

Abstract
The propagation and regulation of an immune response is driven by a network of effector and regulatory T (Treg) cells. The interplay of effector T and Treg cells determines the direction of the immune response towards inflammation or its resolution in an autoimmune disease setting. In autoimmune diseases, this interplay shifts the balance in favor of the development of autoreactive effector T cells, resulting in inflammatory pathology. The objective of an effective therapeutic approach for autoimmune disease is to restore this balance. In this review, we describe the characteristics and development of pathogenic T helper 1 (Th1) and Th17 cells and the beneficial Treg cells in autoimmune diseases and the crucial roles of the cytokine milieu in influencing the balance of these T-cell subsets. Given the importance of cytokines, we discuss current immunotherapeutic strategies using cytokine or cytokine receptor antibodies for the treatment of autoimmune diseases.
Keywords: Autoimmune disease, Cytokine, Regulatory T cell, T helper 17 cell
Introduction
CD4+ T helper (Th) cells are essential effectors of the immune response and play an important role in inflammation. For more than two decades, the Th1/Th2 paradigm, introduced by Mosmann and Coffman, has been used to explain most of the phenomena related to adaptive immunity.1 This paradigm has been modified following the recent discovery of Th17 and regulatory T (Treg) cells, two newly characterized T-cell subsets that are thought to be critically involved in mediating and regulating autoimmune responses.2, 3, 4, 5 An in-depth understanding of the precise pathological mechanisms of these pathogenic T-cell subsets is urgently needed to identify novel targets and to develop specific new treatment approaches for autoimmune diseases, in which interplay or interaction of Th1/Th17 and Treg cells is controlled by the cytokine environment. Pathogenic Th1/Th17 cells secreting signature proinflammatory cytokines, i.e., interferon (IFN)-γ and IL-17, have been shown to promote inflammatory responses and to contribute to the pathogenesis of multiple human autoimmune disorders and their animal models, including experimental autoimmune encephalomyelitis (EAE) for multiple sclerosis (MS) and collagen-induced arthritis (CIA) for rheumatoid arthritis (RA).6, 7, 8 In contrast, Treg cells are crucial for the maintenance of localized immunosuppression and normally keep pathogenic Th1/Th17 cells in check.9 In this review, we discuss recent advances in this area with particular focus given to novel cytokine signaling pathways involved in the regulation of development of Th17 and Treg cells. We also highlight potential new therapeutic targets of these two T-cell subsets for the development of new treatments for MS and other autoimmune diseases.
Roles of Th1/Th17 and Treg cells in autoimmune diseases
Th1 and Th17 cells in autoimmune diseases
The pathogenesis of chronic autoimmune diseases, including MS and RA, arises from the break in tolerance of self-antigens and the development of autoaggressive effector T cells infiltrating the target tissues. Previously, Th1 cells have been regarded as the main pathogenic T cells driving autoimmune tissue damage, which is attributed to the activation of macrophages by the Th1-secreted cytokine IFN-γ.10 Paradoxically, IFN-γ−/− mice were found to be highly susceptible to many organ-specific autoimmune diseases, including EAE, and blocking IFN-γ or its signaling pathway exacerbates autoimmune disease severity in EAE and CIA.11, 12 This dilemma was explained partially by the paradoxical properties of IFN-γ13 but mainly through the discovery of a new lineage of Th subset, IL-17-producing Th17 cells, which are susceptible to inhibition by IFN-γ.13 There is evidence that Th17 cells are activated during the disease process and are responsible for recruiting other inflammatory cell types, especially neutrophils, to mediate pathology in the target tissues.14 The observations accumulated thus far indicate that both Th1 and Th17 cells are important drivers of the inflammatory process in tissue-specific autoimmunity.15 Using EAE as an example, adoptive transfer of myelin oligodendrocyte glycoprotein-specific Th1 or Th17 cells, but not Th2 cells, to naive mice was able to induce EAE.16, 17 Interestingly, the two T-cell subsets caused different central nervous system (CNS) pathologies in the recipients, suggesting that they are functionally distinct. Both Th1 and Th17 cells were observed in the infiltrates of EAE-, CNS- and CIA-inflamed joints. Furthermore, in our study, analysis of myelin oligodendrocyte glycoprotein-specific T cells from the spinal cords of mice with EAE further revealed the presence of substantial numbers of T cells coexpressing IL-17 and IFN-γ,18 raising questions about the function of this double-positive population.
Treg cells in autoimmune diseases
Another lineage of T cells that coexpress CD4, CD25 and the transcription factor Forkhead box p3 (Foxp3) are Tregs.18 Treg cells have the capacity to actively suppress effector cells and to dampen a wide spectrum of immune responses, including those associated with autoimmune diseases.4 Thymic-derived naturally occurring Treg (nTreg) cells are critical for maintaining peripheral immune tolerance and for preventing autoimmunity and tissue injury by inhibiting the proliferation and effector functions of pathogenic Th1/Th17 cells.18, 19, 20 In addition, transforming growth factor (TGF)-β can promote the generation of Treg cells from naive T cells through the induction of Foxp3 expression; these are the so-called iTreg cells.21 Once generated, Treg cells can act as a negative regulator for the control of activation and effector functions of autopathogenic T cells in the immune system.22 Recent studies have suggested that CD4+CD25hi Treg cells were functionally impaired in MS patients.23 Interestingly, it has been shown that Treg cells isolated from the circulation of MS patients, though normal in number, poorly inhibited effector T-cell proliferation in an in vitro coculture system.24, 25 The functional impairment of Treg cells was also reported in a mouse EAE model, in which myelin-specific functional Treg cells accumulate in the CNS but fail to control autoimmune inflammation.26 The situation in RA is different from that in MS, in which reduced number of CD4+CD25hi Treg cells were observed in the peripheral blood of RA patients. It has been reported that Treg cells derived from RA patients can neither regulate proinflammatory cytokine secretion by pathogenic T cells and monocytes nor suppress autoreactive T cells.27, 28 It should be noted that the frequency of CD4+CD25hi Treg cells is higher in rheumatoid synovium than in the peripheral blood of patients with RA, and there were considerable numbers of Treg cells in the CNS lesions of peak disease-stage EAE mice.26, 29, 30, 31, 32, 33 In parallel, our recent study also demonstrated that Treg cells accumulate in the rheumatoid synovium and coexist with pathogenic Th17 and Th1 cells that are highly concentrated at the site of inflammation (data unpublished). Taken together, these findings suggest that functionally impaired Treg cells and increased numbers of autopathogenic Th17 cells contribute to the break in tolerance and shift of the immune system towards a proinflammatory state.
Cytokine environment and signaling pathways for differentiation and maintenance of Th1/Th17 and Treg cells
Roles of cytokines in differentiation of T-cell subsets
Efficient host defense against pathogens is achieved through a complex signaling cross-talk between the innate and adaptive immune systems. In response to the type of pathogen presented by conventional antigen-presenting cells (APCs) in a cytokine milieu produced by activated APCs, naive CD4+ T cells can differentiate to specific lineages, Th1, Th2, Th17 and Treg cells, as categorized by the expression of specific transcription factors, and have distinct effector functions with specific cytokine profiles (Figure 1). In the presence of IFN-γ and IL-12, naive CD4 T cells differentiate towards Th1 in a process that is dependent on the activities of STAT1, the transcription factor T-bet and STAT4. Th1 cells produce large quantities of IFN-γ and play a critical role in protective immunity against intracellular pathogens through the activation of macrophages.1, 34, 35, 36 IL-4 promotes Th2 differentiation through the activation of STAT6 and the transcription factor GATA3. Th2 cells produce IL-4, IL-5, IL-13 and IL-25, which are important for the orchestration of humoral immune responses clearing extracellular pathogens and parasites through the induction of immunoglobulin class switching to immunoglobulin G1 and immunoglobulin E, respectively.34, 35, 37, 38 The newly identified Th17 cells differentiate from naive CD4+ T cells in response to IL-6 and TGF-β, can produce IL-17A, IL-17F and IL-22, play important roles in the clearance of extracellular bacteria and fungi, and have been linked to autoimmune disorders.3, 5 Differentiation of both Th17 and Treg cells from naive CD4+ cells requires TGF-β.39 IL-6 activates STAT3 and, in combination with TGF-β signaling, increases the expression of the retinoid-related orphan receptor (ROR)γt and RORα transcription factors, resulting in the initiation of Th17 differentiation.40, 41, 42, 43 TGF-β also promotes the expression of the transcription factor Foxp3 that, in the absence of IL-6, blocks the activities of RORα and RORγt and allows CD4+ T cells to differentiate into Treg cells.44
Figure 1.
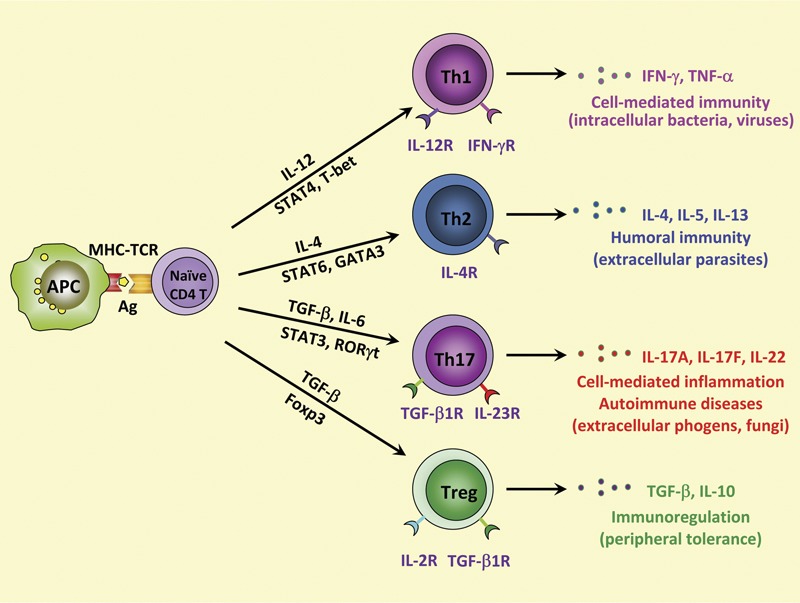
Model for Th cell differentiation from naive CD4+ T cells. In the presence of IL-12, differentiation of naive CD4+ T cells into Th1 cells requires activation of the master regulator transcription factor T-bet through STAT1 and STAT4. Th1 cells produce IFN-γ and are involved in cell-mediated immunity against intracellular bacteria and viruses. IL-4 promotes the activation of STAT6 and GATA3, which are responsible for Th2 cell differentiation. Th2 cells are important in humoral immunity against parasites, an action that is mediated through their production of IL-4, IL-5 and IL-13. The combination of TGF-β and proinflammatory cytokines, such as IL-6 and IL-23, drives the differentiation of naive CD4+ T cells into IL-17-producing Th cells (Th17) through the regulation of STAT3 and RORγt. Th17 cells play a critical role in host protection against extracellular pathogens and in inflammatory autoimmune diseases. In addition, TGF-β can induce differentiation of naive CD4+ T cells into Foxp3+ Treg cells, which produce TGF-β and IL-10 and act as modulators of immune responses. APC, antigen-presenting cell; Foxp3+, forkhead box p3+; IFN, interferon; MHC–TCR, major histocompatibility complex–T-cell receptor; ROR, retinoid-related orphan receptor; TGF, transforming growth factor; Th, T helper; Treg, regulatory T.
Th17 differentiation and maintenance in autoimmune disease
We have recently provided new evidence suggesting that the development of Th17 cells is driven by a complex dichotomous process in autoimmune disease, as characterized by an initial differentiation phase and a latter phase of survival and expansion of committed Th17 cells. Several cytokines, such as TGF-β, IL-6, IL-1β, IL-21 and IL-23, have been demonstrated to induce Th17 cell differentiation, critically contributing to the clinical outcome of autoimmune diseases.45, 46, 47, 48 IL-6 and TGF-β have been reported as the minimal requirements for murine Th17 cell differentiation from naive CD4+ T cells.48 IL-1 signaling in T cells was reported to be critically required for the early programming of Th17 cell lineage in vivo in a mouse EAE model.49 In humans, IL-1β, a proinflammatory cytokine produced predominantly by macrophages, monocytes and dendritic cells, is essential for developing Th17 cells along with IL-6, TGF-β and IL-21.50 IL-1 and IL-6 can induce IL-1 receptor expression in T cells.51 TGF-β and IL-6 also induce the expression of IL-23 receptor on differentiating Th17 cells.52 IL-23 is important to the amplification and stabilization of the Th17 phenotype.53 IL-1 can synergize with IL-6 and IL-23 to regulate Th17 cell differentiation and to maintain cytokine expression in effector Th17 cells.49 Another cytokine, IL-21, produced by Th17 cells, has been shown to provide an additional autocrine amplificatory signal.47 The cytokines IL-6, IL-21 and IL-23 all utilize the JAK–STAT pathway and activate STAT3.40, 42, 54
Our recent study revealed that IL-7 plays a crucial role in the in vivo maintenance of differentiated Th17 cells in EAE. This discovery was initially linked to a polymorphism in the IL-7 receptor (IL-7R) alpha subunit locus, which was identified as an important susceptibility factor for MS.55, 56, 57 It is known that IL-7 binds to IL-7R and activates the JAK/STAT5 and PI3K–AKT signaling pathways.58 As a T-cell growth factor, IL-7 plays a role in the regulation of peripheral homeostasis of the CD4 T-cell pool.59 Upon T-cell receptor activation of naive T cells, IL-7R is downregulated. Cua et al. recently indicated that IL-23 may play an important role in the terminal differentiation of Th17 cells, potentially through its effect on re-expression of IL-7R on Th17 cells.53 Our study shows that IL-7 is essential for the survival and expansion of pathogenic Th17 cells in EAE.60 IL-7 directly triggered expansion of effector Th17 cells in EAE and human Th17 cells in MS subjects, but it was not required for Th17 differentiation.60 IL-7R antagonism rendered differentiated Th17 cells susceptible to apoptosis through altered expression levels of the prosurvival protein Bcl-2 and the proapoptotic protein Bax, leading to decreased EAE severity.60 Our study leads to the new understanding that there are two distinct steps in the development of Th17 cells: induction/terminal differentiation and survival/expansion, in which several cytokines are involved (IL-6, IL-21, IL-23 and IL-7) and play different roles during the two phases of Th17 development (Figure 2).
Figure 2.
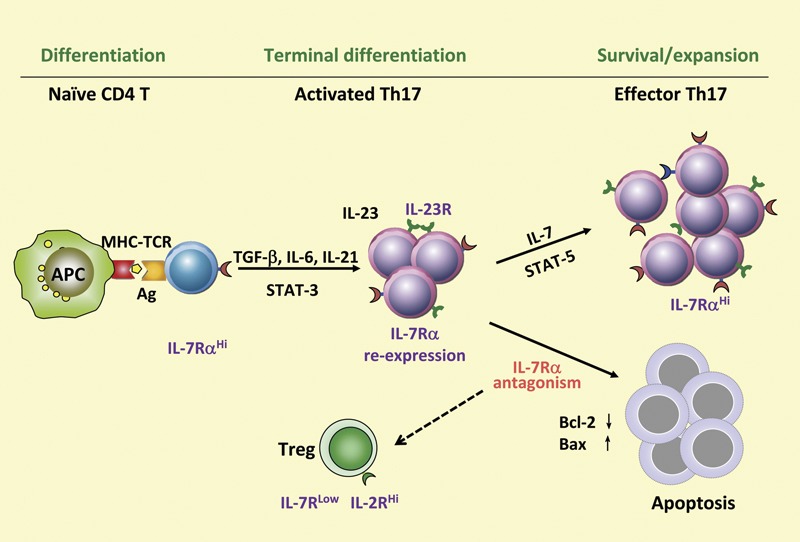
Critical role of IL-7/IL-7R signaling in survival and expansion of differentiated Th17 cells. Th17 cell development is a dichotomous process that is regulated through a complex cytokine network. The differentiation of Th17 cells is mainly mediated by STAT3 signaling through cytokines such as IL-6, IL-21 and IL-23. There is dynamic expression of IL-7R on Th17 cells in the course of T-cell activation/differentiation. In the latter phase, where IL-7R is re-expressed, IL-7 is critically required to sustain survival and expansion of differentiated Th17 cells through STAT5 signaling. IL-7R antagonism renders differentiated Th17 cells susceptible to apoptosis through altered expression of the proapoptotic protein Bax and the antiapoptotic molecule Bcl-2. APC, antigen-presenting cell; IL-7R, IL-7 receptor; MHC–TCR, major histocompatibility complex–T-cell receptor; TGF, transforming growth factor; Th, T helper.
Treg development and function in autoimmune disease
The cytokine milieu not only regulates the Th1 and Th17 response but also affects Treg induction and function. Treg cells express the transcription factor Foxp3, which is not only a lineage specification factor but also a functional marker of Treg cells.61, 62, 63 Genetic defects in the foxp3 gene, which affect the development and function of Treg cells, have been identified as being responsible for X-linked recessive inflammatory disease in Scurfy mice and immunodysregulation polyendocrinopathy enteropathy X-linked syndrome in humans.64, 65, 66, 67 Furthermore, ectopic expression of Foxp3 is sufficient to confer regulatory properties to conventional T cells.61, 68, 69
Multiple signals, such as T-cell receptor and the costimulatory molecule CD28, are required for the thymic development of Treg cells.70, 71, 72 Common γ-chain cytokines, IL-2 and, to a lesser extent, IL-7 and IL-15, are required for Foxp3 expression and Treg cell development in the thymus.73, 74, 75, 76, 77 Gene knockout of γ-chains in mice leads to complete absence of Foxp3+ Treg cells in the thymus and the peripheral immune system.78 In this process, the transcription factor STAT5, which is downstream signal of the activation of γ-chain cytokine receptors, is shown to bind to the Foxp3 promoter.79, 80
There are two types of Foxp3-expressing Treg cells: thymic-derived nTreg cells that prevent autoimmunity and post-thymic-induced iTreg cells that maintain a non-inflammatory environment in the gut, to control immune responses to environmental and food allergens and to decrease chronic inflammation. However, iTreg cells that differentiate from naive CD4+ T cells in the peripheral immune system in response to TGF-β seem less stable than nTreg cells.44, 81 The T-cell receptor repertoire of nTreg cells that are selected by high-avidity interactions in the thymus is mostly against self-antigens, whereas that of iTreg cells that are differentiated from conventional CD4+ is similar to the naive conventional CD4+ T-cell repertoire.82 Cytokines, e.g., IL-4, IFN-γ and IL-6, which induce other Th cell differentiation, mostly dampen iTreg development.83 Inhibition of TGF-β-induced Treg cells by these cytokines or by a high amount of costimulation could be suppressed by retinoic acid.84, 85, 86 CD103+ dendritic cells isolated from the small intestine and the mesenteric lymph node, which produce both TGF-β and retinoic acid, efficiently promote iTreg differentiation.87, 88
Although Treg cells comprise a specific lineage of T-cell subset, a recent study has revealed that in vivo, Foxp3 was transiently expressed in a substantial portion of cells called ‘exFoxp3' T cells.89 These ‘exFoxp3' T cells show a memory phenotype and produce proinflammatory cytokines such as IFN-γ and IL-17. Moreover, an inflammatory milieu in the context of self-antigen can induce ‘exFoxp3' T cells, which could differentiate from nTreg or iTreg cells.89 Furthermore, recent studies have indicated that Foxp3+ Treg cells are not a homogeneous population; they can/cannot co-express T-bet, IFN regulatory factor 4 or STAT3 with Foxp3 to control various subsets of effector T cells.90, 91, 92 IFN-γ induces T-bet expression in Foxp3+ Treg cells, which could promote CXCR3 expression to facilitate migration of Treg cells to Th1 inflammatory sites. T-bet is also critical for Treg cell homeostasis and function during the Th1 response.90 Similarly, Treg cells can also express IFN regulatory factor 4, which is an essential transcription factor for Th2 cell differentiation. IFN regulatory factor 4 deficiency in Treg cells resulted in selective dysregulation of Th2 response in vivo.91 Furthermore, suppression of the Th17 response by Treg cells was inhibited by specific ablation of Stat3 in Treg cells.92 Taken together, Foxp3+ Treg cells are more plastic in vivo; under inflammatory conditions, Treg cells could lose the expression of Foxp3 and become pathogenic T cells contributing to the inflammatory response or could acquire the transcriptional machinery of a particular type of CD4 effector T cells, efficiently regulating the corresponding type of immune response.
Foxp3+ Treg cells control the immune response through multiple mechanisms. Treg cells are potent suppressors of T cells. Many studies have demonstrated that Treg-mediated suppression occurs through inhibition of production of IL-2 mRNA in the responder T cells.93 Although Treg cells exert their suppressive function in a cell–cell contact manner in vitro, it should not be ruled out that Tregs could secrete soluble factor(s). IL-35, a new inhibitory cytokine, contributes to Treg function by directly acting on responder T cells.94 Another candidate factor is galectin-1, which induces cell cycle arrest of responder cells.95 One other mechanism is cytolysis of target cells; Treg cells can express granzyme A or B and kill target cells.96, 97 Furthermore, Treg cells could downregulate or inhibit upregulation of CD80/86 in APCs through CTLA-4.98 Catalytic inactivation of extracellular adenosine triphosphate by CD39 expressed by Treg cells represents a mechanism that prevents the deleterious effects of adenosine triphosphate on APC function.99, 100 Moreover, IL-10 and TGF-β have been shown to be important mediators of Treg-mediated suppression of colitis, type 1 diabetes and EAE.101, 102, 103, 104, 105
Interplay between Th17 and Treg
Distinct populations of Th17 and Treg cells coexist and are reciprocally regulated during differentiation in healthy tissue.39 Imbalances in the ratio of these lymphocytes have been implicated in a wide range of autoimmune disorders including MS and RA. Bettelli et al. have demonstrated a reciprocal developmental relationship between Foxp3+ Treg and Th17 cells; TGF-β triggers the expression of Foxp3 in naive T cells, whereas TGF-β drives Th17 differentiation from naive T cells in combination with IL-6.39 It is well established that inflammatory cytokines such as IL-6 and IL-21 inhibit TGF-β-induced Treg generation.39, 85, 106 However, blocking IL-6 signaling in vivo can generate Foxp3+ Treg cells and can reduce EAE severity.107 Furthermore, Infliximab therapy leads to the generation of CD4+CD25hiFoxp3+ Treg cells that are CD62Llow and suppress effector T cells through TGF-β and IL-10.108 Recent findings showed that Treg cells could be converted to IL-17-producing cells. Proinflammatory cytokines, such as IL-6, IL-1 and IL-21, were able to decrease the Foxp3 expression levels of committed iTreg or nTreg cells and induce IL-17 production by these cells in a TGF-β-dependent manner.81 In addition, the IL-17+/Foxp3+ T cells were functionally impaired.81 These findings indicate that the proinflammatory milieu could convert Treg cells to Th17 cells and shift the balance between immune regulation and inflammation towards inflammation.
In short, proinflammatory cytokines, e.g., IL-6, IL-1β and IL-21, not only support Th17 differentiation but also constrain Treg development by affecting the balance between RORγt and Foxp3.81 In contrast, IL-2 and retinoic acid, which support Treg differentiation, could perturb Th17 development.86, 109 Furthermore, cytokines secreted by Th17 cells, such as TNF-α and IL-21, could diminish Treg function, while IL-10 and IL-35 are able to suppress the proinflammatory response. Taken together, these findings show that the persistence of inflammation in lesion sites is significantly associated with reduced levels and impaired function of CD4+CD25+Foxp3+ Treg cells induced by proinflammatory cytokines. Targeting these cytokines, which restores the functions of Treg cells and induces new Treg cells, holds considerable potential as a treatment for autoimmune disease.
Therapeutic strategies for autoimmune diseases
The understanding of the interplay between pathogenic and regulatory T cells holds great promise in the development of effective therapeutic approaches to autoimmune diseases. Novel therapies are being designed to restore the balance of the two groups of T cells through inhibition of Th17 and promotion of Treg cells. Many companies are targeting cytokines to affect the development and function of Th17 and Treg cells. These approaches are summarized in Table 1.
Table 1. Cytokine therapeutic strategies for autoimmune diseases.
| Cytokine | Therapeutic agent (company: drug name) | Stage of clinical trial | Therapeutic outcome | Reference |
|---|---|---|---|---|
| IL-6 | Anti-IL-6 receptor (Roche: Actemra/Tocilizumab) | RA: efficacious | 110 | |
| IL-12/IL-23 | Anti-P40 (Centocor: Stelara/Ustekinumab) | MS: discontinued | MS: failed | 116 |
| Crohn's: phase IIb | PS: efficacious | |||
| Anti-P40 (Abbot: ABT-874) | Crohn's: phase II | 117, 118 | ||
| PS: phase II | ||||
| MS: phase II | ||||
| Antagonist (SYNTA: Apilimod/STA5326) | RA: phase II | 119, 120 | ||
| Crohn's: phase II | ||||
| IL-23 | Anti-P19 (Shering Plough) | Phase I | ||
| Anti-IL-23 (Eli Lilly) | Phase I | |||
| IL-1β | IL-1β antagonist (Amgen: Kineret/Anakinra) | RA: approved | 121 | |
| Anti-IL-1 (Amgen: AMG108) | RA: phase II | 121 | ||
| IL-17 | Anti-IL-17 (Eli Lilly: LY2439821) | RA: phase II | 122, 123 | |
| PS: phase II | ||||
| Anti-IL-17 (Norvatis: AIN457) | RA: phase II | 122, 123 | ||
| Crohn's: phase II | ||||
| Anti-IL-17 (Amgen) | RA: phase Ib/IIa | 122 | ||
| TNF-α | Soluble receptor (Amgen: Enbrel/etanercept) | RA: approved | 111, 124 | |
| MS: failed | ||||
| Anti-TNF-α (Centocor: Remicade/Infliximab) | Crohn's: phase III | RA: approved | 111, 125 | |
| MS: failed | ||||
| Anti-TNF-α (Abbot: Humira/Adalimumab) | Crohn's: phase III | RA: approved | 111, 126 | |
| Completed | MS: failed | |||
| IL-2 | Anti-IL-2 receptor (Biogen-Idec: Zenapax/Dacliqumab) | MS: phase II | 127 | |
| Completed | ||||
| BAFF/April | Soluble receptor (Zymogenetics/Merck: Atacicept) | MS: failed | 128 |
Abbreviations: Crohn's, Crohn's disease; MS, multiple sclerosis; PS, psoriasis; RA, rheumatoid arthritis.
In general, the various anticytokine approaches are relatively successful in the treatment of autoimmune diseases. Therapeutic agents against IL-6 and TNF-α pathways are efficacious in RA despite the fact that these drugs show side effects, such as the elevation of cholesterol levels in patients treated with anti-IL-6R110 and the development of CNS demyelination in anti-TNF-α treated RA subjects.111 An antibody against the p40 subunit of IL-12 and IL-23 (ustekinumab) is very efficacious in the treatment of psoriasis. A few companies are developing anti-IL-17 antibodies based on knowledge of the pathological role of Th17 cells. Unfortunately, MS treatment is an exception. Ustekinumab and anti-TNF-α agents all failed in MS clinical trials. The possibility that therapeutic agents against the IL-6 and IL-23 pathways that target Th17 differentiation may not work in MS has been suggested by EAE experiments where anti-IL-6 or anti-IL-23 treatments were efficacious when they were applied at the time of immunization but not at the peak of disease.41, 112 Targeting mature instead of differentiating pathogenic Th cell subsets could be a viable alternative. An antibody against the IL-2R subunit CD25 is currently in phase II trials for MS.113 Interestingly, IL-2R and IL-7R were identified as genes associated with MS susceptibility.56 These two cytokine receptors have roles in the survival and expansion of committed pathogenic Th1 and Th17 cells.60, 114 An additional advantage to the therapeutic use of an IL-7R-specific antibody is that Treg cells responsible for resolving inflammation express very low levels of IL-7R and will be unaffected.60, 115
Conclusion
It is important to maintain an appropriate balance between Treg cells and effector Th17/Th1 cells that can ensure effective immunity while avoiding pathological autoimmunity. The proinflammatory cytokine milieu produced during an immune response plays an important role in driving the imbalance between these two T-cell subsets and the outcome of inflammatory autoimmune diseases. The dynamic nature of an immune response was recently highlighted by the demonstration of interconversion between Th17 and Treg cells under the influence of a particular cytokine microenvironment. A better understanding of the molecular mechanisms regulating Treg and Th17 cells in vitro and in vivo will create opportunities for the development of therapeutic approaches, including anticytokine therapies that could be used to treat human autoimmune diseases.
References
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–R128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthay A. How do regulatory T cells work? Scand J Immunol. 2009;70:326–336. doi: 10.1111/j.1365-3083.2009.02308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol. 1988;140:1506–1510. [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, et al. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+CD25− T cells to CD4+ Tregs. J Clin Invest. 2006;116:2434–2441. doi: 10.1172/JCI25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Steinman L. A rush to judgment on Th17. J Exp Med. 2008;205:1517–1522. doi: 10.1084/jem.20072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+2− T lymphocytes. J Immunol. 1981;127:1420–1423. [PubMed] [Google Scholar]
- Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Leung S, Wang C, Tan Z, Wang J, Guo TB, et al. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nat Med. 16:191–197. doi: 10.1038/nm.2077. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Stummvoll GH, DiPaolo RJ, Huter EN, Davidson TS, Glass D, Ward JM, et al. Th1, Th2, and Th17 effector T cell-induced autoimmune gastritis differs in pathological pattern and in susceptibility to suppression by regulatory T cells. J Immunol. 2008;181:1908–1916. doi: 10.4049/jimmunol.181.3.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol. 2008;38:1814–1821. doi: 10.1002/eji.200838346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O'Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183:7602–7610. doi: 10.4049/jimmunol.0901881. [DOI] [PubMed] [Google Scholar]
- Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. 2006;180:178–184. doi: 10.1016/j.jneuroim.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. 2003;33:215–223. doi: 10.1002/immu.200390024. [DOI] [PubMed] [Google Scholar]
- Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6:R335–R346. doi: 10.1186/ar1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. 2005;140:360–367. doi: 10.1111/j.1365-2249.2005.02754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4+CD25+ regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50:2775–2785. doi: 10.1002/art.20499. [DOI] [PubMed] [Google Scholar]
- Chen X, Fang L, Song S, Guo TB, Liu A, Zhang JZ. Thymic regulation of autoimmune disease by accelerated differentiation of Foxp3+ regulatory T cells through IL-7 signaling pathway. J Immunol. 2009;183:6135–6144. doi: 10.4049/jimmunol.0901576. [DOI] [PubMed] [Google Scholar]
- Coffman RL. Origins of the TH1–TH2 model: a personal perspective. Nat Immunol. 2006;7:539–541. doi: 10.1038/ni0606-539. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Szabo SJ, Glimcher LH. Transcriptional regulation of Th1/Th2 polarization. Immunol Today. 2000;21:479–483. doi: 10.1016/s0167-5699(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180:6070–6076. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Ivanov, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- de Jong BA, Huizinga TW, Bollen EL, Uitdehaag BM, Bosma GP, van Buchem MA, et al. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126:172–179. doi: 10.1016/s0165-5728(02)00056-5. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human TH17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Shirakawa F, Tanaka Y, Ota T, Suzuki H, Eto S, Yamashita U. Expression of interleukin 1 receptors on human peripheral T cells. J Immunol. 1987;138:4243–4248. [PubMed] [Google Scholar]
- Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. . Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176:5652–5661. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, de Jager PL, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallstrom E, Khademi M, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Mahajan VS, Trajman LC, Irvine DJ, Lauffenburger DA, Chen J. Interleukin-7 receptor signaling network: an integrated systems perspective. Cell Mol Immunol. 2008;5:79–89. doi: 10.1038/cmi.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med. 2008;205:1701–1714. doi: 10.1084/jem.20071681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Leung S, Wang C, Tan Z, Wang J, Guo TB, et al. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nat Med. 2010;16:191–197. doi: 10.1038/nm.2077. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci USA. 2005;102:5126–5131. doi: 10.1073/pnas.0501701102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Bensinger SJ, Bandeira A, Jordan MS, Caton AJ, Laufer TM. Major histocompatibility complex class II-positive cortical epithelium mediates the selection of CD4+25+ immunoregulatory T cells. J Exp Med. 2001;194:427–438. doi: 10.1084/jem.194.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- Soper DM, Kasprowicz DJ, Ziegler SF. IL-2Rbeta links IL-2R signaling with Foxp3 expression. Eur J Immunol. 2007;37:1817–1826. doi: 10.1002/eji.200737101. [DOI] [PubMed] [Google Scholar]
- Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer AL, Yu A, Adeegbe D, Malek TR. Essential role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal period. J Exp Med. 2005;201:769–777. doi: 10.1084/jem.20041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer AL, Lee JY, de la Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J Immunol. 2008;181:225–234. doi: 10.4049/jimmunol.181.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. . Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced Treg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. . Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109:2058–2065. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri-Payer E, Cantor H. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4+CD25+ T cells. J Autoimmun. 2001;16:115–123. doi: 10.1006/jaut.2000.0473. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini MC, Rizzo A, Fina D, Caruso R, Becker C, Neurath MF, et al. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur J Immunol. 2007;37:3155–3163. doi: 10.1002/eji.200737766. [DOI] [PubMed] [Google Scholar]
- Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–39. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Oldfield V, Dhillon S, Plosker GL. Tocilizumab: a review of its use in the management of rheumatoid arthritis. Drugs. 2009;69:609–632. doi: 10.2165/00003495-200969050-00007. [DOI] [PubMed] [Google Scholar]
- Stubgen JP. Tumor necrosis factor-alpha antagonists and neuropathy. Muscle Nerve. 2008;37:281–292. doi: 10.1002/mus.20924. [DOI] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J, et al. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol. 2009;66:483–489. doi: 10.1001/archneurol.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- Hartigan-O'Connor DJ, Poon C, Sinclair E, McCune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods. 2007;319:41–52. doi: 10.1016/j.jim.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008;371:1675–1684. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- Menter A. The status of biologic therapies in the treatment of moderate to severe psoriasis. Cutis. 2009;84:14–24. [PubMed] [Google Scholar]
- Ding C, Xu J, Li J. ABT-874, a fully human monoclonal anti-IL-12/IL-23 antibody for the potential treatment of autoimmune diseases. Curr Opin Investig Drugs. 2008;9:515–522. [PubMed] [Google Scholar]
- Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R, et al. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn's disease Inflamm Bowel Dis 2009. in press. [DOI] [PubMed]
- Billich A. Drug evaluation: apilimod, an oral IL-12/IL-23 inhibitor for the treatment of autoimmune diseases and common variable immunodeficiency. IDrugs. 2007;10:53–59. [PubMed] [Google Scholar]
- Waugh J, Perry CM. Anakinra: a review of its use in the management of rheumatoid arthritis. BioDrugs. 2005;19:189–202. doi: 10.2165/00063030-200519030-00005. [DOI] [PubMed] [Google Scholar]
- Steinman L. Mixed results with modulation of TH-17 cells in human autoimmune diseases. Nat Immunol; 11:41–44. doi: 10.1038/ni.1803. [DOI] [PubMed] [Google Scholar]
- van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–553. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- Dhillon S, Lyseng-Williamson KA, Scott LJ. Etanercept: a review of its use in the management of rheumatoid arthritis. Drugs. 2007;67:1211–1241. doi: 10.2165/00003495-200767080-00011. [DOI] [PubMed] [Google Scholar]
- Maini RN, Breedveld FC, Kalden JR, Smolen JS, Furst D, Weisman MH, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50:1051–1065. doi: 10.1002/art.20159. [DOI] [PubMed] [Google Scholar]
- Burmester GR, Mariette X, Montecucco C, Monteagudo-Saez I, Malaise M, Tzioufas AG, et al. Adalimumab alone and in combination with disease-modifying antirheumatic drugs for the treatment of rheumatoid arthritis in clinical practice: the Research in Active Rheumatoid Arthritis (ReAct) trial. Ann Rheum Dis. 2007;66:732–739. doi: 10.1136/ard.2006.066761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. Humanized anti-CD25 antibody treatment with daclizumab in multiple sclerosis. Neurodegener Dis. 2008;5:23–26. doi: 10.1159/000109934. [DOI] [PubMed] [Google Scholar]
- Gatto B. Atacicept, a homodimeric fusion protein for the potential treatment of diseases triggered by plasma cells. Curr Opin Investig Drugs. 2008;9:1216–1227. [PubMed] [Google Scholar]