

Abstract
Interleukin-17 (IL-17) and IL-17-producing cells have been shown to play important roles in inflammation and the immune response. IL-17 is believed to be mainly produced by T helper 17 (Th17) cells, a unique helper T-cell subset different from Th1 and Th2 cells. Other subsets of T cells such as γδT and natural killer T (NKT) cells have also been found to produce IL-17 in response to innate stimuli. IL-17 acts as a proinflammatory cytokine that can induce the release of certain chemokines, cytokines, matrix metalloproteinases (MMPs) and antimicrobial peptides from mesenchymal and myeloid cells. This leads to the expansion and accumulation of neutrophils in the innate immune system and links innate and adaptive immunity in vivo. Furthermore, increasing evidence indicates that IL-17 and IL-17-producing cells are involved in the pathogenesis of various diseases such as allergies, autoimmune diseases, allograft transplantation and even malignancy. They may also play protective roles in host defense against infectious diseases and promote induction of cytotoxic T lymphocyte (CTL) responses against cancer. Targeting of the IL-17 axis is under investigation for the treatment of inflammatory disorders.
Keywords: autoimmune disease, immunity, interleukin-17, neutrophil, T cells
Introduction
Interleukin-17A (IL-17A), discovered in 1993, is a prototypic member of the newest subclass of cytokines, which differ from the known cytokine families.1, 2, 3 It is recognized as an inflammatory cytokine and exerts its function mainly on myeloid cells and mesenchymal cells to induce the expression of granulocyte colony-stimulating factor (G-CSF), IL-6 and certain kinds of chemokines, which in turn increase granulopoiesis and recruit neutrophils to the infectious site.4 However, it was originally thought to be of minimal importance because it lacked immediate effects on T and B cells.
Mosmann and Coffman introduced the concept of distinct types of helper T cells in 1986, which was based on the types of cytokines that T cells preferentially produce when they are stimulated5 (Figure 1). When naive T cells are activated in the presence of IL-12, they differentiate into T helper 1 (Th1) cells that produce large amounts of interferon (IFN)-γ and activate macrophages; these cells are responsible for host defenses against intracellular pathogens. Cultured under IL-4 conditions, naive T cells differentiate into Th2 cells, producing IL-4, IL-5 and IL-13 and activating eosinophils; these cells are responsible for host defenses against extracellular pathogens.5 More recently, transforming growth factor (TGF)-β and IL-6 together were shown to trigger the production of IL-17A from CD4 T cells, thus designating a third helper T-cell subset as Th17 cells. Since then, IL-17A, the canonical cytokine of Th17 cells, has attracted much more attention. In addition to Th17 cells, several cell types are described as sources for IL-17, including γδT, natural killer T (NKT), CD8 T and lymph tissue inducer cells. IL-17 and IL-17-producing cells exert various functions in host defense and pathological conditions. Here, we will summarize the biological characteristics of IL-17 and its roles in allergic diseases, autoimmune disorders, host defense and malignancy.
Figure 1.
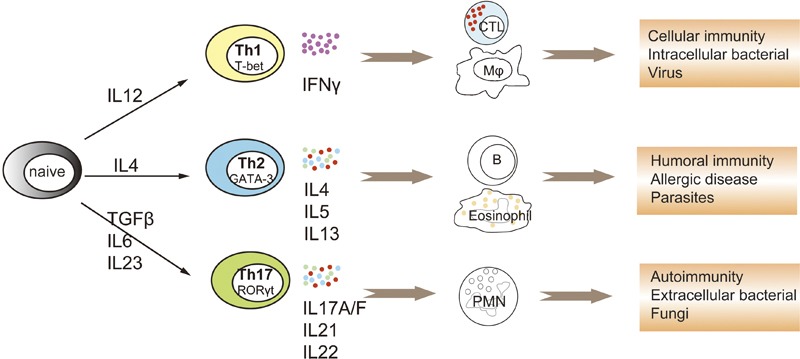
Helper T-cell subsets in host defense and immune responses. After being activated by antigen-presenting cells, naive CD4 T cells undergo clonal expansion and differentiate into different subsets of functional effector Th cells. IL-12 initiates the differentiation of Th1 cells, which is characterized by high production of IFN-γ, which act on macrophages and CTLs and are indispensable for cellular immunity and clearing intracellular pathogens. IL-4 triggers the differentiation of Th2 cells, which is characterized by production of IL-4, IL-5 and IL-13, which act on B cells, eosinophils and mast cells, and are responsible for humoral immunity and clearing of parasites. Recently, TGF-β plus IL-6 was demonstrated to initiate the programming of the third Th cell subset, Th17 cells, which are characterized by the production of IL-17A/F, IL-21 and IL-22. Th17 cytokines can stimulate the expansion and recruitment of PMN, and are critical in autoimmune diseases such as MS and RA and in host defenses against fungi and some extracellular pathogens. CTL, cytotoxic T lymphocyte; IFN, interferon; MS, multiple sclerosis; PMN, polymorphonuclear leukocytes; RA, rheumatoid arthritis; TGF, transforming growth factor; Th, T helper.
IL-17 and its receptors
IL-17 is a proinflammatory cytokine that was identified almost two decades ago. The gene encoding IL-17 was first described and cloned by Rouvier1 from a murine cytotoxic T lymphocyte hybridoma cDNA library in an attempt to screen for cytotoxic T lymphocyte (CTL)-associated transcripts, and thus named CTLA-8 (CTL antigen-8). Subsequently, the homologous T lymphotrophic viral Herpesvirus saimiri gene 13 was recognized to have 58% homology with CTLA-8.2 Human and rat homologs were identified later.3 Both have cytokine like activity but no homology to other known cytokine families. At the same time, a CTLA-8-binding receptor was cloned and shown to be unique compared with other known cytokine receptors.2 Thus, these factors represent a new cytokine family, and were designated IL-17 or IL-17A. Human IL-17 is a homodimeric glycoprotein consisting of 155 amino acids with a molecular weight of about 35 kDa.2 Homology-based cloning has revealed an additional five homologous cytokines, termed IL-17B to IL-17F,4 which all form homodimers to exert their biological functions.
Among the IL-17 family members, IL-17F has the highest homology (60%) with IL-17A.6 The genes encoding IL-17A and IL-17F are closely clustered on chromosome 1A4 in mouse and 6p12 in human.5 There are no reports of discordant expression of IL-17A and IL-17F,9 strongly implying similarity both in regulation and biological function. The function and regulation of IL-17F has been well reviewed elsewhere by Chang and Dong.10 Recent studies have revealed that IL-17A and IL-17F can be secreted as homodimers as well as heterodimers, and the IL-17A/F heterodimer is more potent than IL-17F but less than IL-17A in inducing chemokine expression.7, 8 The functions of other members of this family remain poorly characterized, with IL-17E (or IL-25) shown to be mainly a Th2-promoting cytokine.16
The IL-17 receptors also constitute a distinct family of cytokine receptors; the family includes IL-17RA, IL-17RB, IL-17RC, IL-17RD and IL-17RE,17 all of which are type I transmembrane proteins. IL-17RA (or IL-17R) was the first described IL-17 receptor,2, 19 and it binds IL-17A with higher affinity than IL-17F in human. IL-17RA appears to be ubiquitously expressed in hematopoietic tissues,2, 18 various myeloid cells, epithelial cells, fibroblasts, endothelial cells, epithelial cells and osteoblasts.11 Unlike other cytokine receptors, the IL-17RA subunits are preassembled on the plasma membrane before ligand binding, enabling it to respond rapidly and specifically to its ligand.20 Although the precise receptor complex of IL-17A has not been clearly elucidated, the IL-17A receptor consists of at least two IL-17RA subunits and one IL-17RC subunit.13, 14 IL-17F and IL-17A/F also exert their activity via the IL-17RA and IL-17RC heteromeric complex, although the binding affinity of IL-17F to IL-17RC is much stronger than to IL-17RA.12 IL-17RC has several splice isoforms and cannot induce signaling in the absence of IL-17RA.21 Depletion of IL-17RA completely abrogates the activity of IL-17A and IL-17F in mice.15 However, the way in which these receptors are paired to mediate signal or whether there are unknown subunits that cooperate with IL-17RA is not known. Thoroughly defining the cooperation of IL-17 family cytokines and their receptors is therefore important to fully understand their biological functions and to allow their application in clinical therapy.
Signal transduction of IL-17
Analysis of precise mechanisms for IL-17 signaling has been very difficult, as IL-17 is a unique cytokine with no homology to any other known cytokine families. Early studies showed that IL-17 could activate the nuclear factor (NF)-κB pathway,2 but the proximal activator of NF-κB was unknown for a long time. IL-17A induces proinflammatory gene expression just resembling TLR ligands, and TLRs are preassembled before ligand binding, implying that they may share some characteristics. Tumor-necrosis factor receptor-associated factor 6 (TRAF6), which is a key adaptor in the TLR- and IL-1R-signaling cascades, was shown to be indispensable in IL-17A-mediated NF-κB activation,22 as fibroblasts from Traf6−/− mice are unresponsive to IL-17A stimuli. Nonetheless, the intermediate adaptor between IL-17RA and TRAF6 remained unknown.
The key breakthrough occurred in 2003 by use of a bioinformatics algorithm. A conserved ‘SEFIR' (short for SEF/IL-17R) domain in the cytoplasmic tail of all IL-17Rs was identified,23 which has similarity to the ‘Toll/IL-1R (TIR)' domain in TLRs and IL-1Rs and is critical for the recruitment of myeloid differentiation factor 88, TIR domain-containing adaptor protein-inducing IFN-β and other factors. Deletion or point mutation of this domain in IL-17RA impairs the activation of NF-κB by IL-17A.24 Further analysis revealed that SEFIR lacks the BB-loop,23 the crucial specificity component of TIR domains, perhaps explaining why it cannot engage TLR-associated adaptors24, 25 and suggesting the existence of different intermediates. However, a region called TIR-like loop is only found at the C-terminal of SEFIR of IL-17RA.24 This may explain why IL-17RA functions as a common subunit to all other IL-17Rs in the family. Subsequently, ACT1 (also known as CIKS), an activator of NF-κB that previously linked to B cell-activating factor and CD40L signaling, was found to contain a SEFIR domain.23 It is recruited within minutes after IL-17A stimulation and binds IL-17RA through SEFIR-dependent interactions.25, 26 Moreover, ACT1 contains a TRAF6-binding motif and thus has the ability to bind TRAF6 and TGF-β-activated kinase 1 to deliver downstream signals, resulting in activation of the canonical NF-κB pathway. Deficiency in Act1 renders cells unresponsive to IL-17A,26 strongly suggesting its essential role in downstream signaling of IL-17RA. Consequently, the ACT1/TRAF6/NF-κB pathway has now been elucidated and may be the most important signal pathway of IL-17A (Figure 2).
Figure 2.
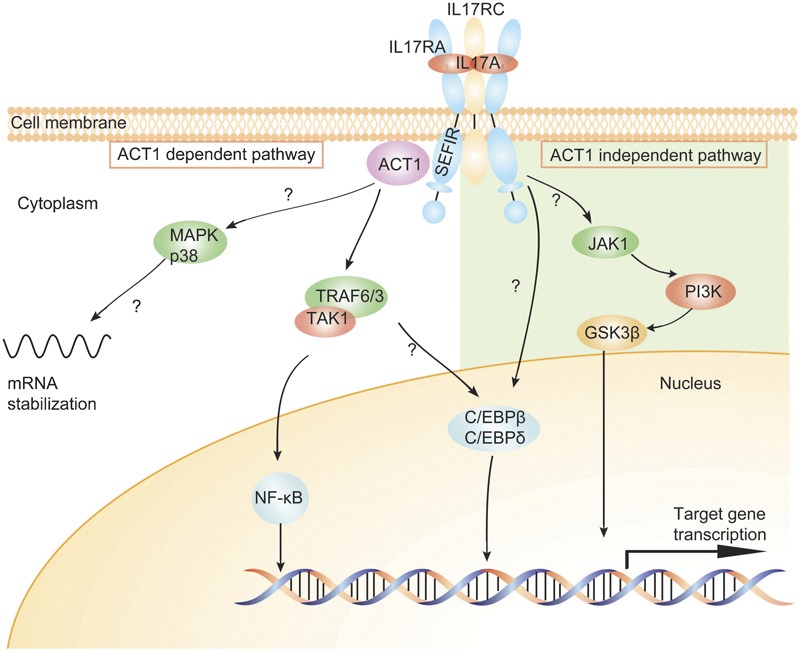
Signal pathways of IL-17. The IL-17R complex is composed of two IL-17RA and one IL-17RC; both subunits encode SEFIR domains. After activation, the intracellular IL-17 signaling includes ACT1-dependent and -independent downstream pathways. Left: the ACT1-dependent pathway: IL-17RA engages its SEFIR domain to recruit the adaptor protein ACT1. ACT1 contains a TRAF6-binding motif and can bind TRAF6, TRAF3 and TAK1, which subsequently leads to activation of the canonical NF-κB pathway. ACT1 is also required for the activation of MAPK p38, and this pathway leads to the stabilization of mRNAs, particularly those encoding chemokines and cytokines. Right: the ACT1-independent pathway involves JAK1 and PI3K, followed by subsequent inactivation of GSK-3β. Both ACT1-dependent and -independent pathways contribute to the activation of transcription factors C/EBP-β and C/EBP-δ. ACT1, nuclear factor-κB activator 1; C/EBP, CCAAT/enhancer-binding protein; GSK-3β, glycogen synthase kinase-3β JAK1, Janus kinase 1; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; SEFIR, SEF/IL-17R; TAK1, TGF-β-activated kinase 1; TRAF6, tumor-necrosis factor receptor-associated factor 6.
ACT1 can also activate the mitogen-activated protein kinase pathway. Generally, extracellular signal-regulated kinase is the most strongly and rapidly phosphorylated mitogen-activated protein kinase member following ACT1 activation, and these extracellular signal-regulated kinases as well as p38 act to stabilize several mRNAs, especially those encoding proinflammatory cytokines and chemokines,29, 31 through the inhibition of destabilizing proteins. Surprisingly, TRAF6 was shown to be dispensable in this process.27 CCAAT/enhancer-binding protein (C/EBP) transcription factors, specifically C/EBP-β and C/EBP-δ, are involved in the induction of IL-6 expression.28 ACT1 is required for the transcription of C/EBP-δ but is not essential for C/EBP-β.24 Additional mechanisms independent of ACT1 have also been recently implicated in IL-17A signaling in human airway epithelial cells. In this pathway, IL-17A activates Janus kinase-mediated phosphatidylinositol 3-kinase and subsequently inactivates glycogen synthase kinase-3β.30 Thus, the IL-17 signaling pathway is far from been fully understood (Figure 2).
Biological function of IL-17 in innate immunity
The biological functions of IL-17 have been extensively studied since its discovery (Figure 3). Mesenchymal cells and myeloid cells were shown to be the major targets of IL-17. Its target genes include proinflammatory cytokines, hematopoietic cytokines, chemokines, antimicrobial peptides and tissue-remodeling substances, depending on the cell type and disease model. Its effects on neutrophil expansion (through G-CSF) and chemotaxis (through CXC chemokines) are considered its characteristic roles, though less is known of its direct actions on lymphocytes.
Figure 3.
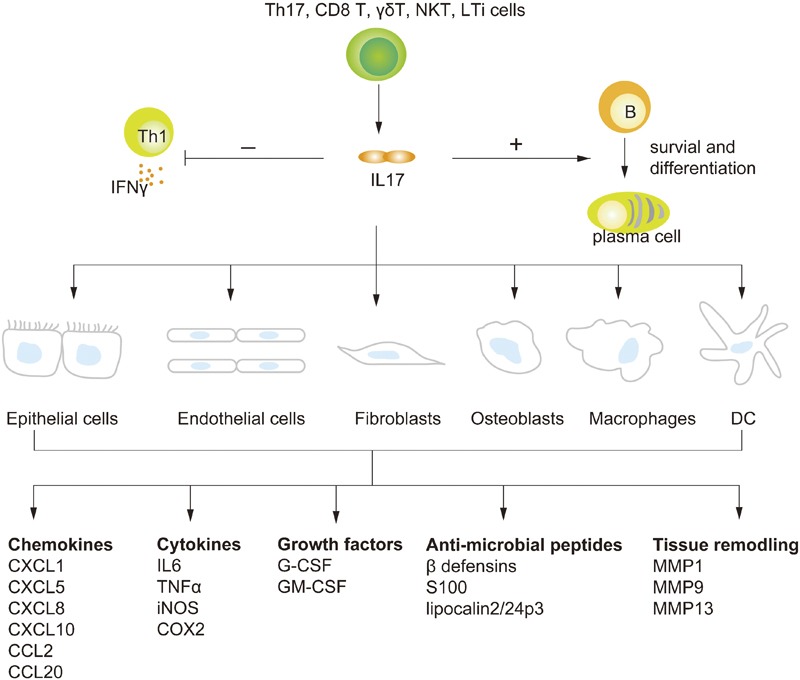
Biological activities of IL-17. Th17, γδT, NKT, CD8 T, and LTi cells are cellular sources of IL-17. IL-17, in turn, can induce the production of various molecules (including chemokines, cytokines, growth factors, antimicrobial peptides and tissue remodeling enzymes) in epithelial cells, endothelial cells, fibroblasts, osteoblasts, macrophages and DCs. IL-17 can also exert its functions on B and T cells. For B cells, IL-17 can promote the survival and expansion of B cells and the differentiation of B cells into antibody producing plasma cells. IL-17 can also inhibit IFN-γ production by targeting T-bet expression. COX2, cyclooxygenase 2; DC, dendritic cell; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFN, interferon; iNOS, inducible nitric oxide synthase; LTi, lymph tissue inducer; MMP, matrix metalloproteinase; NKT, natural killer T; Th, T helper; TNF, tumor-necrosis factor.
Most experiments suggest that IL-17 induces tissue inflammation mainly by stimulating expression of several proinflammatory cytokines including IL-6, TNF-α, G-CSF, granulocyte–macrophage CSF (GM-CSF)and others. IL-6 was the first identified gene target of IL-17,2 and this mechanism has since been used as a bioassay for IL-17A. IL-6 is also essential for the de novo differentiation of Th17 cells, which suggests a positive feedback circuit induced by IL-17. In addition to IL-6, IL-17 also induces production of other proinflammatory cytokines, such as TNF-α and IL-1β,32 and, by targeting cyclooxygenase 2 and inducible nitric oxide synthase, can trigger an increase in prostaglandin E2 and NO in various cell types.35, 36 Stimulation with IL-17 also induces the production or release of at least two different CSFs, G-CSF and GM-CSF.33, 34 Ectopic expression of IL-17 caused a strong expansion of neutrophil lineage or neutrophilia through G-CSF, and neutralization of IL-17 is associated with granulopenia defects and susceptibility to infection.
In addition to inflammatory cytokines, another major group of IL-17 target genes are chemokines, especially the CXC chemokines, including CXCL1 (keratinocyte/growth-regulated oncogene-α), CXCL2 (macrophage inflammatory protein 2), CXCL5, CXCL8 (IL-8), CXCL10 (IFN-inducible protein 10), and others.5, 31 These chemokines potentially mediate the biological function of IL-17 by attracting neutrophils in vivo. In mice, the key target chemokines are CXCL1 and CXCL5,37, 38, 39 whereas CXCL8 may be the most important IL-17-induced neutrophil chemokine in humans.40 Overexpression of IL-17 in the lung leads to neutrophil infiltration mediated by both chemotaxis and G-CSF-induced granulopoiesis.46 IL-17 can also stimulate the expression of some CC chemokines such as CCL2 (monocyte chemotactic protein 1) and CCL20 (macrophage inflammatory protein 3α).41, 42 Monocyte chemotactic protein 1 enables IL-17 to cause accumulation of monocytes, but this mechanism remains to be characterized. CCL20 is the ligand of CCR6, which is selectively expressed by Th17 cells,43 indicating another positive feedback loop for IL-17 by recruiting more IL-17-producing cells to inflammatory sites. Additionally, it is interesting that the ability of IL-17 to induce expression of chemokines and cytokines can be enhanced by costimulation with TNF-α or IL-1β.28, 44 We (data not published) and others45 have also demonstrated that IL-17 exhibits potent synergic function when stimulated with TLR ligands such as lipopolysaccharide.
In addition to proinflammatory cytokines and chemokines, IL-17 also promotes expression of various antimicrobial peptides. For example, β-defensin and some S100 proteins, which act as natural antibiotics in the lung, skin and gut,48 are significantly induced by IL-17 in epithelial cells,47, 49 providing protection against a broad spectrum of microorganisms. Recent reports revealed that β-defensin can function as a ligand for CCR6, linking innate and adaptive immunity by recruiting dendritic cells (DCs) and T cells.52 Furthermore, acute phase proteins such as lipocalin 2/24p3 are induced by IL-17.50, 51 Lipocalin 2/24p3 exerts its antimicrobial function by binding to bacterial siderophores that are necessary for bacterial survival in vivo.53
Certain kinds of tissue remodeling-associated molecules may also be induced by IL-17 stimulation. Elevated levels of IL-17 are found in synovial fluids from patients with rheumatoid arthritis.54 IL-17 can increase membrane expression of receptor activator of NF-κB ligand in osteoblasts,55 which in turn promotes osteoclastogenesis and subsequent bone destruction. In addition, Th17 cells themselves produce higher levels of receptor activator of NF-κB ligand than other T-cell subsets. Inflammatory cytokines such as TNF-α and IL-1β augment the activity of IL-17 and further cause bone erosion. Overexpression of IL-17 at the articular site results in joint erosion, whereas IL-17-deficient mice are protected from bone destruction.56 Matrix metalloproteinases (MMPs), including MMP1, MMP3, MMP9 and MMP13, are also targets of IL-17. They play important roles in extracellular matrix destruction and tissue damage and are essential in tumor angiogenesis.
Besides these functions of IL-17 on non-lymph linage cells, IL-17 was recently also shown to regulate functions of lymphocytes. In a mouse model of autoimmune BXD, exacerbated IL-17 secretion caused spontaneous development of germinal centers before the production of pathogenic autoantibodies. Blocking IL-17 signaling disrupts germinal center formation and reduces humoral responses.60 IL-17 alone or in combination with B cell-activating factor promotes human B-cell survival, proliferation and differentiation into immunoglobulin-secreting cells. This process is dependent on the NF-κB-activated transcription factor Twist-1.61 On the other hand, T cells are not only the source but also a target of IL-17. IL-17 can modulate Th1 cell polarization both in vitro and in vivo by directly acting on CD4 T cells to suppress T-bet expression.62
Cell sources and regulation of IL-17
Although its mRNA was discovered in a CTL hybridoma, IL-17A was originally associated with CD4 T cells, where IL-23 stimulates its production from memory CD4 T cells.57 This led to the discovery of IL-17-producing Th17 cells, a distinct helper T-cell subset different from Th1 and Th2 cells.58, 59 This Th cell linage was recognized as the major source of IL-17 in vivo. Subsequently, three independent studies demonstrated that a combination of TGF-β and IL-6 is required for their de novo differentiation from naive T cells in mice.65, 66, 67 In addition, even induced regulatory T (Treg) cells could be reprogrammed to the Th17 lineage in the presence of IL-6 plus TGF-β after 5 days.81 Retinoic acid-related orphan receptor-γt (ROR-γt) was identified as the linage specific transcription factor for Th17 cells,69 as T-bet and GATA-3 are the lineage specific transcription factors for Th1 and Th2 cells, respectively.70, 71 Another ROR family member, ROR-α, had a similar and redundant effect in promoting Th17 cell differentiation, as mutation of both ROR-α and ROR-γt completely inhibited Th17 cell generation in vitro and in vivo.74 Importantly, loss of STAT3, which is downstream of IL-6 and IL-21 signals, markedly decreased ROR-γt and ROR-α expression and impaired Th17 differentiation. By contrast, a hyperactive form of STAT3 facilitated IL-17 production in CD4 T cells.77, 107 Other transcription factors are also involved in the full differentiation of Th17 cells, including IFN regulatory factor 4 and runt-related transcription factor 1.75, 76 Further studies revealed that IL-23 is not required for Th17 differentiation, but can promote the growth, survival and effector function of this lineage.72, 73 IL-21 can substitute for IL-6, and constitutes an alternative pathway to generate Th17 cells in IL-6-deficient mice.68 In addition, IL-21 can be secreted by Th17 cells themselves, thus comprising an autocrine amplification loop in Th17 cell differentiation. Th17 cells also produce a certain amount of other cytokines such as IL-17F and IL-22. Development of human Th17 cells is analogous to mice,79 although IL-1β rather than IL-6 plays a more important role.78 Coexpression of the chemokine receptors CCR4 and CCR6, which mediate cell homing to mucosal surfaces such as the lung and gut, appears to define human Th17 cells.80
Almost all of the Th17 polarizing cytokines such as IL-1β, IL-6, TGF-β and IL-23 are induced in DCs activated by different kinds of stimulation. Which subset of DCs or what kind of stimulation orchestrates Th17 cell programming is still largely unknown. In vivo, the IL-17 producing cells mainly exist in the lamina propria. A unique subset of DCs that express high levels of CD70, which are selectively present in the lamina propria but not in spleen or lymph node, have been shown to be responsible for Th17 differentiation.63 However, other CD103+ DC subsets in the lamina propria inhibit Th17 but facilitate Treg generation in a retinoic acid-dependent mechanism.64 β-glucans, a component of fungus cell walls, preferentially induce IL-23 over IL-12 from DCs,109 which specifically drives Th17 differentiation.
As Th17 cells and IL-17 have pathogenic properties due to the induction of inflammatory pathways, the generation of Th17 cells is tightly regulated. First, both Th1 and Th2 cytokines negatively regulate Th17 development.58, 59 Type I IFNs suppress the generation of Th17 through STAT1 signaling. Similarly, STAT5 signaling induced by IL-2 also inhibits differentiation of Th17 cells, while facilitating the induction of Treg cells.83 Other inhibitory molecules include IL-10, IL-27, retinoic acid and the transcription factor Ets-1.82 Finally, Th17 cells were shown to produce the inhibitory cytokine IL-10 with IL-17, which prevents Th17-mediated immune pathogenesis.89
Although most of the recent studies have focused on IL-17 produced by T-cell antigen receptor-αβ+ Th17 cells, γδT cells have been shown to be another potent producer of IL-17, especially during the early innate immune responses. IL-23 alone can induce IL-17 from γδT cells, while TGF-β plus IL-6 cannot.84 Two subsets of γδT cells were shown to preferentially produce IL-17: Vγ6Vδ1 and Vγ4Vδ4.84, 85, 86 Recently, Sutton et al. have shown that DCs, after activation by TLRs, could secrete large amounts of IL-1 and IL-23, which could subsequently induce innate IL-17 production from γδT cells without engagement of T-cell antigen receptor signals.87 These IL-17-producing γδT cells also share characteristic features with Th17 cells, such as expression of CCR6, ROR-γt and the IL-23 receptor.88 They also express TLRs such as TLR1 and TLR2, and thus can directly respond to pathogen products. When under inducing signals, γδT cells can be rapidly induced to proliferate and produce innate IL-17 much earlier than the formation of adaptive Th17 cells. These γδT cells show no antigen specificity, implying a different role to Th17 cells in the immune response. Our unpublished data further show that the Th1 cytokine IFN-γ negatively regulates IL-17 production from γδT cells.
A variety of other T cells can also produce IL-17, including CD8 T cells and NKT cells. IL-17 production from CD8 T cells appears to be dependent on TGF-β plus IL-6.91 NKT cells constitutively express the IL-23 receptor and ROR-γt and rapidly produce IL-17 in an IL-6-independent manner.90 Furthermore, other innate immune cells such as neutrophils and macrophages also produce IL-17.92, 93 Recently, lymph tissue inducer cells, which are essential in the formation of lymph tissue and are now defined as a subset of natural killer cells, also constitutively express ROR-γt and IL-17.94 The biological functions of these IL-17-producing cells have yet to be fully determined.
IL-17 in diseases
Roles of IL-17 in allergy
Accumulating data suggest that IL-17 or Th17 play important roles in the development of various allergic diseases that have classically been considered to be Th1- or Th2-mediated disorders. Allergic asthma is classified into two types: atopic and non-atopic asthma. Atopic asthma is a Th2-dominated chronic inflammatory disease in the lungs, which is characterized by accumulation and activation of Th2 cells, eosinophils and mast cells, and increased immunoglobulin E (IgE) levels in the serum. Non-atopic asthma is characterized by accumulation of IL-8+ cells, neutrophils and mast cells without an increased serum IgE. IL-17 was reported to be increased in the lung, sputum, bronchoalveolar lavage fluid (BALF) and serum in asthmatic patients, and IL-17 levels correlated well with the severity of airway hypersensitivity.113 However, the type of asthma was not clearly differentiated.
Immunization of a mouse with ovalbumin (OVA), fungal antigens and cockroach antigens before challenge is a well-established model for Th2-dominated atopic asthma. In IL-17-deficient mice or mice with IL-17 neutralized, the airway hyperreactivity and pulmonary eosinophilia induced by OVA plus aluminum were shown to be normal,114, 115 although an increase in Th2 cytokines and decreased neutrophil infiltration was also observed. Repeated OVA immunization without adjuvant in IL-17-neutralized mice also resulted in a normal number of eosinophils in the BALF,117 though with reduced neutrophil influx. However, when a subcutaneous sensitization route was used, IL-17RA-deficient mice had reduced Th2 cytokines and impaired eosinophil recruitment.116 This was shown to be due to reduced antigen-specific Th cells during the sensitization phase. These disparate experimental outcomes may be due to different adjuvants, immunization routes and mouse backgrounds. Nonetheless, it is generally recognized that IL-17 makes little contribution to the induction of Th2-mediated and eosinophil-dominated atopic asthma.
By contrast, IL-17 has been clearly shown to play a key role in neutrophil-dominated non-atopic asthma. DO11.10 or OT II transgenic mice exhibited neutrophil- rather than eosinophil-dominated airway inflammation and airway hyperreactivity after OVA inhalation without prior sensitization. Th1 and Th17 cells were increased in the BALF, but neutralizing IFN-γ did not affect neutrophil recruitment.119 IFN-γ-deficient OT II mice even showed increased neutrophilila,118 suggesting a protective role of IFN-γ in this model. Nonetheless, the neutrophilia was profoundly suppressed in IL-17-deficient OT II or DO11.10 mice,118 and adoptive transfer of DO11.10 Th17 cells also resulted in recruitment of neutrophils to the lung.7 These observations revealed that IL-17 or Th17 cells are responsible for the airway inflammation in a mouse model of non-atopic asthma.
Contact hypersensitivity (CHS) has long been regarded as a delayed-type hypersensitivity reaction; recently, however, they have been discriminated. CD8 T cells were shown to be the effector cells in CHS, while CD4 T cells only contribute to delayed-type hypersensitivity responses. Chemically induced CHS developed normally in IFN-γ- and IFN-γR1-deficient mice,120, 121 indicating that IFN-γ is not essential for CHS. However, the responses are impaired in IL-17-deficient mice.114 Furthermore, IL-17-producing CD8 T cells rather than T cytotoxic type 1 cells are suggested to be involved in the process.122 Th clones established from allergic CHS patients can also produce IL-17. Thus, IL-17 may, at least partially, contribute to the development of CHS.
Roles of IL-17 in autoimmune diseases
The concept of Th17 or IL-17 immunity emerged from studies in two models of autoimmune diseases: experimental autoimmune encephalomyelitis (EAE), a well-established model for human multiple sclerosis, and collagen-induced arthritis (CIA), which models human rheumatoid arthritis. These were thought to be Th1 cell-mediated diseases, as IL-12p40 deficiency dramatically attenuated disease severity. However, mice treated with anti-IFN-γ or mice deficient in IFN-γ or IFN-γR1 also developed aggravated EAE and CIA. This paradox was resolved when Cua123 and Murphy124 both independently used p35 (IL-12−/−)-, p19 (IL-23−/−)- and p40 (IL-12−/−/IL-23−/−)-deficient mice to induce EAE or CIA. IL-23-deficient (p19−/−) mice showed dramatically attenuated disease development, indicating that IL-23 rather than IL-12 is critically linked to the pathogenesis of EAE and CIA. Exacerbated IL-17 producing CD4 T cells were also observed in the central nervous system in the EAE model.125 Further studies revealed that neutralization of IL-17 or IL-17 deficiency rendered the mice resistant to EAE induction126 and impaired joint inflammation.127, 128 Adoptive transfer of pathological Th17 cells, but not Th1 cells, from established EAE mice also re-established EAE in recipient mice.125 Taken together, these data clearly suggested a central role of IL-17 or Th17 cells in autoimmune disease development. Recently, IL-17 production from γδT cells was also suggested to play an important role in EAE induction.87 γδT cells provide the early source of innate IL-17, which could facilitate later adoptive Th17 cell generation by induction of IL-6 and IL-23 secretion. Consistent with these observations, IL-17 expression has also been reported to be upregulated in the cerebrospinal fluids from multiple sclerosis patients and in the synovial fluid of rheumatoid arthritis patients.129, 132 Additionally, T cells derived from these patients produced more IL-17 than healthy controls.
The roles of IL-17 or Th17 in experimental autoimmune uveitis130 and experimental autoimmune myocarditis131 may be similar to that in EAE. However, the conditions in another autoimmune disease—inflammatory bowel disease, are more controversial. IL-17 levels were found to be increased in the inflamed gut of patients with Crohn's disease and ulcerative colitis.133 The major sources of this IL-17 are CD3+ T cells and CD68+ macrophages. In chemically induced experimental colitis models, IL-17RA-deficient mice show resistance to colitis induced by intrarectal administration of trinitrobenzenesulfonic acid.134 Additionally, administration of an IL-17RA/IgG1 fusion protein in wild-type mice significantly attenuated trinitrobenzenesulfonic-induced colonic inflammation and prevented weight loss.135 These data suggest a non-redundant role of IL-17A or IL-17F in this model. Another chemical dextran sulfate sodium-induced colitis model showed reduced pathology in IL-17F-deficient mice but a more severe disease in IL-17A-deficient mice,136 indicating a protective role for IL-17A and a pathogenic role for IL-17F. Naive CD4 T cell transfer can also establish inflammatory bowel disease in lymphopenic mice. In this model, adoptive transfer of Th17 cells resulted in more significant gut inflammation than with Th1 cell transfer.137 Administration of an anti-p19 monoclonal antibody prevented active colitis and reduced inflammatory cytokines in the gut.137 Adoptive transfer of naive CD8 T cells into Rag−/− mice caused proliferation and IL-17 and IFN-γ production from these cells, resulting in severe colitis. Lack of IL-17 or IFN-γ both attenuated the disease.138 Thus, IL-17 definitely orchestrates pathogenesis of inflammatory bowel disease, either by facilitation of or resistance to inflammation, depending on the different models used.
Roles of IL-17 in host defense
Components from various pathogens can induce IL-17 production from various cell types, especially Th17 cells and γδT cells, implying an indispensable role of IL-17 in host defense against infectious diseases. IL-17 exerts its protective function mainly through the effective recruitment and expansion of neutrophils mediated by CXC chemokines and G-CSF induction.11 In addition, chemokines induced by IL-17 can recruit other immune cells to the infection site, which may provide another protection mechanism as well. Antimicrobial molecules such as β-defensin, S100 and others can enhance host defense at the mucosal site. Recent advances expanded our understanding of the broad effects of IL-17 in protection against various bacteria, fungi and virus infection.102 However, enhanced inflammation is a double-edged sword, and under certain infectious conditions, IL-17 may not provide protection, but it could also function to exacerbate the pathogenic process. Thus, the exact role of IL-17 in host defenses may depend on the pathogen species.
Emerging evidence supports the concept that IL-17 plays a protective role in extracellular bacterial infections. The first clues were generated in a respiratory Klebsiella pneumonia model.15, 95 Although IL-12 and IFN-γ signaling have been shown to be critical for K. pneumonia infection, mice deficient in IL-17 signaling showed impaired neutrophil recruitment and reduced bacterial clearance in the lung. Overexpression of IL-17 by a recombinant adenovirus in IL-17-deficient mice reversed the disease phenotype resulting in elevated chemokines, cytokines and increased neutrophil recruitment.96 These conclusions can be extended to gut infection with Citrobacter rodentium, as IL-23 rather than IL-12 is required for host defense in the early phase, and Th17 cells in the lamina propria are indispensable for protection.66, 97 Although IL-22 produced by Th17 cells was shown to be more protective than IL-17, IL-17RA-deficient mice also showed higher susceptibility to infection by porphyromonas gingivalis, an anaerobic periodonatal pathogen of the oral mucosa.99
The biological functions of IL-17 in intracellular infections are not well understood. Mycobacterium is the most commonly studied model for host defense against intracellular infection. IL-17RA−/−, IL-17A−/− and IL-23−/− mice are no more susceptible than their wide-type controls to Mycobacterium tuberculosis infection,97, 100, 101 suggesting protection mainly by the IL-12–IFN-γ axis and the Th1 pathway. However, the induction of IL-17 after Salmonella typhimurium infection may contribute to neutrophil recruitment and host defense.98 The absence of IL-17 signals increased bacterial dissemination from the intestine. In addition, the mice were protected from respiratory infection with Mycoplasma pneumonia in the presence of IL-17 signals.104 We (unpublished data) and others105 revealed that intraperitoneal infection with Listeria monocytogens could induce IL-17 production, with γδT cells being the main source of IL-17, which was secreted 1 day post-infection. Deficiency of IL-17 substantially reduced bacterial clearance associated with decreased neutrophil infiltration. These studies show that in models in which neutrophils are involved in protection, IL-17 may be critical for the full induction of immune responses and influx of neutrophils leading to bacterial control, but it may have no role in other models of intracellular infection.
Accumulating data suggest that IL-17 or Th17 cells may be preferentially induced in fungal infections and may play important roles in orchestrating host defenses against these fungi. Candida albicans is one of the most studied models. It is a commensal pathogen in the oral cavity and gastrointestinal tract but can cause pathogenic disease in immunodeficient individuals. Interestingly, IL-17-producing memory CD4 T cells in humans were found to have a high frequency of Candida-specific T-cell antigen receptors,80 suggesting a preferential induction of IL-17 by this fungal infection. Higher susceptibility to mucocutaneous Candida infections is observed in patients with hyper-IgE syndrome, which results from a mutation in the STAT3 gene locus and has reduced numbers of Th17 cells.107 In mice, IL-17 signals, but not IFN-γ or Th1 cells, are highly protective in oral C. albicans infection.106 In pulmonary defenses against Aspergillus fumigatus, the IL-23–IL-17 axis has also been shown to be required for clearance of the pathogen.108 β-glucans, a component of fungus cell walls, recognize a C-type lectin receptor Dectin-1 on DCs and macrophages and preferentially induce IL-23 over IL-12 production,109 resulting in Th17 differentiation and IL-17 secretion from γδT cells. Viral specific Th17 cells are also found in HIV and cytomegalovirus infection in humans, suggesting a role for IL-17 in viral infections.110
Nonetheless, IL-17 or Th17 cells are not always protective in pathogen infection, as uncontrolled production of IL-17 may exaggerate damage in infected tissues. For example, Helicobacter pylori infection in the stomach induces robust IL-17 production and infiltration of neutrophils in the gastric mucosa, which leads to pathogenic inflammatory responses and gastritis.103 In addition, Bordetella pertussis, a Gram-negative bacteria in the respiratory system, may preferentially induce IL-23 and inhibit IL-12 production,111 resulting in severe inflammation, respiratory destruction and persistent cough. In chronic granulomatous disease, heightened IL-17 production causes lethal lung pathology.112 These findings indicate that the early induction of IL-17 during pathogen infection is protective. However, in chronic infectious diseases, IL-17 appears to be dispensable for protection, and uncontrolled IL-17 production may even results in pathology rather than protection.
Roles of IL-17 in malignancy
Rudolph Virchow postulated that chronic inflammation may facilitate carcinogenesis and tumor growth in the 1800s. IL-17 is associated with chronic diseases, suggesting a role in promoting carcinogenesis and tumor growth. Indeed, human cervical cancer and murine fibrosarcoma cells transfected with IL-17 show significantly increased tumor formation in vivo in nude mice and C57BL/6 mice;139, 140 however, IL-17 has no direct effect on tumor proliferation in vitro. Exogenous IL-17 expression in vivo can stimulate the secretion of vascular endothelial growth factor, prostaglandin E1 and prostaglandin E2 in murine fibroblasts and other cells, thus enhancing tumor angiogenesis.141 In addition, IL-17 can stimulate MMP expression, leading to the extracellular matrix destruction that is necessary for angiogenesis. Consistent with this, the growth of B16 melanoma and MB49 bladder carcinoma cells is reduced in IL-17-deficient mice,142 and increased numbers of intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients.147 IL-17 can induce IL-6 production, which in turn upregulates prosurvival and proangiogenic genes in tumor cells.142 In another study, IL-17 derived from CD8+ T cells was shown to exert an antiapoptotic function directly on breast cancer cells.148 These studies together suggest that IL-17 promotes cancer via its antiapoptotic and angiogenic activities.
However, the growth of IL-17-transfected P815 and J558L cells in immunocompetent mice was inhibited more significantly than mock controls.143 This phenomenon is abrogated in nude mice, suggesting that T cells contribute to the antitumor action of IL-17. Adoptive transfer of in vitro generated tumor-specific Th17-polarized cells can eradicate large established melanomas.144 Dong's group145 elucidated the mechanism of this action, showing that Th17 cells induce expression of chemokines in the tumor site that recruit CD8α+ DCs and generate more tumor antigen-targeted CTLs. The growth of MC38 colon adenocarcinomas was also suppressed in wild-type mice compared with mice deficient in IL-17A.146 These studies showed an opposite role of IL-17; specifically, they suggest an antitumor effect through promoting effector CTL generation.
Thus, IL-17 plays multifactorial roles in tumor immunity. The prosurvival effect on tumor growth or antitumor effects might depend on the immunogenicity of the tumor, the immune status of the host and the phase of the disease. We postulate that in the acute phase of tumor, IL-17 may exert antitumor activity via the stimulation of tumor specific CTLs; however, when the disease reaches a chronic stage, the angiogenic activity of IL-17 begins to emerge, and the balance of CTL formation and angiogenesis determines the outcome of tumors.
Roles of IL-17 in transplantation
Allograft transplantation is the best available treatment option for many end-stage diseases of solid organs. However, the long-term success of this procedure is limited due to allograft rejection. Allograft rejection has been shown to be driven by Th1 subsets, but contradictory results have been obtained. Loss of IFN-γ in rodent heart and kidney transplant recipients results in accelerated graft rejection and increased parenchymal necrosis.149, 150 Recent data suggest that IL-17/Th17 cells may be the ‘missing piece' in this process, especially during acute rejection. In humans, increased IL-17 protein levels have been detected in rejected renal allografts but undetectable in renal transplant tissue without signs of rejection.151 In a rat renal allograft model, IL-17 mRNA expression appeared on day 2 after operation, peaked at day 5 and then declined, becoming undetectable by day 9.151 In cardiac allograft transplantation, neutralization of IL-17 by an IL-17RA/IgG-Fc fusion protein prevented acute rejection and prolonged both non-vascularized and vascularized heart graft survival.153, 154 By contrast, IL-17 antagonism does not appear to prevent ensuing chronic graft vascular diseases.155 More recently, IL-17 has been implicated in the rejection of lung transplants. Elevated IL-17 is found in BALF from patients with acute lung allograft rejection compared with those without rejection.156 In addition, a population of collagen type V-specific T cells expressing IL-17 was predisposed to obliterative bronchiolitis in human lung transplants.157 Together with studies in liver153 and corneal152 transplantation, these data reveal a disease-promoting role of IL-17 or Th17 cells in the early stage of allograft rejection. IFN-γ is a negative regulator of Th17, and IFN-γ deficiency may lead to increased IL-17 production and neutrophil infiltration. These observations may explain why IFN-γ-deficient mice show accelerated allograft rejection.
Allogeneic bone marrow transplantation (BMT) is a potentially curative therapy for various hematopoietic malignancies. Graft versus host disease (GVHD) is the major complication of allogeneic BMT and causes significant morbidity and mortality. Both Th1 and Th2 cells are involved in the pathology of GVHD. Significant numbers of alloreactive CD4+ donor T cells expressing IL-17 have been found in the lymphoid organs of recipients of an allogeneic bone marrow transplant.158 Although recipients of IL-17−/− CD4 T cells developed GVHD at a slower rate than recipients of wild-type CD4 T cells, these mice eventually succumbed to GVHD, and there was no difference in overall survival by day 90 after BMT. This suggests that IL-17 only mediates acute GVHD but does not influence the latent phase. However, another group observed a protective role of IL-17.159 An absence of donor IL-17 leads to augmented Th1 differentiation and exacerbated acute GVHD. Thus, the real function of IL-17 in GVHD remains largely unknown due to a limited number of reports in this field.
Perspectives and implications
Since the identification of Th17 cells, much attention and progress has been achieved in the understanding of the developmental and biological functions of the cytokine IL-17, the canonical product of Th17 cells. It is clear that IL-17 exerts a complicated role in allergenic disease, host defense, autoimmune diseases, malignancy and transplantation rejection. The impact of the Th17 pathway is likely to be more profound than previously thought. The findings in preclinical animal models have supported therapeutic approaches targeting the IL-17A pathway, although these were limited to autoimmune disorders. In principle, there are three strategies for modulating the IL-17 pathway: blocking upstream of the pathway, blocking downstream of IL-17 or neutralizing IL-17.
With respect to targeting IL-17, clinical trials are now being performed or planned to evaluate an anti-IL-17A monoclonal antibody in psoriasis, Crohn's disease and rheumatoid arthritis.162 Additional approaches being considered to inhibit IL-17 include using a soluble IL-17RA/Ig fusion protein to block IL-17 ligands, using monoclonal antibodies to block IL-17RA or IL-17RC, or a combination of the two.
We can also target upstream of IL-17 production, perhaps by inhibiting IL-23 or IL-6 signals and thus reducing IL-17 production. Monoclonal antibodies against the p40 subunit are being tested in phase-II clinical trials for patients with Crohn's disease160 or psoriasis,161 and have proven to be efficacious. All-trans retinoic acid and its analogs can be tested to block IL-17 production in vivo in humans, as this inhibits Th17 but facilitates Treg generation in mouse models. In addition, using small molecules to inhibit the intracellular signaling pathways of IL-17 is another approach to inhibit its biological activity. The key adaptor ACT1 may be the best target, but these molecules have not been identified.
In conclusion, we should always keep in mind the limitations of targeting IL-17 in clinical therapy. The link between IL-17 targeting and neutropenia is important, as this may affect the host defenses against some pathogens. At least in mice, inhibition of IL-17 has been associated with increased mortality from bacterial lung infections. Cellular immunity has also been shown to be hindered by neutralization of IL-17. Thus, IL-17 sits at the center of many diseases that integrate innate and adaptive immune responses, and the efforts to decrease the risk of infectious complications will allow effective therapies for treating inflammatory disorders.
References
- Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. 2007;282:13447–13455. doi: 10.1074/jbc.M700499200. [DOI] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Dong C. IL-17F: regulation, signaling and function in inflammation. Cytokine. 2009;46:7–11. doi: 10.1016/j.cyto.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol. 2007;179:5462–5473. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- Kramer JM, Yi L, Shen F, Maitra A, Jiao X, Jin T, et al. Evidence for ligand-independent multimerization of the IL-17 receptor. J Immunol. 2006;176:711–715. doi: 10.4049/jimmunol.176.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Regulation and pro-inflammatory function of interleukin-17 family cytokines. Immunol Rev. 2008;226:80–86. doi: 10.1111/j.1600-065X.2008.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, et al. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci USA. 2007;104:7506–7511. doi: 10.1073/pnas.0611589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM, Yi L, Shen F, Maitra A, Jiao X, Jin T, et al. Evidence for ligand-independent multimerization of the IL-17 receptor. J Immunol. 2006;176:711–715. doi: 10.4049/jimmunol.176.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1239. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, et al. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci USA. 2007;104:7506–7511. doi: 10.1073/pnas.0611589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Hartupee J, Liu C, Novotny M, Sun D, Li X, Hamilton TA. IL-17 signaling for mRNA stabilization does not require TNF-receptor-associated factor 6. J Immunol. 2009;182:1660–1666. doi: 10.4049/jimmunol.182.3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-κB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol. 2007;179:6504–6513. doi: 10.4049/jimmunol.179.10.6504. [DOI] [PubMed] [Google Scholar]
- Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. 2008;41:92–104. doi: 10.1016/j.cyto.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC. IL-17 stimulates the production and expression of pro-inflammatory cytokines, IL-β and TNF-α, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce pro-inflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan M, Prause O, Miyamoto M, Sjöstrand M, Hytönen AM, Kaneko T, et al. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-α. Eur Respir J. 2003;21:387–393. doi: 10.1183/09031936.03.00303503. [DOI] [PubMed] [Google Scholar]
- Trajkovic V, Stosic-Grujicic S, Samardzic T, Markovic M, Miljkovic D, Ramic Z, et al. Interleukin-17 stimulates inducible nitric oxide synthase activation in rodent astrocytes. J Neuroimmunol. 2001;119:183–191. doi: 10.1016/s0165-5728(01)00391-5. [DOI] [PubMed] [Google Scholar]
- LeGrand A, Fermor B, Fink C, Pisetsky DS, Weinberg JB, Vail TP, et al. Interleukin-1, tumor necrosis factor α, and interleukin-17 synergistically up-regulate nitric oxide and prostaglandin E2 production in explants of human osteoarthritic knee menisci. Arthritis Rheum. 2001;44:2078–2083. doi: 10.1002/1529-0131(200109)44:9<2078::AID-ART358>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GROα chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- Ruddy MJ, Shen F, Smith J, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- Laan M, Cui A-H, Hoshino H, Lötvall J, Sjöstrand M, Gruenert DC, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- Takaya H, Andoh A, Makino J, Shimada M, Tasaki K, Araki Y, et al. Interleukin-17 stimulates chemokine (interleukin-8 and monocyte chemoattractant protein-1) secretion in human pancreatic periacinar myofibroblasts. Scand J Gastroenterol. 2002;37:239–245. doi: 10.1080/003655202753416948. [DOI] [PubMed] [Google Scholar]
- Kao C-Y, Huang F, Chen Y, Shimada M, Tasaki K, Araki Y, et al. Upregulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK-NFκB-dependent signaling pathway. J Immunol. 2005;175:6676–6685. doi: 10.4049/jimmunol.175.10.6676. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, et al. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, et al. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-κB signaling pathways. J Immunol. 2004;173:3482–3491. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- Ganz T. Defensins and host defense. Science. 1999;286:420–421. doi: 10.1126/science.286.5439.420. [DOI] [PubMed] [Google Scholar]
- Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17 and TNF-α-induced genes in bone cells. J Leukoc Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. β-defensins: linking innate immunity and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-κ B ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–2662. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A. Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Ann Rheum Dis. 2000;59:529–532. doi: 10.1136/ard.59.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- O'Connor W, Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce pro-inflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of pro-inflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. . Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmedby orphan nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüstle A, Heink S, Huber M, Rosenplänter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- Zhang F, Meng G, Strober W. Inter-actions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC. Ets-1 is a negative regulator of Th17 differentiation. J Exp Med. 2007;204:2825–2835. doi: 10.1084/jem.20070994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, et al. IL-17A produced by γδ T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–3463. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O'Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing γδ T cells. J Immunol. 2007;179:5576–5583. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting edge: NKT cells constitutively express IL-23 receptor and RORγt and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Tsai JP, Shen CR, Sher YP, Hsieh CL, Yeh YC, et al. Induction of a distinct CD8 Tnc17 subset by transforming growth factor-β and interleukin-6. J Leukoc Biol. 2007;82:354–360. doi: 10.1189/jlb.0207111. [DOI] [PubMed] [Google Scholar]
- Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–2112. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- Gu Y, Yang J, Ouyang X, Liu W, Li H, Yang J, et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38:1807–1813. doi: 10.1002/eji.200838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. . J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–340. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JJ, Ruddy MJ, Conti HR, Boonanantanasarn K, Gaffen SL. The interleukin-17 receptor plays a gender-dependent role in host protection against Porphyromonas gingivalis-induced periodontal bone loss. Infect Immun. 2008;76:4206–4213. doi: 10.1128/IAI.01209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-γ responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009;2:403–411. doi: 10.1038/mi.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, et al. IL-17A produced by γδ T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–3463. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, et al. Requisite role for the dectin-1 β-glucan receptor in pulmonary defense against Aspergillus fumigatus. . J Immunol. 2009;182:4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat Immunol. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue FY, Merchant A, Kovacs CM, Loutfy M, Persad D, Ostrowski MA. Virus-specific interleukin-17-producing CD4+ T cells are detectable in early human immunodeficiency virus type 1 infection. J Virol. 2008;82:6767–6771. doi: 10.1128/JVI.02550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele G, Nasso M, Spensieri F, Palazzo R, Frasca L, Watanabe M, et al. Lipopolysaccharides from Bordetella pertussis and Bordetella parapertussis differently modulate human dendritic cell functions resulting in divergent prevalence of Th17-polarized responses. J Immunol. 2008;181:208–216. doi: 10.4049/jimmunol.181.1.208. [DOI] [PubMed] [Google Scholar]
- Romani L, Fallarino F, de Luca A, Montagnoli C, D'Angelo C, Zelante T, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med. 2003;97:726–733. doi: 10.1053/rmed.2003.1507. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385–393. doi: 10.1084/jem.20071507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- Nakae S, Suto H, Berry GJ, Galli SJ. Mast cell-derived TNF can promote Th17 celldependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood. 2007;109:3640–3648. doi: 10.1182/blood-2006-09-046128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott PG, Gater PR, Bertrand CP. Airway inflammation driven by antigen-specific resident lung CD41 T cells in αβ-T cell receptor transgenic mice. Am J Respir Crit Care Med. 2000;161:1340–1348. doi: 10.1164/ajrccm.161.4.9906078. [DOI] [PubMed] [Google Scholar]
- Saulnier M, Huang S, Aguet M, Ryffel B. Role of interferon-γ in contact hypersensitivity assessed in interferon-γ receptor-deficient mice. Toxicology. 1995;102:301–312. doi: 10.1016/0300-483x(95)03101-k. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Narumi S, Sudo K, Horai R, Tagawa Y, et al. IL-1-induced tumor necrosis factor-α elicits inflammatory cell infiltration in the skin by inducing IFN-γ-inducible protein 10 in the elicitation phase of the contact hypersensitivity response. Int Immunol. 2003;15:251–260. doi: 10.1093/intimm/dxg028. [DOI] [PubMed] [Google Scholar]
- He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. CD81 IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177:6852–6858. doi: 10.4049/jimmunol.177.10.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, Oppers B, van den Bersselaar L, Coenen-de Roo CJ, Kolls JK, et al. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. doi: 10.4049/jimmunol.167.2.1004. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderegger I, Röhn TA, Kurrer MO, Iezzi G, Zou Y, Kastelein RA, et al. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36:2849–2856. doi: 10.1002/eji.200636484. [DOI] [PubMed] [Google Scholar]
- Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human interleukin-17: a T cell-derived pro-inflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–388. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- Tajima M, Wakita D, Noguchi D, Chamoto K, Yue Z, Fugo K, et al. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J Exp Med. 2008;205:1019–1027. doi: 10.1084/jem.20071133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999;59:3698–3704. [PubMed] [Google Scholar]
- Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- Numasaki M, Lotze MT, Sasaki H. Interleukin-17 augments tumor necrosis factor-α-induced elaboration of pro-angiogenic factors from fibroblasts. Immunol Lett. 2004;93:39–43. doi: 10.1016/j.imlet.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautès-Fridman C, et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114–2121. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–359. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, et al. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol. 2009;50:980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- Nam JS, Terabe M, Kang MJ, Chae H, Voong N, Yang YA, et al. Transforming growth factor β subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68:3915–3923. doi: 10.1158/0008-5472.CAN-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halloran PF, Miller LW, Urmson J, Ramassar V, Zhu LF, Kneteman NM, et al. IFN-γ alters the pathology of graft rejection: protection from early necrosis. J Immunol. 2001;166:7072–7081. doi: 10.4049/jimmunol.166.12.7072. [DOI] [PubMed] [Google Scholar]
- Miura M, El-Sawy T, Fairchild RL. Neutrophils mediate tissue necrosis and accelerate rejection of cardiac allografts in the absence of IFNγ. Am J Pathol. 2003;162:509–519. doi: 10.1016/s0002-9440(10)63845-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh HG, Loong CC, Lui WY, Chen A, Lin CY. IL-17 expression as a possible predictive parameter for subclinical renal allograft rejection. Transpl Int. 2001;14:287–298. doi: 10.1007/s001470100344. [DOI] [PubMed] [Google Scholar]
- Chen H, Wang W, Xie H, Xu X, Wu J, Jiang Z, et al. A pathogenic role of IL-17 at the early stage of corneal allograft rejection. Transpl Immunol. 2009;21:155–156. doi: 10.1016/j.trim.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Fábrega E, López-Hoyos M, San Segundo D, Casafont F, Pons-Romero F. Changes in the serum levels of interleukin-17/interleukin-23 during acute rejection in liver transplantation. Liver Transpl. 2009;15:629–633. doi: 10.1002/lt.21724. [DOI] [PubMed] [Google Scholar]
- Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, et al. Evidence for a role of IL-17 in alloimmunity: a novel IL-17 antagonist promotes heart graft survival. Transplant Proc. 1999;31:93. doi: 10.1016/s0041-1345(98)01453-5. [DOI] [PubMed] [Google Scholar]
- Tang JL, Subbotin VM, Antonysamy MA, Troutt AB, Rao AS, Thomson AW. Interleukin-17 antagonism inhibits acute but not chronic vascular rejection. Transplantation. 2001;72:348–350. doi: 10.1097/00007890-200107270-00035. [DOI] [PubMed] [Google Scholar]
- Vanaudenaerde BM, DuPont LJ, Wuyts WA, Verbeken EK, Meyts I, Bullens DM, et al. The role of interleukin-17 during acute rejection after lung transplantation. Eur Respir J. 2006;27:779–787. doi: 10.1183/09031936.06.00019405. [DOI] [PubMed] [Google Scholar]
- Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117:3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, et al. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood. 2009;113:945–952. doi: 10.1182/blood-2008-08-172155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Zhao D, Lin CL, Zhang C, Chen Y, Todorov I, et al. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood. 2008;112:2101–2110. doi: 10.1182/blood-2007-12-126987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–592. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- Ivanov S, Lindén A. Interleukin-17 as a drug target in human disease. Trends Pharmacol Sci. 2009;30:95–103. doi: 10.1016/j.tips.2008.11.004. [DOI] [PubMed] [Google Scholar]