 sequence was amplified using a forward primer targeting the CDR3 region and a reverse primer targeting the 3'-UTR. The overlapped PCR products were used as templates for the generation of a δ2 sequence containing the tumor antigen-specific CDR3 sequence. The full-length γ9 sequence was inserted into a BglII and XhoI-treated pMSCVhyg vector and was named pMSCVhyg-γ9. The PCR product from the full-length δ2 chain containing the tumor antigen-specific CDR3 region was subcloned into the XhoI and BglII-site of the pMSCVneo vector and was named pMSCVneo-δ2 (OT3). All plasmids were sequenced using ABI 3770 automatic sequencer (Applied Biosystems, Foster City, CA, USA), which confirmed the sequences had been cloned correctly. The pMSCVhyg-γ9, pMSCVneo-δ2 (OT3), pMSCV-hyg and pMSCV-neo vectors were transfected into a RetroPack PT67 cell line by using the Lipofectamine 2000 kit (Invitrogen, Carlsbad, CA, USA). Hygromycin and neomycin were added to the transfected packaging cells for 2 weeks. The retroviral supernatant was concentrated and stored at −80 °C.
Peripheral blood lymphocytes transfected with retrovirus
PBLs isolated from healthy donors were stimulated for 3 days and transfected using a modified version of the method described by Bunnell et al27Briefly, 1×106 cells/ml in media containing retrovirus and 200 IU/ml IL-2 were transferred into non-tissue culture 24-well plates precoated with Retronectin (recombinant human fibronectin fragment CH-296, 20 µg/cm2; Takahara, Otsu, Japan). Plates were centrifuged at 300g for 90 min at 32 °C and incubated overnight at 37 °C in a humidified 5% CO2 incubator. This transfection procedure was then repeated two more times. On day 8, PBLs were selected in geneticin (0.5 mg/ ml) and hygromycin B (0.1 mg/ ml) for 3 days. The live cells were adjusted to 1×106 cells/ml and maintained in culture medium supplemented with 200 IU/ml IL-2 until day 14.
Reverse transcription (RT)-PCR analysis of γδTCR expression
The mock-transfected and TCR-transfected cells were harvested after retroviral transfection. Total RNA was prepared using TRIzol regent (Invitrogen) according to the manufacturer's protocol. The RNA quality was evaluated with 1% agarose gel electrophoresis. Single-strand cDNA was synthesized using oligo-dT (Promega, Madison, WI, USA) and moloney murine leukemia virus reverse transcriptase (Promega). Single-strand cDNA was used as a template for PCR amplification of the full-length γ9 chain, the full-length δ2 chain, the γ9 chain CDR3 region, the δ2 chain CDR3 region and β-actin used for normalization. The following primer sequences were used for amplification: γ9-5: GAAGATCTGCCACCATGCTGTCACTGCTCCACACAT; γ9-3: CCGCTCGAGTGATTTCTCTCCATTGCAGCAG; δ2-5: CCGCTCGAGGCCACCATGCAGAGGATCTCCTCCCTCATCC; δ2-3: GAAGATCTCTTACAAGAAAAATAACTTGGCAGTC; CDR3γ9-5: AATGTAGAGAAACAGGAC; CDR3γ9-3: ATCTGTAATGATAAGCTTT; CDR3δ2-5: GCCATTGAGTTGGTGCCTGAACAC; CDR3δ2-3: AAACGGATGGTTTGGTATGAGGC; β-actin-5: GCTCGTCGTCGACAACGGCT; β-actin-3: CAAACATGATCTGGGTCATCTTCTC. PCR products were resolved using a 1% agarose gel.
Western blot analysis of γδTCR expression
The transfected cells were harvested and lysed in sodium dodecyl sulfate–polyacrylamide gel electrophoresis sample buffer. Total cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrically transferred onto a nitrocellulose membrane. After blocking with 5% nonfat milk, the blot membrane was incubated with an anti-γ9 antibody (Immunotech, Marseille, France) as the primary antibody. After incubation with a horseradish peroxidase-conjugated secondary antibody, enhanced chemiluminescence was performed using the SuperSignal West Pico Trial Kit (Pierce Biotechnology Inc., Rockford, IL, USA). The glyceraldehyde 3-phosphate dehydrogenase antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Flow cytometric analysis
Cells (1×106) were harvested, washed and suspended in 50 µl phosphate-buffered saline (PBS) containing 1% bovine serum albumin FITC- and phycoerythrin-conjugated antibodies were added to each reaction. After incubation for 30 min at 4 °C, cells were washed twice, resuspended in 500 µl PBS containing 1% formaldehyde and analyzed on a FACSort flow cytometer (Becton Dickinson, Mountain View, CA, USA). FITC-conjugated anti-TCRγδ (IMMU510), phycoerythrin-conjugated anti-TCRαβ (BMA031) and the respective isotype control monoclonal antibodies (mAbs) were purchased from Immunotech.
Cytokine release assay
The transfected cells were tested for antitumor cell reactivity using a cytokine release assay. Twenty-four-well plates were treated with 40 µg/ml SKOV3, HO8910, HeLa, Daudi and Raji cell extracts for 2 h at 37 °C. After three washes, 1×106 cells transfected cells were plated in complete medium containing 200 IU/ml IL-2. After 24 or 72 h of coculture, the supernatants were harvested. Interferon (IFN)-γ and tumor necrosis factor (TNF)-α secretion were measured using enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. For the blocking assay, effector cells were incubated with 10 µg/ml anti-TCRγδ mAbs (B1.1; eBioscience, San Diego, CA, USA) or isotype control for 2 h at 37 °C and then cocultured with SKOV3, HO8910 and HeLa extracts. Supernatant IFN-γ production was measured using ELISA.
Cytotoxicity and blocking assay
The cytotoxicity of the transfected cells was assessed using a colorimetric 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. SKOV3, HeLa and HO8910 tumor cells were added as target cells to 96-well plates at a concentration of 1×104 cells/well. Transfected effector cells were incubated with target cells in varying concentrations for 8 h Subsequently, MTT solution (5 mg ml–1) was added to each well (10 µl/well) and incubated at 37 °C for an additional 4 h. The reaction was stopped by adding 100 µl dimethyl sulfoxide to dissolve the tetrazolium crystals. The plate was read at 570nm with 630nm correction using a Multiskan Microplate Reader (Thermo Labsystems, Helsinki, Finland) and cytotoxicity was calculated using the following formula: TOD–[(E+T)OD–EOD]/TOD×100%, where TOD=optical density (OD) value of target cells, EOD=OD value of effector cells and (E+T)OD=OD value of effector cells cultured with target cells.
For the blocking assay, effector cells were incubated with 10 µg/ml anti-TCRγδ mAbs (B1.1; eBioscience) or isotype control for 2 h at 37 °C and then cocultured with the target cells. For the anti-Fas antibody blocking assay, target cells were incubated with a saturating concentration of anti-Fas mAbs (ZB4; Immunotech) for 2 h at 37 °C and then cocultured with the effector cells. In other experiments, the effector cells were treated with a saturating concentration of concanamycin A (CMA; Sigma-Aldrich, Munich, Germany) and brefeldin A (BFA; Sigma-Aldrich) for 2 h, mixed with target cells at an effector/target (E/T) ratio of 10∶1 and then subjected to cytotoxicity assays.
Mice
Athymic BALB/c nu/nu mice (female, 4–6 weeks) were purchased from the Laboratory Animal Center of the Chinese National Institute for the Control of Pharmaceutical and Biological Products. Mice were maintained in micro-isolator cages in a specific pathogen-free facility at the Animal Center, Institute of Basic Medical Sciences, CAMS. The care and use of animals were approved by the Animal Use Committees of the CAMS Institute.
Animal tumor model
To test the antitumor effects of the CDR3δ-grafted γ9δ2T cells in vivo, SKOV3 cells (1×106) were injected subcutaneously in the back right flank of BALB/c nu/nu mice. When the majority of tumors grew to a size of approximately 100 mm3, mice bearing tumors were randomly divided into three groups and injected intratumorally with CDR3δ-grafted γ9δ2T cells (1×106, n=6), mock-transfected cells (1×106, n=6) or PBS (1×106, n=6). Human IL-2 was administrated intraperitoneally at a dose of 5000 IU for all mice in the CDR3δ-grafted γ9δ2T cells and mock-transfected cell-treated groups. The treatment was repeated every 3 days for a total of four times. The general condition of the nude mice and tumor progression were observed every 2 days. Tumor volumes were estimated using the formula 3.14×[largest diameter×(perpendicular diameter)2]/6. Mouse survival was also observed.
Statistical analysis
Data were presented as means±s.d. Repeated measures analysis of variance and independent samples t-tests were used to analyze all data as indicated in the results and in the figure legends. The log rank test was used in Kaplan–Meier survival analysis. When P was <0.05, the difference was considered statistically significant.
Results
Construction of CDR3δ-grafted γ9δ2TCR-transfected lymphocytes
The γ9 and δ2 chains were PCR-amplified from PBMC cDNA from healthy donors. The CDR3 region of the δ2 chain was replaced with the tumor antigen-specific CDR3 sequence of the TILs from the OEC (Figure 1a). The full-length γ9 and δ2 sequences with the tumor antigen-specific CDR3 sequences were cloned into retroviral vectors. Stimulated PBLs from healthy donors were transfected with a retrovirus containing the γ9 and δ2 chain. After transfection, the γδTCR expression on the expanded cells was assessed using RT-PCR, western blotting and flow cytometric analysis. The RT-PCR assay showed that the γ9 and δ2 chain cDNA containing the tumor antigen-specific CDR3 sequences were successfully expressed in PBLs (Figure 1b). The expression of the γ9 and δ2 chains was confirmed by western blot (Figure 1c). After transfection with the γ9 and δ2 chains about 70% of PBLs successfully expressed the γδTCR on the cell surface, while only 3% of PBLs transfected with mock vector did so (Figure 1d).
Figure 1.
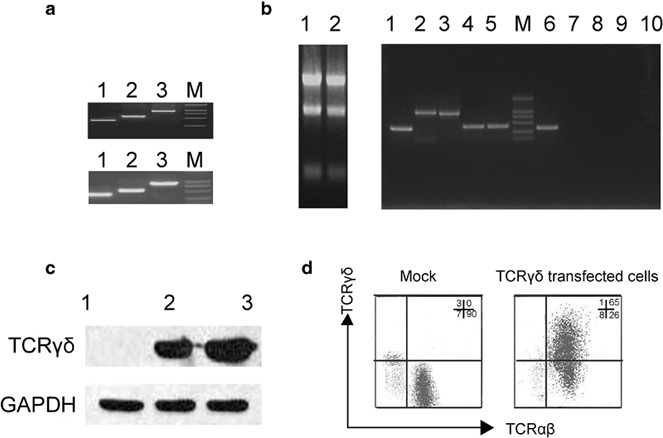
Transfected PBLs expressed the γδTCR with a tumor antigen-specific CDR3δ sequence. (a) PCR products of the full-length γ9 and δ2 chains containing the tumor-specific CDR3 sequence. The upper image displays the PCR products of the full-length γ9 chain. Lane 1: the first half sequence of the full-length γ9 chain; lane 2: the second half of the full-length γ9 chain; lane 3: the full-length γ9 chain. The lower image shows the PCR products of the full-length δ2 chain containing the tumor antigen-specific CDR3 sequence derived from TILs isolated from OEC. Lane 1: the first half sequence of the full length δ2 chain containing the OEC-derived CDR3 sequence; lane 2: the second half of the full length δ2 chain with the OEC-derived CDR3 sequence; lane 3: the full length δ2 chain with the OEC-derived CDR3 sequence. (b) PBLs isolated from a healthy donor were stimulated for 3 days and then transfected with a retrovirus containing pMSCVhyg-γ9 and pMSCVneo-δ2 (OT3) in culture media containing 200 IU/ml IL-2. The expression of the full-length γ9 and δ2 chains and their respective CDR3 region mRNA in transfected cells was analyzed by RT-PCR. Left: the mRNA extracted from mock-transfected cells (lane1) and TCR-transfected cells (lane2); right: the β-actin from TCR-transfected cells (lane1);, the full-length γ9 chain from the TCR-transfected cells (lane 2); the full-length δ2 chain from the TCR-transfected cells (lane 3); the CDR3 region of the γ9 chain from the TCR-transfected cells (lane 4); the CDR3 region of the δ2 chain from the TCR-transfected cells (lane 5); β-actin expression in the mock-transfected cells (lane 6); the full-length γ9 chain from the mock-transfected cells (lane 7); the full-length δ2 chain from the mock-transfected cells (lane 8); the CDR3 region of the γ9 chain from the mock-transfected cells (lane 9); and the CDR3 region of the δ2 chain from the mock-transfected cells (lane 10). (c) Western blot analysis of γδTCR expression. Lane 1: mock control; lane2: TCR-transfected cells; lane 3: γδT cells as a positive control. (d) Surface expression of the γδTCR on transfected cells was analyzed by FCM. The cells were stained with FITC-conjugated anti-γδTCR mAbs and polyethylene-conjugated anti-αβTCR mAbs. γδTCR and αβTCR cell surface expression on mock-transfected cells (left) and TCRγδ-transfected cells (right) was determined by flow cytometry. One representative result from three independent experiments is shown. FCM, flow cytometry; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; OEC, ovarian epithelial carcinoma; PBL, peripheral blood lymphocyte; RT, reverse transcription; TCR, T-cell receptor; TIL, tumor-infiltrating lymphocyte.
Recognition and cytotoxicity of CDR3δ-grafted γ9δ2TCR-transfected lymphocytes (CDR3δ-grafted γ9δ2T cells)
After transfection with γ9 and δ2 chain, the CDR3δ-grafted γ9δ2T cells were incubated with tumor cell extracts to investigate cytokine production. After 72 h, IFN-γ concentration in the supernatant was evaluated. As shown in Figure 2a, CDR3δ-grafted γ9δ2T cells secreted more IFN-γ than mock-transfected cells after stimulation with Daudi cell extract. CDR3δ-grafted γ9δ2T cells and mock-transfected T cells had no significant differences in IFN-γ secretion after stimulation with Raji cell extract. CDR3δ-grafted γ9δ2T cells produced much more IFN-γ than mock-transfected cells after stimulation with HeLa, SKOV3 and HO8910 cell extracts (P<0.01). TNF-α production by CDR3δ-grafted γ9δ2T cells after stimulation with various tumor cell extracts was also measured. CDR3δ-grafted γ9δ2T cells produced more TNF-α than mock vector-transfected T cells after stimulation with HeLa, SKOV3 and HO8910 cell extracts. An MTT assay was used to test the transfected T-cell cytotoxicity. The results showed that CDR3δ-grafted γ9δ2T cells effectively killed HeLa and HO8910 cells and were particularly effective at killing SKOV3 cells (Figure 2c). The CDR3δ-grafted γ9δ2T cell cytotoxicity was as twofold higher than mock-transfected cells at an E/T ratio of 20∶1 (P<0.01). The above results suggest that the observed antitumor effects of CDR3δ-grafted γ9δ2T cells is γδTCR-dependant. As shown in Figure 3, CDR3δ-grafted γ9δ2T cell IFN-γ production after stimulation with tumor cell extracts was inhibited by an anti-TCR mAb (P<0.05) (Figure 3a), and the transfected T-cell killing of tumor cells was significantly blocked by an anti-TCR mAb (Figure 3b–d).
Figure 2.
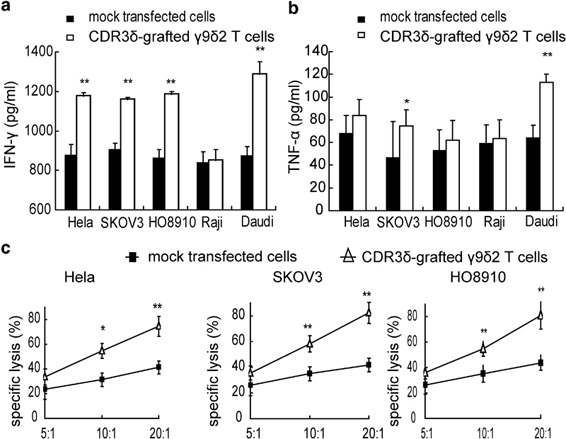
Tumor cell recognition and cytotoxicity by CDR3δ-grafted γ9δ2T cells. (a) The CDR3δ-grafted γ9δ2T cells or mock-transfected cells were incubated with tumor cell extracts from HeLa, SKOV3, HO8910, Daudi and Raji cell lines for 72 h, after which the supernatant was harvested and IFN-γ production was measured by ELISA. (b) The CDR3δ-grafted γ9δ2T cells or mock-transfected cells were incubated with HeLa, SKOV3, HO8910, Daudi and Raji cell extracts for 24 h, after which the supernatant was harvested for detection of TNF-α production by ELISA. (c) The MTT assay was used to evaluate the transfected cell cytotoxicity against tumor cells. The transfected effector cells were incubated with HeLa, SKOV3 and HO8910 target cells at different effector-to-target ratios for 8 h MTT solution (5 mg/ml) was added to each well (10 µl/well) and cells were incubated at 37 °C for an additional 4 h. The reaction was stopped by the addition of 100 µl DMSO to dissolve the tetrazolium crystals. The plate was examined at 570nm with 630nm correction in a Multiskan Microplate Reader. Asterisks indicate significant differences compared to the mock control: *P<0.05, **P<0.01. All data are representative of three independent experiments. DMSO, dimethylsulfoxide; IFN, interferon; MTT, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Figure 3.

The antitumor effects of CDR3δ-grafted γ9δ2T cells were blocked by an anti-TCRγδ mAb. (a) CDR3δ-grafted γδT cells were incubated with an anti-TCRγδ mAb, a mouse IgG1 isotype control or no mAb for 2 h and then stimulated with tumor cell extracts for 72 h. IFN-γ secretion in the supernatants was detected by ELISA. (b–d)The CDR3δ-grafted γ9δ2T cells were incubated with an anti-TCRγδ mAb, mouse IgG1 isotype control or no treatment for 2 h and then cocultured with tumor target cells at different E/T ratios. The MTT assay was used to evaluate cytotoxicity. Asterisks indicated significant differences compared to the mock control, *P<0.05, **P<0.01. The results are representative of three independent experiments. E/T, effector/target; IFN, interferon; mAb, monoclonal antibody; MTT, 3-(4,5-dimethylHSthiazol-2-yl)-2,5-diphenyltetrazolium bromide; TCR, T-cell receptor.
Cytotoxic mechanism of CDR3δ-grafted γ9δ2T cells
To further investigate the CDR3δ-grafted γ9δ2T cell antitumor cytotoxic mechanisms, tumor cells were treated with an anti-Fas mAb at several different concentrations before incubation with effector cells. The effector cell killing ability decreased as anti-Fas mAb concentration increased. As shown in Figure 4a, the killing inhibition rate of HO8910, SKOV3 and HeLa cell lines with 5000 ng/ml anti-Fas mAb added to culture was 29.41%, 18.13% and 22.91%, respectively. Similar results were measured after treatment with BFA, a protein transport inhibitor that can block Fas ligand surface expression on effector cells. When CDR3δ-grafted γ9δ2T cells were treated with BFA, the antitumor killing ability decreased in a dose-dependant manner and peaked with the addition of 25 µM BFA to culture (Figure 4b). This suggested that the CDR3δ-grafted γ9δ2T cells cytotoxic mechanism was Fas–FasL-related, but did not entirely depend on Fas–FasL pathway. Therefore, we theorized that the perforin–granzyme B pathway might be involved.
Figure 4.
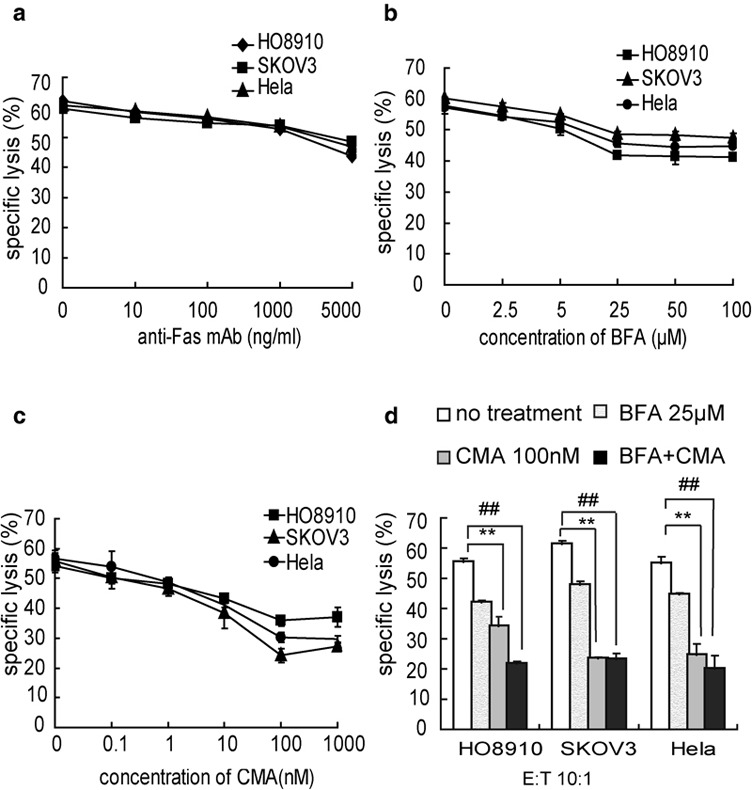
The CDR3δ-grafted γ9δ2T antitumor cytotoxicity mechanism. (a) Influence of treatment with an anti-Fas mAb on cytotoxicity of CDR3δ-grafted γ9δ2T cells against various tumor cell lines. Tumor cells were treated with an anti-Fas mAb at saturating concentrations for 2 h before being incubated with effector cells. (b) Effects of BFA on CDR3δ-grafted γ9δ2T-cell antitumor cytotoxicity. CDR3δ-grafted γ9δ2T cells were treated with BFA at saturating concentrations for 2 h before being incubated with target tumor cells (HeLa, SKOV3 and HO8910). (c) Effects of CMA on CDR3-grafted γ9δ2T-cell antitumor cytotoxicity. CDR3δ-grafted γ9δ2T cells were incubated with CMA at saturating concentrations for 2 h before the cytotoxicity assay. (d) CDR3δ-grafted γ9δ2T cells were pretreated with a combination of CMA (100 nM) and BFA (25 µM) before being cocultured with tumor cells at an E/T ratio of 10:1. **P<0.01, no treatment group vs. CMA-treated group; # # P<0.01, no treatment group vs. CMA+BFA-treated group. The results are representative of three independent experiments. BFA, brefeldin A; CMA, concanamycin A; E/T, effector/target; mAb, monoclonal antibody.
To test if this pathway played a role, CDR3δ-grafted γ9δ2T cells were treated with CMA, an inhibitor of the perforin–granzyme B pathway, at different concentrations before incubation with tumor cells. This treatment inhibited killing in a dose-dependant manner, reaching the peak level of inhibition at a concentration of 100 nM. Killing of HO8910, SKOV3 and HeLa cell lines was inhibited by 33.93%, 56.71% and 46.52%, respectively(Figure 4c). When CMA and BFA treatment were combined with a concentration of 100 nM and 25 µM, respectively, CDR3δ-grafted γ9δ2T-cell cytotoxicity was inhibited by over 60% against all three tumor cell lines (Figure 4d). These results suggested that both the Fas–FasL and perforin–granzyme B pathways play a role in CDR3δ-grafted γ9δ2T-cell killing of tumor cells, and that the perforin–granzyme B pathway could play a major role.
In vivo antitumor activity of CDR3δ-grafted γ9δ2T cells
We further investigated the antitumor efficacy of the CDR3δ-grafted γ9δ2T cells in vivo BALB/c nude mice were subcutaneously inoculated with SKOV3 cells. When the tumor mass grew to approximately 100 mm3, CDR3δ-grafted γ9δ2T cells, mock-transfected cells, or PBS were injected intratumorally every 3 days for a total of four injections. Mice treated with CDR3δ-grafted γ9δ2T cells showed slowed tumor growth compared to mice treated with mock-transfected cells or PBS (P<0.05, Figure 5a), whereas mock-transfected T cell-treated mice and PBS-treated mice displayed similar tumor progression (P>0.05). Intratumoral injections of CDR3δ-grafted γ9δ2T cells not only delayed the early stage tumor development but also prolonged the survival of the treated mice. As shown in Figure 5b, the median survival time of mice in the PBS treated group was 37 days and the maximum survival time was 49 days. The median survival time of mice treated with mock-transfected cells was 40 days. In contrast, the median survival time of mice treatment with CDR3δ-grafted γ9δ2T cells was 47 days, significantly longer than the PBS-treated and mock-transfected T cell-treated groups (P<0.05).
Figure 5.

In vivo antitumor effects of CDR3-grafted γ9δ2T cells. Mice were injected subcutaneously 1×106 SKOV3 cells in the back right flank. When the tumor mass grew to approximately 100 mm3, CDR3δ-grafted γ9δ2T cells, mock-transfected cells or PBS were injected intratumorally every 3 days for a total of four injections. Human IL-2 was administered intraperitoneally at a dose of 5000 IU for all mice with the exception of the PBS group. The general condition of nude mice and tumor progression were observed every 2 days. Mouse survival was observed every day. (a) Mice treated with CDR3δ-grafted γ9δ2T cells showed slow tumor mass growth compared to mice treated with mock-transfected cells or PBS (P<0.05), whereas mock-transfected cells-treated mice and PBS-treated mice displayed similar tumor progression (P>0.05). (b) Survival of nude mice bearing SKOV3 cells was prolonged by treatment with CDR3-grafted γ9δ2T cells plus IL-2. Treatment with CDR3-grafted γ9δ2T cells resulted in longer survival when compared to PBS treated group or mock-transfected group (P<0.05). PBS, phosphate-buffered saline.
Discussion
γδT cells have unique characteristics that are not seen in αβT cells. γδT cells can directly recognize antigens without MHC restriction,12 have a wide antigen recognition spectrum, which includes protein antigens and phosphoantigens13 and can directly kill several tumor types including colorectal, lung, prostate, ovarian and renal cell carcinomas.16, 18, 25, 28, 29 Because of these features, γδT cells are an interesting candidate effector for antitumor immune therapy. There are two commonly available strategies for using γδT cells in cancer therapy. One is to stimulate γδT cells in vivo and the other is to expand γδT cells in vitro.30 Both strategies require a large number of γδT cells with high avidity and potent antitumor activity. However, the quantity and specificity of resulting γδT cells correlate with the original repertoire of γδT cells isolated from patients. Because of cancer patients' immune suppressive tumor microenvironment, seed γδT cells are difficult to isolate and expand in vitro. Because γδT cells show natural antitumor activity, we hypothesized that their tumor reactivity could be used to construct genetically modified γδT cells, overcoming the previously mentioned problems. Moreover, genetically modified γδT cells may avoid the disadvantage of genetically modified αβT cells in which hybridization can result in TCRs with potentially autoreactive specificities.11
To generate genetically modified γδT cells, we needed to find a γδTCR that could both recognize tumor antigens and kill tumor cells. In our previous studies, we confirmed that the CDR3δ region played an important role in antigen recognition by the TCRγδ. We found that synthesized OEC-derived CDR3δ-peptides could mimic the TCRγδ and bind specifically to tumor cell lines and tissues25 A CDR3δ-grafted antibody also showed specific binding to several tumor cells19 We used synthesized OEC-derived CDR3δ peptides as probes to search for putative TCR-binding epitopes by screening a 12-mer random peptide phage-displayed library and identified several putative TCR ligands within tumor cell extracts by affinity chromatography and liquid chromatography/electrospray ionization tandem mass spectrometry analysis. Nine peptides and two proteins that bound the γδTCR been identified including human mutS homolog 2, a novel γδTCR ligand15, 31 Another research group also found that the CDR3δ region was responsible for the T22 protein binding specificity of the Vδ2 T cells clone G832 We had previously constructed full-length PBMCs-derived γ9 and δ2 chains using overlapping PCR. The full-length γ9 and δ2 chains with tumor antigen-specific CDR3 regions were stably transfected into J.RT3-T3.5 by electroporation and expressed functional γ9δ2TCR26 In this study, we induced γ9 and δ2 (OT3) expression in PBLs using a retroviral vector and generated efficient tumor reactive T cells. We chose the retroviral transfection system instead of electroporation to avoid the damage that electroporation causes to primary cells. The resulting CDR3δ-grafted γ9δ2T cells could produce inflammatory cytokines after stimulation with tumor cell extracts and secretion of IFN-γ was impaired by treatment with an anti-γδTCR mAb. CDR3δ-grafted γ9δ2T cells were effective at killing tumor cells, and their cytotoxicity was blocked by an anti-γδTCR mAb. These data demonstrated that the CDR3δ-grafted γ9δ2T cell antitumor response depended on the engineered TCR.
Moreover, in a mouse adoptive transfer model, treatment with CDR3δ-grafted γ9δ2T cells plus IL-2 significantly impaired tumor growth and prolonged the survival of tumor-bearing mice.
Immune effector cells recognize and destroy tumor target cells via two main mechanisms: the death receptor-dependent pathway and the perforin-dependent pathway. The death receptor-dependent pathway acts through interactions between Fas with its ligand FasL. One study investigating the mechanism behind Vγ1T-cell cytotoxicity showed that FasL expression and Fas–FasL interactions were necessary for γδT-cell killing of infection-elicited activated macrophages33 The expression of FasL on effector cells is an inducible process. After antigen recognition, the cytotoxic cells synthesized FasL mRNA de novo, translated it into protein and transported it to the cell surfaces through the Golgi.
Pore-forming proteins are constitutively expressed by peripheral blood γδT cells,34 suggesting that these proteins play a vital role in γδT-cell cytotoxicity. Moreover, Vγ9δ2T lymphocytes were reported to kill microorganisms residing in intracellular compartments in a perforin and granzyme-dependant manner.35
In our study, we used an anti-Fas mAb, BFA and CMA to block CDR3δ-grafted γ9δ2T-cell killing of tumor cells. CMA functions as an inhibitor of the perforin and granzyme pathway by imcreasing the pH of lytic granules to accelerate the degradation of perforin. The anti-Fas mAb blocks the Fas–FasL pathway by blocking ligation of Fas protein expressed on tumor cells. BFA, a protein transport inhibitor, blocks trafficking of FasL to the surface of effector cells. We found that the cytolytic activity of genetically modified γ9δ2T cells was mediated through both the Fas-dependent and perforin-dependent pathways, but the perforin–granzyme pathway was dominant. These results demonstrated that cells transfected with the CDR3δ-grafted TCR might naturally acquire γ9δ2TCR-like cytotoxicity towards tumor cells.
Upon activation, γδT cells primarily produce Th1-associated cytokines including IFN-γ and TNF-α36 IFN-γ has been described to inhibit tumor proliferation in some models and plays a central role in immune surveillance of tumors. TNF-α has also been reported to play a pivotal role in tumoricidal activity of perforin/IFN-γ double knockout effector T cells37 We found that CDR3δ-grafted γ9δ2T cells secreted IFN-γ and TNF-α after antigen stimulation with tumor cell extracts. Moreover, IFN-γ secretion was inhibited by treatment of CDR3-grafted γ9δ2T cell with anti-γδTCR mAbs. These results indicated that genetically modified γδT cells responded directly to tumor antigens and were activated after stimulation with tumor cells.
In conclusion, we have demonstrated that PBLs from healthy donors can be transfected with a γ9δ2TCR-containing tumor-specific CDR3δ regions isolated from OEC TILs. The resulting CDR3δ-grafted γ9δ2T cells secreted high levels of cytokines upon stimulation with tumor cell extracts and exhibited cytotoxicity against tumor cells. Adoptive transfer of CDR3-grafted γ9δ2T cells slows tumor progression and prolongs the survival of mice inoculated with human tumor cells. These data suggest that CDR3-grafted γ9δ2T cells are a promising tumor immunotherapy strategy. Further research will focus on optimizing the transfection method and evaluating the mechanism of tumor rejection in greater detail.
Acknowledgments
This work is supported by two grants, no. 2006AA02Z480 and no. 2007AA021109, from the National Program for Biomedical Hightech, the Ministry of Science and Technology, China. The authors gratefully acknowledged Dr Keng Shen for providing tumor cell line SKOV3 and HO8910.
References
- Peng L, Shu SKrauss JC. Treatment of subcutaneous tumor with adoptively transferred T cells. Cell Immunol. 1997;178:24–32. doi: 10.1006/cimm.1997.1124. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- Plautz GE, Bukowski RM, Novick AC, Klein EA, Kursh ED, Olencki TE, et al. T-cell adoptive immunotherapy of metastatic renal cell carcinoma Urology 199954617–623.discussion 623–614. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513. [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol. 2001;2:962–970. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Wolkers MC, Schumacher TN. Adoptive transfer of T-cell immunity. Trends Immunol. 2002;23:264–269. doi: 10.1016/s1471-4906(02)02219-6. [DOI] [PubMed] [Google Scholar]
- Hayday AC. Gammadelta cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. 2000;18:975–1026. doi: 10.1146/annurev.immunol.18.1.975. [DOI] [PubMed] [Google Scholar]
- Kabelitz D, Wesch D, He W. Perspectives of gammadelta T cells in tumor immunology. Cancer Res. 2007;67:5–8. doi: 10.1158/0008-5472.CAN-06-3069. [DOI] [PubMed] [Google Scholar]
- Scotet E, Martinez LO, Grant E, Barbaras R, Jeno P, Guiraud M, et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. 2005;22:71–80. doi: 10.1016/j.immuni.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Chen H, He X, Wang Z, Wu D, Zhang H, Xu C, et al. Identification of human T cell receptor gammadelta-recognized epitopes/proteins via CDR3delta peptide-based immunobiochemical strategy. J Biol Chem. 2008;283:12528–12537. doi: 10.1074/jbc.M708067200. [DOI] [PubMed] [Google Scholar]
- Kang N, Zhou J, Zhang T, Wang L, Lu F, Cui Y, et al. Adoptive immunotherapy of lung cancer with immobilized anti-TCRgammadelta antibody-expanded human gammadelta T-cells in peripheral blood. Cancer Biol Ther. 2009;8:1540–1549. doi: 10.4161/cbt.8.16.8950. [DOI] [PubMed] [Google Scholar]
- Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, et al. Gammadelta T cells for immune therapy of patients with lymphoid malignancies. Blood. 2003;102:200–206. doi: 10.1182/blood-2002-12-3665. [DOI] [PubMed] [Google Scholar]
- Dieli F, Vermijlen D, Fulfaro F, Caccamo N, Meraviglia S, Cicero G, et al. Targeting human gammadelta T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007;67:7450–7457. doi: 10.1158/0008-5472.CAN-07-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang T, Hu H, Zhang H, Yang Z, Cui L, et al. Targeting solid tumors via T cell receptor complementarity-determining region 3delta in an engineered antibody. Cancer Lett. 2008;272:242–252. doi: 10.1016/j.canlet.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Sato K, Ashihara E, Takeuchi M, Maita S, Tsuchiya N, et al. Intravesical administration of gammadelta T cells successfully prevents the growth of bladder cancer in the murine model. Cancer Immunol Immunother. 2009;58:493–502. doi: 10.1007/s00262-008-0571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JH, Heemskerk MH. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006;66:3331–3337. doi: 10.1158/0008-5472.CAN-05-4190. [DOI] [PubMed] [Google Scholar]
- van der Veken LT, Coccoris M, Swart E, Falkenburg JH, Schumacher TN, Heemskerk MH. Alpha beta T cell receptor transfer to gamma delta T cells generates functional effector cells without mixed TCR dimers in vivo. . J Immunol. 2009;182:164–170. doi: 10.4049/jimmunol.182.1.164. [DOI] [PubMed] [Google Scholar]
- Rock EP, Sibbald PR, Davis MM, Chien YH. CDR3 length in antigen-specific immune receptors. J Exp Med. 1994;179:323–328. doi: 10.1084/jem.179.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams EJ, Strop P, Shin S, Chien YH, Garcia KC. An autonomous CDR3delta is sufficient for recognition of the nonclassical MHC class I molecules T10 and T22 by gammadelta T cells. Nat Immunol. 2008;9:777–784. doi: 10.1038/ni.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zhang H, Hu H, He H, Wang Z, Xu Y, et al. Gammadelta T cells recognize tumor cells via CDR3delta region. Mol Immunol. 2007;44:302–310. doi: 10.1016/j.molimm.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Xi X, Guo Y, Chen H, Xu C, Zhang H, Hu H, et al. Antigen specificity of gammadelta T cells depends primarily on the flanking sequences of CDR3delta. J Biol Chem. 2009;284:27449–27455. doi: 10.1074/jbc.M109.011684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci USA. 1995;92:7739–7743. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvaisier M, Moreau-Aubry A, Diez E, Bennouna J, Mosnier JF, Scotet E, et al. V gamma 9V delta 2 T cell response to colon carcinoma cells. J Immunol. 2005;175:5481–5488. doi: 10.4049/jimmunol.175.8.5481. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, et al. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol Immunother. 2007;56:469–476. doi: 10.1007/s00262-006-0199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Kimura S, Segawa H, Yokota A, Matsumoto S, Kuroda J, et al. Cytotoxic effects of gammadelta T cells expanded ex vivo by a third generation bisphosphonate for cancer immunotherapy. Int J Cancer. 2005;116:94–99. doi: 10.1002/ijc.20987. [DOI] [PubMed] [Google Scholar]
- He X, Chen H, Wu D, Cui L, He W. Tandem-epitope peptide: a novel stimulator for gammadeltaT cells in tumor immunotherapy. Cancer Lett. 288:86–93. doi: 10.1016/j.canlet.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Shin S, El-Diwany R, Schaffert S, Adams EJ, Garcia KC, Pereira P, et al. Antigen recognition determinants of gammadelta T cell receptors. Science. 2005;308:252–255. doi: 10.1126/science.1106480. [DOI] [PubMed] [Google Scholar]
- Dalton JE, Howell G, Pearson J, Scott P, Carding SR. Fas–Fas ligand interactions are essential for the binding to and killing of activated macrophages by gamma delta T cells. J Immunol. 2004;173:3660–3667. doi: 10.4049/jimmunol.173.6.3660. [DOI] [PubMed] [Google Scholar]
- Nakata M, Smyth MJ, Norihisa Y, Kawasaki A, Shinkai Y, Okumura K, et al. Constitutive expression of pore-forming protein in peripheral blood gamma/delta T cells: implication for their cytotoxic role in vivo. . J Exp Med. 1990;172:1877–1880. doi: 10.1084/jem.172.6.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieli F, Troye-Blomberg M, Ivanyi J, Fournie JJ, Krensky AM, Bonneville M, et al. Granulysin-dependent killing of intracellular and extracellular Mycobacterium tuberculosis by Vgamma9/Vdelta2 T lymphocytes. J Infect Dis. 2001;184:1082–1085. doi: 10.1086/323600. [DOI] [PubMed] [Google Scholar]
- Christmas SE, Meager A. Production of interferon-gamma and tumour necrosis factor-alpha by human T-cell clones expressing different forms of the gamma delta receptor. Immunology. 1990;71:486–492. [PMC free article] [PubMed] [Google Scholar]
- Poehlein CH, Hu HM, Yamada J, Assmann I, Alvord WG, Urba WJ, et al. TNF plays an essential role in tumor regression after adoptive transfer of perforin/IFN-gamma double knockout effector T cells. J Immunol. 2003;170:2004–2013. doi: 10.4049/jimmunol.170.4.2004. [DOI] [PubMed] [Google Scholar]