

Abstract
Inflammation, demyelination, and neurodegeneration are pathological features of multiple sclerosis (MS). In the brains of MS patients, tissue plasminogen activator (tPA) mRNA and protein are upregulated, and changes in the levels of tPA correlate with progression of the disease. However, the role of tPA in MS is as yet unknown. tPA functions in the CNS in neuronal plasticity and cell death. tPA also mediates the activation of microglia, the CNS “immune cells.” In this study, we establish that tPA activity increases during major oligodendrocyte glycoprotein-induced experimental allergic encephalomyelitis (EAE) in normal mice. To explore the role of tPA in this disease as a model for MS, we have examined the EAE course and expression of histopathological markers in mice lacking tPA (tPA−/−). We find that tPA−/−mice have a delayed onset of EAE but then exhibit increased severity and delayed recovery from the neurological dysfunction. Demyelination and axon degeneration are delayed, microglial activation is attenuated, and the production of chemokines is decreased. Our results suggest that tPA and activated microglia have complex roles in MS/EAE, and that these roles are harmful during the onset of the disease but beneficial in the recovery phase. A temporally restricted attenuation of tPA activity could have therapeutic potential in the management of MS.
Keywords: proteolysis, multiple sclerosis, microglia, mice, plasmin, cytokine
Multiple sclerosis (MS) is an autoimmune disease of the CNS. It results in neurological impairments that range from sensory defects to difficulties in movement and paralysis. The cause of MS remains unknown, although viral, environmental, and genetic factors have been proposed to contribute to its development (Martin et al., 1992). Several animal models have been developed to explore causes of and treatments for MS. One of these is experimental allergic encephalomyelitis (EAE), which is characterized by paralysis, weight loss, and mononuclear cell infiltration in the CNS. The pathology of MS is similar and includes multicentric inflammation and demyelination (Opdenakker and Damme, 1994; Steinman, 1996). Despite anti-inflammatory and immunosuppressive therapy, most patients exhibit progressive neurological deterioration. Several groups have now shown that MS is more than just a demyelinating disease; axonal degeneration has been shown to be present throughout active lesions even early in the course of the disease and at the active borders of less acute lesions (Trapp et al., 1998; Waxman, 1998). These findings indicate that axonal loss, in addition to demyelination, occurs early and may also contribute to the development of MS. Therefore, therapeutic modalities aimed at conferring protection to axons in early stages of the disease could reduce the irreversible neurological deficits. Recent reports support this possibility; the AMPA/kainate antagonist 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) protected against oligodendroglial and axonal damage and ameliorated EAE symptoms (Pitt et al., 2000; Smith et al., 2000). Therefore, the degenerative pathway may involve excitotoxicity, although it is not clear whether the NBQX benefit comes from a direct effect on neurons or whether it protects oligodendrocytes and myelin.
In the CSF of MS patients, tissue plasminogen activator (tPA) activity is increased >10-fold compared with reference subjects. Moreover, the increase in tPA activity correlates with the disease progression (Akenami et al., 1997). tPA is also detected in macrophages of inflammatory cuffs in the spinal cord of EAE rats and in degenerating axons during the third relapse of symptoms (Kreutzberg, 1995). tPA mRNA and protein expression are upregulated, and at least some of the increase takes place specifically in neurons (Cuzner et al., 1996;Akenami et al., 1999). tPA is a serine protease that converts the zymogen plasminogen (plg) to the active protease plasmin and thus initiates a potent proteolytic cascade. Mice lacking functional tPA (Carmeliet et al., 1994) are resistant to excitotoxin-induced neurodegeneration (Tsirka et al., 1995). tPA also plays a role in the CNS in neuronal plasticity and reorganization (Seeds et al., 1995; Wu et al., 2000).
Neurons and microglia in the CNS express tPA. Microglia are phagocytic brain cells that resemble tissue-specific macrophages and are considered to be the “immune” cells of the CNS. When microglia sense injury to the brain, they migrate to the wound site, proliferate locally, and undergo a series of changes that characterize their activation (Kreutzberg, 1995). Activated microglia are inextricably linked to neurodegeneration. They are present around the lesions of many neuropathologies and are participants in many experimental models of CNS injury, including excitotoxin injection, ischemia, and autoimmune inflammation (Dickson et al., 1993; London et al., 1996).
To investigate the role of tPA and activated microglia in MS, we used the myelin oligodendrocyte glycoprotein (MOG)-induced EAE model in tPA-deficient (tPA−/−) mice. Our results indicate that tPA affects both the onset of and recovery from acute EAE. We find that tPA contributes to neuronal degeneration at the early stages of EAE, and that its absence causes a delay in the onset of EAE. However, at later stages of EAE, the presence of tPA may be beneficial in neuronal regeneration, because tPA−/−mice exhibit sustained symptoms.
MATERIALS AND METHODS
Mice. C57BL/6(H-2b), C57BL/6-tPA+/−, and C57BL/6-tPA−/− mice were bred in-house under specific pathogen-free conditions [Division of Laboratory Animal Resources at the State University of New York (SUNY) Stony Brook], controlled for temperature (21°C), and maintained with a daily light period of 12 hr. Adult (6- to 8-week-old) female mice were used in all experiments.
MOG peptide. MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was synthesized by Quality Controlled Biochemicals and purified using reverse-phase (C18) HPLC.
Induction of EAE with MOG35–55peptide. EAE was induced actively, as described previously (Bernard et al., 1997), by subcutaneous injection in the flank on day 0 with 300 μg of MOG35–55 peptide thoroughly emulsified in complete Freund's adjuvant (CFA) containing 500 μg of heat-inactivated Mycobacterium tuberculosis (Difco, Detroit, MI). One week later (day 7), mice were boosted with 300 μg of MOG35–55 peptide subcutaneously in the other flank. Pertussis toxin (500 ng; List Biologicals, Campbell, CA) in 200 μl of PBS was injected intraperitoneally on days 0 and 2.
Evaluation of the EAE clinical course. After immunization with MOG, mice were observed and weighed daily. The disease severity was scored on a scale of 0–5 with graduations of 0.5 for intermediate clinical signs. The score is designated as follows (Hjelmstrom et al., 1998): 0, no detectable clinical signs; 1, weakness of the tail; 2, hindlimb weakness or abnormal gait; 3, complete paralysis of the hindlimbs; 4, complete hindlimb paralysis with forelimb weakness or paralysis; 5, moribund or death. Half-scores were assigned when disease signs were intermediate. Paralyzed mice were given easy access to food and hand watered at least twice daily. A mean clinical score was assigned to each group using this scale.
Immunohistochemistry. At different time points during the course of EAE, the mice were killed, and the spinal cords were removed. The dissected spinal cords were embedded in Tissue-Tek (Miles, Elkhart, IN) optimal cutting temperature compound, frozen on dry ice, and stored at −80°C until use. Cross sections (10 μm) were cut on a cryostat (Leica, Nussloch, Germany) at −20°C. Sections were processed for detection of multiple markers to compare the differences between wild-type (wt) and tPA−/− EAE mice. Primary antibodies were chosen that detect axonal damage [amyloid precursor protein (APP), a gift from Dr. W. van Nostrand, SUNY Stony Brook], myelin [myelin basic protein (MBP); Roche Products, Hertforshire, UK], activated macrophage/microglia (F4/80; Serotec, Indianapolis, IN), and activated microglia (5-D-4; Seikagaku Kogyo, Tokyo, Japan) (Ferguson et al., 1997; Kennedy et al., 1998; Kiefer et al., 1998; Wilms et al., 1999). To perform immunohistochemistry, sections were fixed with 4% paraformaldehyde in PBS, washed in 0.3% H2O2 to block endogenous peroxidase, and then incubated overnight at 4°C in primary antibodies at the appropriate concentrations. After washing in PBS, sections were incubated with biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA). The avidin–biotin complex was visualized with diaminobenzidine and H2O2(Vector Laboratories), as described previously (Tsirka et al., 1997).
Quantitative Western blot analysis. Spinal cord extracts were prepared in 0.25% Triton X-100 in PBS. Cell debris was removed by centrifugation, and total protein concentration was measured using the Bio-Rad (Richmond, CA) Bradford Dc assay. Twenty-five micrograms of protein was separated by 12% SDS-PAGE and transferred on a polyvinylidene difluoride membrane. Membranes were blocked using 5% nonfat dry milk in PBS containing 0.05% Tween 20 and incubated overnight at 4°C with sheep anti-goat plasminogen activator inhibitor (PAI)-1 antibody (1:1000; American Diagnostic); goat polyclonal monocyte chemotactic protein (MCP)-1 antibody; goat polyclonal regulated on activation, normal T cell expressed and secreted (RANTES) antibody (1:500); monoclonal mouse anti-human B cell/CD22 antibody (1:500; Dako, Carpinteria, CA); monoclonal rat anti-human T cell/CD3 antibody (1:500; Serotec); or monoclonal mouse anti-rat osteopontin (OPN) (1:500; obtained from Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Then the immunocomplex was detected with biotinylated anti-rabbit IgG (Vector Laboratories). For quantification of the bands, the biotinylated secondary antibody was detected using FITC-labeled ExtrAvidin (1:200; Sigma, St. Louis, MO). Emitted fluorescence was visualized by FluorImager (Molecular Devices, Palo Alto, CA), which has an extended dynamic linear range, and quantified using ImageQuant software. Sample loading was visualized by Ponceau S red staining.
Amidolytic assay for tPA activity. For the quantitative determination of tPA activity, amidolytic assays were performed as described previously (Andrade-Gordon and Strickland, 1986). Briefly, spinal cord was lysed in 0.25% Triton X-100 and incubated in a mix containing either 0.3 mm S-2251 and 0.42 μm plg or 0.3 mm S-2288 (which is specific for tPA) in 0.1 m Tris HCl, pH 8.1, 0.1% Tween 80. The samples were incubated at 25°C. The change in absorbency (ΔA) at 405 nm was measured at different time points. Known concentrations of recombinant tPA were used as controls. tPA activity was calculated from the initial rates in the amidolytic assay. Total protein content in the aliquots of each sample was determined using the Bio-Rad Bradford Dc assay. These concentrations were used to normalize the amount of tPA in each sample. The measurements were performed in triplicate.
RNA isolation and reverse transcriptase-PCR. Mice were killed at different time points during the course of EAE, and the lumbar spinal cords were removed immediately. Total RNA was extracted using the TriPure isolation reagent (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions. The concentration of RNA was determined by UV spectroscopy at 260 nm. From each sample, 5 μg of RNA was used for synthesis of the first oligo-dT-primed cDNA strand with Moloney murine leukemia virus reverse transcriptase (RT; Invitrogen, San Diego, CA). cDNA (50 ng) from each spinal cord was then used for PCR in a final volume of 50 μl using the primers for mouse CD8α, tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), or β-actin. The cDNA templates were denatured for 5 min at 94°C and then amplified using 35 cycles of denaturing (94°C, 30 sec), annealing (60–65°C, 30 sec), and extension (74°C, 45 sec). The final step was incubation for 7 min at 42°C. Each sample was amplified using a FastStart DNA Master SYBR Green I kit in a real-time LightCycler system (Roche Products). The level of each specific cDNA was quantified in the exponential phase of PCR product accumulation and normalized by the level of actin expression in each individual sample. Primers used for PCR amplification are listed in Table1.
Table 1.
Oligonucleotide primers for RT-PCR
| Primer | Product size (bp) | Sequence (5′ to 3′) |
|---|---|---|
| TNF-α (forward) | 150 | GCTTTCCGAATTCACTGGAG |
| TNF-α (reverse) | TGCAACTCAAGGGAGGAATC | |
| iNOS (forward) | 148 | CTTCGGTGCAGTCTTTTCCT |
| iNOS (reverse) | GGATTGCATTTCGCTGTCTC | |
| CD8α (forward) | 273 | TCTGTCGTGCCAGTCCTTC |
| CD8α (reverse) | CCTTCCTGTCTGACTAGCGG | |
| β-Actin (forward) | 150 | GTCCCTGTATGCCTCTGGTC |
| β-Actin (reverse) | GGATCTTCATGAGGTAGTCTGTC |
RESULTS
Levels of tPA activity reflect the clinical course of EAE in wt mice
The spinal cords from MOG-induced EAE mice were removed at different time points. As shown in Figure1A, the level of tPA activity increased up to fourfold by day 20 (3.2 ng of tPA per microgram of protein vs 0.8 for the control mice). The tPA activity was increased during the period that the EAE mice were observed to be symptomatic (see below) but returned almost to control levels after recovery (1.2 ng of tPA per microgram of protein). Increases in tPA release are generally accompanied by release in parallel of its inhibitors (PAIs) to ensure stringent regulation of potentially deleterious proteolytic activity. In situ upregulation of tPA and PAI-1 and elevated tPA and PAI-1 antigen levels in the CSF of MS patients have been reported previously (Akenami et al., 1996, 1997,1999). Accordingly, we assessed whether PAI-1 expression was altered during the clinical course of EAE. As shown in Figure1B, the level of PAI-1 expression was increased during the period of EAE clinical symptoms and returned almost to normal levels after recovery. However, PAI-1 expression increased more slowly and to a lesser extent than was observed for the increase in tPA activity. Given the stoichiometric interaction between tPA and PAI-1 (Kiefer et al., 1998), the increase in tPA would appear to be prevalent, further suggesting that (nonsequestered) tPA activity increases in vivo during EAE.
Fig. 1.

Levels of tPA activity increase during the clinical course of MOG-induced EAE in wt mice. Lumbar spinal cord lysates were prepared from wt EAE mice at different time points after MOG injection. Uninjected adult female mice were used as controls.Top, To quantitatively determine tPA activity, amidolytic assays were performed as described in Materials and Methods. Total protein content in aliquots of each sample was determined using the Bradford assay. Note that tPA activity significantly increased during the disease but returned to near normal levels after recovery. Although this assay does not discriminate between uPA and tPA activity, uPA is unlikely to have contributed to the increased activity observed for two reasons: (1) previous reports (Akenami et al., 1996; Cuzner et al., 1996) using tPA-specific assays have demonstrated that it is the activity of tPA rather than uPA that becomes upregulated, and (2) we performed in situ zymographic assays on spinal cord sections in the presence or absence of amiloride, a specific uPA inhibitor, and observed no differences in activity (data not shown). This result is consistent with our previous report that uPA mRNA and protein are not detected in the mouse CNS (Tsirka et al., 1997).Bottom, PAI-1 expression was determined by Western blotting. FluorImager was used for the quantification of the bands. Note that the level of PAI-1 expression also increased during this period, although less quickly or dramatically than tPA, and returned to baseline levels after recovery. The data are presented as mean ± SEM (n = 3 mice). *p < 0.05; Student's t test.
Altered progression of EAE in tPA−/− mice
To evaluate the role of tPA in EAE, the clinical course of MOG-induced EAE was assessed in tPA−/−mice. C57BL/6(H-2b) wt mice exhibited signs of disease on average at day 7.8 ± 0.5 after immunization and developed a chronic course (Fig. 2) that was accompanied by histopathological hallmarks of EAE, such as spinal cord inflammation and demyelination (Fig.3), in agreement with the literature (Suen et al., 1997). By day 40, the clinical score observed for wt mice was 1 (flaccid tail) (Fig. 2), and they exhibited no other motor dysfunction. In contrast, the tPA−/−mice, which otherwise were genetically quite similar to the wt mice because they had been back crossed for 10 generations to the C57BL/6 background, showed a significant delay in disease onset (day 11.5 ± 0.6). However, they exhibited more severe symptoms at later time points (e.g., day 50) (Table 2). The tPA-deficient mice had a much slower recovery; their neurological and motor dysfunction continued over extended periods of time, ≤100 d after immunization (data not shown). To evaluate whether the altered progression of EAE was subject to a dosage effect, we subjected heterozygous (tPA+/−) animals to EAE. These mice exhibited clinical symptoms at approximately day 8, which was similar to wt mice, and the subsequent clinical symptomatology of the disease also followed the wt time course (data not shown). Accordingly, a 50% reduction in the amount of tPA present does not suffice to alter the progression of EAE; instead, a more dramatic reduction is required. This result is in agreement with data obtained from other experimental paradigms (Dickson et al., 1993; London et al., 1996). The altered progression (delay in onset) of EAE in tPA−/− mice indicates that tPA contributes to neuronal degeneration during the early stage of EAE. At later stages of EAE, however, tPA appears be beneficial in neuronal regeneration, because the tPA−/− mice exhibited slower recovery and more severe and sustained symptoms.
Fig. 2.

Altered progression of EAE in tPA−/− mice. The wt and tPA−/− mice were injected with MOG35–55 peptide in CFA and pertussis toxin (PT) to induce EAE. The disease severity was scored on a clinical scale from 0 to 5 as described in Materials and Methods. The average score for each day was calculated by averaging the clinical score for that day for each mouse in the group (n = 12 mice for each group). tPA−/− mice showed a significant delay in the onset of EAE, followed by a delay in recovery. Table 2 presents the statistical significance in the day of onset, average maximum clinical score, and score at day 50.
Fig. 3.
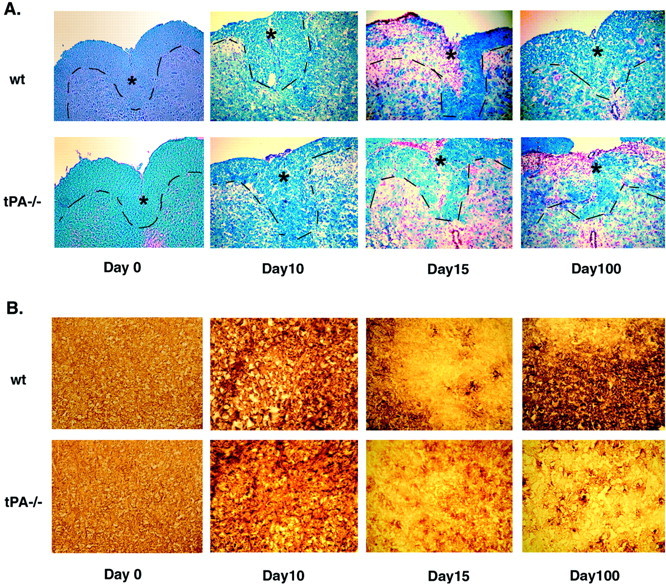
Delayed demyelination in tPA−/− mice during EAE. Frozen cross sections of spinal cords from wt and tPA−/− mice at different time points during the EAE course were stained with LFB and Nuclear Red (A). Note the extensive demyelination and infiltration of inflammatory cells evident in sections of wt mice at day 15 and in sections of tPA−/− mice until day 100. The dashed line demarcates the border of the dorsal horn (as denoted by asterisks), where demyelination is observed. B, Immunohistochemistry using an antibody to visualize MBP revealed similar patterns of demyelination (600× magnification).
Table 2.
tPA−/− mice exhibit a significant delay in the onset of EAE as well as a deficiency in recovery
| Groups | Incidence | Day of onset2-a | Mean maximal clinical score2-b | Score at day 502-a |
|---|---|---|---|---|
| C57BL/6 | 12 /12 | 7.8 ± 0.5 | 2.8 ± 0.1 | 0.8 ± 0.1 |
| tPA−/− | 12 /12 | 11.5 ± 0.6 | 3.3 ± 0.3 | 2.5 ± 0.4 |
Data are presented as mean ± SEM in all cases. Single comparisons of two means were performed in each case using Student'st test. The average day of disease onset was calculated by averaging the first day of clinical signs for each mouse in the group. The mean maximal clinical score was calculated by averaging the highest individual score for each mouse.
p < 0.01.
p < 0.1.
Delayed demyelination in tPA−/− EAE mice
Inflammation and demyelination are two well defined characteristics of EAE. Luxol Fast Blue (LFB) histological stain was used to stain for myelin, and Nuclear Red was used to show influx of inflammatory cells. Myelination was examined at the dorsal horn of the spinal cord (to which the myelinated axons normally extend). At days 0 and 10, there were no significant differences between wt and tPA−/− mice. In contrast, severe demyelination was detected in wt but not in tPA−/− EAE mice at day 15. As shown in Figure 3, a significant decrease in LFB intensity staining (indicating demyelination) is obvious in the dorsal horn (as indicated by asterisks) of the spinal cord of wt mice, but only minimal demyelination is seen in tPA−/− animals. Both strains exhibited extensive inflammatory cell infiltrates. By day 100, the clinical symptoms of EAE were no longer evident in the wt mice, and they appeared to have recovered from the inflammation and demyelination. However, the clinical symptoms and inflammatory pathology were still quite severe in the tPA−/− EAE mice. Demyelination was also evaluated by MBP immunohistochemistry (Fig. 3B); extensive demyelination was observed at early time points in the wt mice (day 15), whereas for the tPA−/− mice, the onset of demyelination was again observed to be delayed (minimal demyelination was observed at day 15) but then continued beyond day 100.
Delayed axonal damage in tPA−/− mice during EAE
Early neuronal degeneration is a newly identified pathological characteristic of MS. Using histopathological markers that specifically detect damaged axons, axonal degeneration has been shown to occur throughout active lesions even early in the course of the disease and to be present at the active borders of less acute lesions (McDonald et al., 1992; Ferguson et al., 1997; Trapp et al., 1998; Waxman, 1998). An antibody to APP was used to detect axonal damage in EAE. APP is normally expressed in neurons but only at low levels that are not detectable by standard immunocytochemistry. Its detection at sites of axonal injury is thought to represent accumulation attributable to failure of axonal transport (Ferguson et al., 1997; Bitsch et al., 2000; Bjartmar and Trapp, 2001). We found that in EAE wt mice, APP could be detected on day 10, had reached its peak at day 15, and decreased to negligible levels by day 100. In contrast, however, in EAE tPA−/− mice, APP was not observed until day 15, and then it continued to be detectable through day 100 (Fig.4). At day 0, there was no difference in APP immunoreactivity between wt and tPA−/− animals, indicating that there are no intrinsic differences between the two genotypes.
Fig. 4.

Delayed axonal damage in tPA−/− mice during EAE. Using an antibody directed against APP, neuronal degeneration was detected in the white matter of spinal cord sections from wt mice as early as day 10 and more strongly at day 15. For the tPA−/− mice, in contrast, only minimal APP was detected, and even that was not observed until day 15 (600× magnification).
Attenuated microglial activation in tPA−/−mice during the course of EAE
Activated microglia accumulate and presumably play a role in MS/EAE (Benveniste, 1997; Diemel et al., 1998). In the kainic acid-induced excitotoxic neurodegeneration model, tPA has been shown to mediate microglial activation (microglial activation in tPA−/− mice is attenuated) (Tsirka et al., 1995; Rogove et al., 1999; Siao and Tsirka, 2002). We evaluated the levels of microglial activation at different time points (days 0, 10, 15, and 100) during EAE, using antibodies either to the mature macrophage/microglial-specific antigen F4/80 or to the microglial surface antigen 5-D-4 (Fig. 5). Microglial activation, assessed by immunohistochemistry for both markers of microglia/macrophages, was not detectable at day 0 and was very limited at day 10 (data not shown) in both wt and tPA−/− animals. Highly activated macrophage/microglial cells were found in wt mice at day 15, and this activation persisted through day 100 (Fig. 5, arrows). In contrast, microglial activation in tPA−/− EAE mice was delayed and attenuated, with minimal presence of F4/80+ and 5-D-4+ cells (and decreased intensity of staining for the two markers) at day 15. At day 100, activated macrophages/microglia were present in tPA−/− mice, but even then the cells were not fully activated judging from the lack of extensive branching of their processes and amoeboid morphology. These results suggest a possible mechanism of action through which altering the levels of tPA might affect EAE progression (i.e., through interference with microglial activation).
Fig. 5.
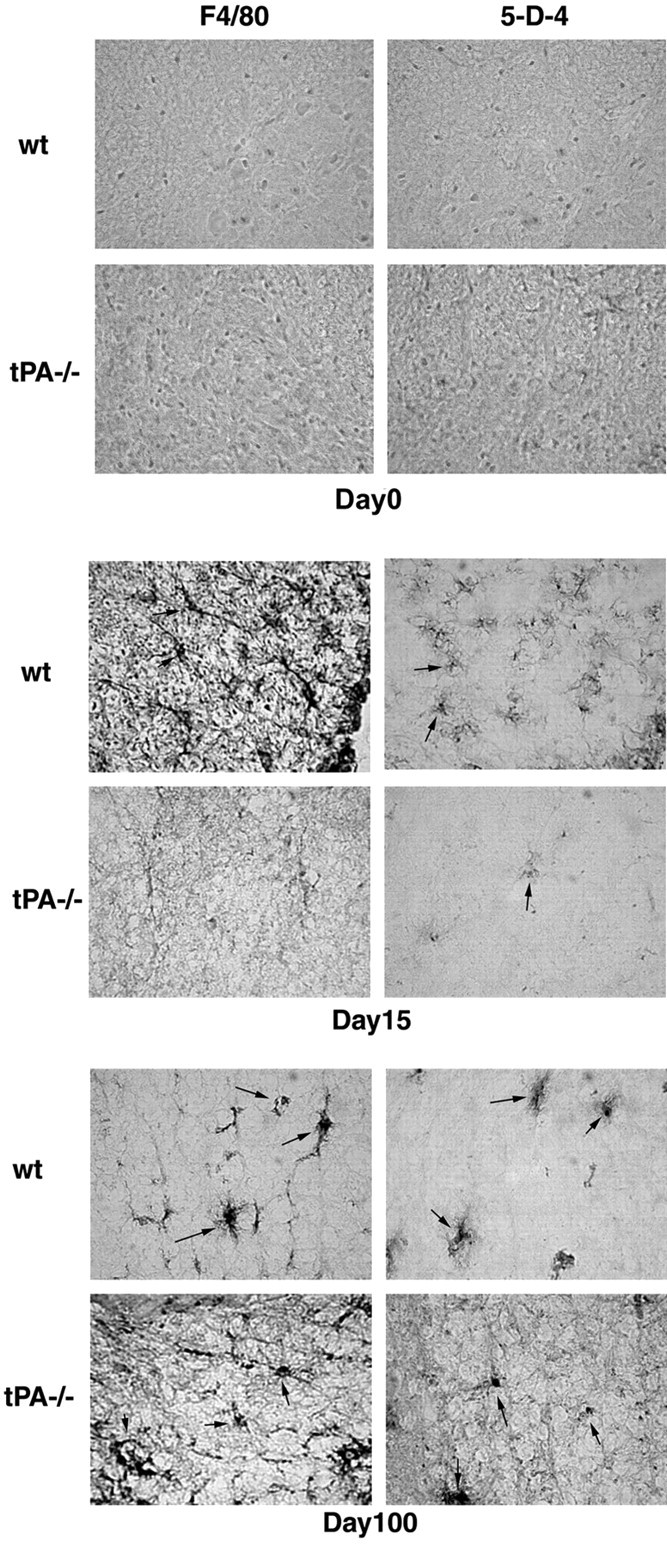
Attenuated microglial activation in tPA−/− mice during the course of EAE. Frozen cross sections of spinal cords from wt and tPA−/− mice at different time points of EAE were probed using antibodies directed against either the mature macrophage/microglia-specific antigen F4/80 or the microglial-specific cell surface antigen 5-D-4. Highly activated microglial cells were found in wt mice at day 15. In contrast, only attenuated microglial activation was noted in tPA−/− mice, and even that was only seen at day 100. Arrows point to individual activated macrophage/microglial cells (600× magnification).
Alteration in cytokine/chemokine expression in tPA−/− mice during EAE
Members of the CC chemokine family have been implicated in the immunopathology of EAE (Kuchroo et al., 1993). Moreover, the production of MCP-1 and RANTES in the CNS has been associated with disease symptoms in EAE models (Hulkower et al., 1993; Godiska et al., 1995;Kennedy et al., 1998; Asensio et al., 1999). Lysates of spinal cords from wt and tPA−/− mice collected at different time points during the course of EAE (days 0, 10, 15, 24, and 45) were prepared and used to perform quantitative Western blotting analysis. As shown in Figure6A, delayed expression of MCP-1 was found in the tPA−/− EAE mice. From days 10 to 15, MCP-1 expression increased twofold in wt mice. The increase was less dramatic in tPA−/− mice. However, from days 15 to 24, MCP-1 expression was still increasing (more than twofold) in tPA−/− mice but had already decreased in wt mice. The increase in RANTES expression (Fig. 6A) was not as robust as that of MCP-1, but the trend was similar.
Fig. 6.

Altered expression of chemokines and cytokines in tPA−/− mice during the course of EAE.A, Quantitative Western blotting analysis for chemokines was performed using spinal cord lysates from wt and tPA−/− mice at days 0, 10, 15, 24, and 45 of EAE, using either anti-MCP-1 antibody, anti-RANTES antibody, or anti-OPN. Note the delayed expression of all three chemokines and persistent expression of MCP-1 and OPN in the tPA−/− mice during the course of EAE. B, Quantitative RT-PCR analysis for TNF-α and iNOS. RNA from lumbar spinal cords of wt and tPA−/− mice at days 0, 10, 24, and 45 after MOG injection was used to perform RT-PCR in a real-time LightCycler system. Specific primer sets were designed to detect the cytokines iNOS and TNF-α (described in Table 1). The level of specific mRNAs was quantified in the exponential phase of PCR product accumulation and normalized by comparison with standard curves obtained from serial dilutions of plasmids encoding cDNAs for each gene.
OPN, also called early T cell activator gene-1, costimulates T cell proliferation and is classified as a T helper cell-1 (Th1) cytokine. It was reported very recently that OPN transcripts are elevated in EAE, and that OPN-deficient mice are resistant to EAE (Chabas et al., 2001). Accordingly, we performed a Western blotting analysis to evaluate the OPN expression level in both wt and tPA−/− mice. In wt mice, the level of OPN increased by day 15 and then returned to control levels (Fig.6A). In tPA−/− mice, the OPN expression level peaked at day 24 and remained high at day 45. Because interleukin-10 production is upregulated by OPN and has been associated with remission from EAE, the sustained OPN expression in tPA−/− EAE mice may partly explain the sustained symptoms in the later stages of EAE.
The levels of TNF-α and iNOS, other cytokines known to be upregulated during EAE, were evaluated using quantitative RT-PCR. Rapid and dramatic upregulation of both TNF-α and iNOS was observed at days 10, 24, and 45 in wt mice (Fig. 6B). In the tPA−/− mice, in contrast, the expression of the two cytokines did increase, but more slowly and to a significantly lesser degree. Because both cytokines are synthesized and secreted primarily by activated microglia (Kreutzberg, 1996), these data are in agreement with the observation (Fig. 5) that microglial activation is attenuated in tPA−/−mice.
Alteration of a T cell proliferation/activation marker but not a B cell marker in tPA−/− mice during EAE
EAE is a T cell-mediated autoimmune disease. Therefore, we set out to assess whether possible differences in the tPA−/− immune response could account for the altered disease progression. CD3 and CD22 were used as markers for T and B cells, respectively. Using quantitative Western blotting analysis, T cell proliferation during EAE was found to occur but to be delayed in tPA−/− mice (Fig.7A). However, no significant difference was detected for the B cell marker (Fig. 7A). This observation supports the notion that T cells play a very important role in this disease, and that this role is affected in the absence of tPA. The expression levels of CD3 and CD22 did not differ between tPA−/− and wt mice at days 0 and 10, suggesting that there are not obvious baseline differences in the tPA−/− immune system. We also assessed the levels of expression of CD8α (as a marker for cytotoxic T cells). Quantitative RT-PCR was used to determine these levels. As shown in Figure 7B, the expression of CD8α is very low in tPA−/− animals compared with wt ones, possibly leading to lower levels of neurodegeneration in the tPA−/− mice (Fig. 4).
Fig. 7.

Alteration of a T cell proliferation/activation marker but not a B cell marker in tPA−/− mice during EAE. A, Quantitative Western blotting analysis was performed using spinal cord lysates from wt and tPA−/− mice at days 0, 10, 15, 24, and 45 of EAE, using either anti-T cell/CD3 or anti-B cell/CD22. Note that the expression of the T cell marker (but not that of the B cell marker) showed a delayed rise in tPA−/− mice during EAE. No differences in marker expression were observed at day 0.B, Quantitative RT-PCR analysis of CD8α, a marker for cytotoxic T cells. RNA from lumbar spinal cords of wt and tPA−/− mice at days 0, 10, 24, and 45 after MOG injection was used to perform RT-PCR in a real-time LightCycler system. Specific primer sets were designed as described in Table 1. The level of specific mRNAs was quantified in the exponential phase of PCR product accumulation and normalized by comparison with standard curves obtained from serial dilutions of a plasmid containing the CD8α cDNA.
DISCUSSION
In this study, using mice lacking tPA, we examined changes in the clinical course and histopathology of MOG35–55-induced EAE, an animal model for MS. We found that tPA−/− mice exhibit (1) altered EAE progression characterized by delayed onset but then increased severity at later time points, resulting in delayed recovery of neurological and motor dysfunction; (2) delayed demyelination, which then persists >100 d after the EAE onset; (3) attenuated microglial activation; (4) delayed axonal degeneration; and (5) delayed expression of chemokines and altered/reduced expression of cytokines. We also found that levels of tPA activity increase during the clinical course of EAE in wt mice.
Our data indicate that tPA is involved in the pathogenesis of EAE. Reviewing the literature, several possible mechanisms through which tPA may be acting can be proposed. First, tPA may promote demyelination, because plasmin (which is generated by the action of tPA on plasminogen) can directly degrade MBP (Cammer et al., 1978). In addition, plasmin is the key initiator of the matrix metalloproteinase (MMP) activation cascade. MMP activity has been documented to have an important role in the breakdown of myelin membranes (Cuzner and Opdenakker, 1999). Second, tPA may alter inflammatory reactions in the CNS by increasing the permeability of the blood–brain barrier (Paterson et al., 1987). Third, because tPA can promote excitotoxic cell death, tPA may also play a role in the early stages of MS by contributing to glutamate-induced oligodendrocyte injury and neuronal death (Pitt et al., 2000; Smith et al., 2000). Fourth, tPA may help neuronal regeneration by reducing local fibrin deposition (Herbert et al., 1996; Akassoglou et al., 2000) or by promoting migration of oligodendrocyte progenitors through the extracellular matrix (Uhm et al., 1998). It is probable that tPA plays both harmful and beneficial roles in MS, and that its involvement is complex.
There is precedent for the involvement of tPA, and plasminogen activators in general, in different autoimmune diseases. In rheumatoid arthritis, elimination of tPA or urokinase plasminogen activator (uPA) results in exacerbation of the disease and very severe clinical symptoms (Yang et al., 2001). In other studies, this finding has been attributed to disease-specific conformational changes of its remaining assembled substrate, plasminogen, on cell or fibrin surfaces, which results in the presentation of new epitopes recognized by autoantibodies (Dominguez et al., 2001). Furthermore, in patients with systemic lupus erythematosus, decreased levels of tPA have been measured along with increases in the levels of PAI-1. The changes in the levels of tPA were shown to be caused by the presence of auto-antibodies to fibrin-bound tPA (Salazar-Paramo et al., 1996). Changes in the levels of tPA and tPA activity have also been observed in antiphospholipid syndrome (Ieko et al., 2000), a disease that occasionally is indistinguishable from MS (Cuadrado et al., 2000). Such fluctuations in the expression of plasminogen activators are thought to be the primary reason for the frequent incidence of thromboses observed in these diseases (Satoh et al., 1998; Munoz-Rodriguez et al., 2000).
More interestingly, activated microglia also appear to play complex roles in MS/EAE. Microglia, the immunocompetent cells of the CNS (Kreutzberg, 1996), are thought to contribute to MS/EAE through several mechanisms, including production of proinflammatory cytokines, proteases, and free radicals (Benveniste, 1997). However, microglia may also potentially contribute to recovery from MS/EAE by expressing a wide variety of growth factors and cytokines. These include several that directly affect oligodendrocyte survival, proliferation, and differentiation, such as insulin-like growth factor-1, platelet-derived growth factor, and fibroblast growth factor (Diemel et al., 1998). The microglia also express immunosuppressive TGF-β1, which may promote MS/EAE remission (Kiefer et al., 1998). We assessed the expression of TGF-β1 during MOG35–55-induced EAE; in wt mice, TGF-β1 is expressed as reported previously (Kiefer et al., 1998) (i.e., it was increased late in the course of EAE). In contrast, decreased expression was found in the tPA−/− EAE mice (data not shown). Consistent with this, delayed and decreased microglial activation was noted in tPA−/− EAE mice.
The activation/proliferation of T cells and the infiltration of cytotoxic T cells (which is evident by the increased expression of CD8α) were delayed in tPA−/− EAE mice. However, whether this is the cause of the delayed onset and recovery or just a component or consequence of the clinical course will need to be addressed. Apoptosis of T lymphocytes is thought to be a key element in the downregulation of autoimmune CNS inflammation. This apoptosis affects both autoreactive T-cell populations and secondarily recruited lymphocytes. Elimination of T cells may depend at least in part on intact Fas–Fas ligand (FasL) signaling (Bauer et al., 1998). In the CNS, Fas+ T cells and FasL+ microglial cells and macrophages have been found in the brains of MS patients, suggesting that Fas-induced apoptosis of T cells by microglial cells may occur. In our quantitative Western blot analysis, CD3 was expressed at higher levels in tPA−/− mice at unexpectedly late time points (days 24 and 45), possibly suggesting a defect in T cell apoptosis. Intriguingly, we have observed that in a different model of apoptosis (in the spontaneous neurodegeneration- and premature apoptosis-prone lurcher mouse), tPA does mediate the c-JunP, caspase-8 apoptotic pathway (Lu and Tsirka, 2002). This pathway is initiated via Fas–FasL interactions and signaling. It is therefore possible that the persistence of T cells and deficient T cell elimination in tPA−/− mice is a potential cause or primary component of the prolonged EAE clinical symptoms and inflammation.
The pattern of delay/failure of the tPA-deficient animals with regard to recovery from acute MOG-induced EAE is very reminiscent of the EAE progression pattern in interferon-γ knock-out mice primed with MOG35–55, for which the disease appears to be mediated by Th2 (Chu et al., 2000). The decreased expression of TNF-α in the tPA−/− animals would also suggest that this disease may be Th2 in nature (Fig. 6B).
It is probable that tPA and microglia play both harmful and beneficial roles in MS/EAE, and that there is a balance between injury and recovery. This balance may be regulated via the expression and secretion of cytokines and proteolytic enzymes. Our results suggest that attenuation of tPA activity in a temporally restricted manner could prove beneficial in combination with the existing therapeutic management of MS.
Footnotes
This work was supported by a grant from the National Institutes of Health, a research grant from the Wadsworth Foundation, and a Targeted Research Opportunities award (S.E.T.). We thank Drs. K. Akassoglou and M. Frohman for advice and critical reading of this manuscript. We also thank members of the Tsirka laboratory for helpful suggestions.
Correspondence should be addressed to Dr. Stella Tsirka, Department of Pharmacological Sciences, University Medical Center at Stony Brook, Stony Brook, NY 11794-8651. E-mail: stella@pharm.sunysb.edu.
REFERENCES
- 1.Akassoglou K, Kombrinck K, Degen J, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166. doi: 10.1083/jcb.149.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akenami F, Sirén V, Koskiniemi M, Siimes M, Teräväinen H, Vaheri A. Cerebrospinal fluid activity of tissue plasminogen activator in patients with neurological diseases. J Clin Pathol. 1996;49:577–580. doi: 10.1136/jcp.49.7.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akenami F, Koskiniemi M, Mustjoki S, Siren V, Farkkila M, Vaheri A. Plasma and cerebrospinal fluid activities of tissue plasminogen activator and urokinase in multiple sclerosis. Fibrinolysis Proteolysis. 1997;11:109–113. [Google Scholar]
- 4.Akenami F, Siren V, Wessman M, Koskiniemi M, Vaheri A. Tissue plasminogen activator gene expression in multiple sclerosis brain tissue. J Neurol Sci. 1999;165:71–76. doi: 10.1016/s0022-510x(99)00080-5. [DOI] [PubMed] [Google Scholar]
- 5.Andrade-Gordon P, Strickland S. Interaction of heparin with plasminogen activators and plasminogen: effects on the activation of plasminogen. Biochemistry. 1986;25:4033–4040. doi: 10.1021/bi00362a007. [DOI] [PubMed] [Google Scholar]
- 6.Asensio VC, Lassmann S, Pagenstecher A, Steffensen SC, Henriksen SJ, Campbell IL. C10 is a novel chemokine expressed in experimental inflammatory demyelinating disorders that promotes recruitment of macrophages to the central nervous system. Am J Pathol. 1999;154:1181–1191. doi: 10.1016/S0002-9440(10)65370-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer J, Bradl M, Hickey W, Forss-Peter S, Breitschopf H, Linington C, Wekerle H, Lassmann H. T cell apoptosis in acute inflammatory lesions. Am J Pathol. 1998;153:715–724. doi: 10.1016/s0002-9440(10)65615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benveniste E. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 9.Bernard CC, Johns TG, Slavin A, Ichikawa M, Ewing C, Liu J, Bettadapura J. Myelin oligodendrocyte glycoprotein: a novel candidate autoantigen in multiple sclerosis. J Mol Med. 1997;75:77–88. doi: 10.1007/s001090050092. [DOI] [PubMed] [Google Scholar]
- 10.Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- 11.Bjartmar C, Trapp B. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Cammer W, Bloom BR, Norton WT, Gordon S. Degradation of basic protein in myelin by neutral proteases secreted by stimulated macrophages: a possible mechanism of inflammatory demyelination. Proc Natl Acad Sci USA. 1978;75:1554–1558. doi: 10.1073/pnas.75.3.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord J, Collen D, Mulligan R. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 14.Chabas D, Baranzini SE, Mitchell D, Bernard CCA, Rittling SR, Denhardt DT, Sobel RA, Lock C, Karpuj M, Pedotti R, Heller R, Oksenberg JR, Steinman L. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–1735. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 15.Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuadrado MJ, Khamashta MA, Ballesteros A, Godfrey T, Simon MJ, Hughes GR. Can neurologic manifestations of Hughes (antiphospholipid) syndrome be distinguished from multiple sclerosis? Analysis of 27 patients and review of the literature. Medicine (Baltimore) 2000;79:57–68. doi: 10.1097/00005792-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94:1–14. doi: 10.1016/s0165-5728(98)00241-0. [DOI] [PubMed] [Google Scholar]
- 18.Cuzner M, Gveric D, Strand C, Loughlin A, Paemen L, Opdenakker G, Newcombe J. The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol. 1996;55:1194–1204. doi: 10.1097/00005072-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Dickson D, Lee S, Mattiace L, Yen S, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 20.Diemel LT, Copelman CA, Cuzner ML. Macrophages in CNS remyelination: friend or foe? Neurochemical Res. 1998;23:341–347. doi: 10.1023/a:1022405516630. [DOI] [PubMed] [Google Scholar]
- 21.Dominguez M, Cacoub P, Garcia de la Torre I, Piette TJ, Salazar-Paramo M, Godeau P, Anglees-Cano E. Autoantibodies to receptor induced neoepitopes of fibrinolytic proteins in rheumatic and vascular diseases. J Rheumatol. 2001;28:302–308. [PubMed] [Google Scholar]
- 22.Ferguson B, Matyszak M, Esiri M, Perry V. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 23.Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- 24.Herbert CB, Bittner GD, Hubbell JA. Effects of fibrinolysis on neurite growth from dorsal root ganglia cultured in two- and three-dimensional fibrin gels. J Comp Neurol. 1996;365:380–391. doi: 10.1002/(SICI)1096-9861(19960212)365:3<380::AID-CNE4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol. 1998;161:4480–4483. [PubMed] [Google Scholar]
- 26.Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshretha S, Guida MP, Rapoport DA, Berman JW. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. 1993;150:2525–2533. [PubMed] [Google Scholar]
- 27.Ieko M, Ichikawa K, Atsumi T, Takeuchi R, Sawada KI, Yasukouchi T, Koike T. Effects of β2-glycoprotein I and monoclonal anticardiolipin antibodies on extrinsic fibrinolysis. Semin Thromb Hemost. 2000;26:85–90. doi: 10.1055/s-2000-9808. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus W. Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1a and monocyte chemotactic protein-1. J Neuroimmunol. 1998;92:98–108. doi: 10.1016/s0165-5728(98)00187-8. [DOI] [PubMed] [Google Scholar]
- 29.Kiefer R, Schweitzer T, Jung S, Toyka K, Hartung H-P. Sequential expression of transforming factor-b1 by T-cells, macrophages, and microglia in rat spinal cord during autoimmune inflammation. J Neuropathol Exp Neurol. 1998;57:385–395. doi: 10.1097/00005072-199805000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Kreutzberg G. Microglia, the first line of defense in brain pathologies. Arzneimittelforschung. 1995;45:357–360. [PubMed] [Google Scholar]
- 31.Kreutzberg G. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 32.Kuchroo VK, Martin CA, Greer JM, Ju S-T, Sobel RA, Dorf ME. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- 33.London J, Biegel D, Pachter J. Neurocytopathic effects of β-amyloid-stimulated monocytes: a potential mechanism for central nervous system damage in Alzheimer's disease. Proc Natl Acad Sci USA. 1996;93:4147–4152. doi: 10.1073/pnas.93.9.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu W, Tsirka S. Partial rescue of neural apoptosis in the Lurcher mutant mouse through elimination of tissue plasminogen activator. Development. 2002;129:2043–2050. doi: 10.1242/dev.129.8.2043. [DOI] [PubMed] [Google Scholar]
- 35.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 36.McDonald W, Miller D, Barners D. The pathological evolution of multiple sclerosis. Neuropathol Appl Neurobiol. 1992;18:319–334. doi: 10.1111/j.1365-2990.1992.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 37.Munoz-Rodriguez FJ, Reverter JC, Font J, Tassies D, Cervera R, Espinosa G, Carmona F, Balasch J, Ordinas A, Ingelmo M. Prevalence and clinical significance of antiprothrombin antibodies in patients with systemic lupus erythematosus or with primary antiphospholipid syndrome. Haematologica. 2000;85:632–637. [PubMed] [Google Scholar]
- 38.Opdenakker G, Damme J. Cytokine-regulated proteases in autoimmune disease. Immunol Today. 1994;15:103–107. doi: 10.1016/0167-5699(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 39.Paterson PY, Koh C-S, Kwaan HC. Role of the clotting system in the pathogenesis of neuroimmunologic disease. Fed Proc. 1987;46:91–96. [PubMed] [Google Scholar]
- 40.Pitt D, Werner P, Raine C. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 41.Rogove A, Siao C-J, Keyt B, Strickland S, Tsirka S. Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci. 1999;112:4007–4016. doi: 10.1242/jcs.112.22.4007. [DOI] [PubMed] [Google Scholar]
- 42.Salazar-Paramo M, Garcia de la Torre I, Fritzler MJ, Loyau S, Angles-Cano E. Antibodies to fibrin-bound tissue-type plasminogen activator in systemic lupus erythematosus are associated with Raynaud's phenomenon and thrombosis. Lupus. 1996;5:275–278. doi: 10.1177/096120339600500406. [DOI] [PubMed] [Google Scholar]
- 43.Satoh N, Abe T, Nakajima A, Ohkoshi M, Koizumi T, Tamada H, Sakuragi S. Analysis of uveitogenic sites in phosducin molecule. Curr Eye Res. 1998;17:677–686. [PubMed] [Google Scholar]
- 44.Seeds N, Williams B, Bickford P. Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- 45.Siao C-J, Tsirka S. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci. 2002;22:3352–3358. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 47.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 48.Suen W, Bergman C, Hjelmstroem P, Ruddle N. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186:1233–1240. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapp B, Peterson J, Ransohoff R, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 50.Tsirka S, Gualandris A, Amaral D, Strickland S. Excitotoxin induced neuronal degeneration and seizure are mediated by tissue-type plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 51.Tsirka S, Rogove A, Bugge T, Degen J, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uhm J, Dooley N, Oh L, Yong V. Oligodendrocytes utilize a matrix metalloproteinase, MMP-9, to extend processes along an astrocyte extracellular matrix. Glia. 1998;22:53–63. doi: 10.1002/(sici)1098-1136(199801)22:1<53::aid-glia5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 53.Waxman S. Demyelinating disease–new pathological insights, new therapeutic targets. N Engl J Med. 1998;338:323–325. doi: 10.1056/NEJM199801293380610. [DOI] [PubMed] [Google Scholar]
- 54.Wilms H, Wollmerb M, Sievers J. In vitro-staining specificity of the antibody 5-D-4 for microglia but not for monocytes and macrophages indicates that microglia are a unique subgroup of the myelomonocytic lineage. J Neuroimmunol. 1999;98:89–95. doi: 10.1016/s0165-5728(99)00066-1. [DOI] [PubMed] [Google Scholar]
- 55.Wu Y-P, Siao C-J, Lu W, Sung T-C, Frohman M, Milev P, Bugge T, Degen J, Levine J, Margolis R, Tsirka S. The tPA/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J Cell Biol. 2000;148:1295–1304. doi: 10.1083/jcb.148.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang YH, Carmeliet P, Hamilton JA. Tissue-type plasminogen activator deficiency exacerbates arthritis. J Immunol. 2001;167:1047–1052. doi: 10.4049/jimmunol.167.2.1047. [DOI] [PubMed] [Google Scholar]