

Abstract
Non-structural protein 1 (NS1) is an important virulence factor of the highly pathogenic H5N1 avian influenza virus. A five-amino-acid (5 aa) deletion at position 80–84 and an aspartic acid to glutamic acid substitution at position 92 (D92E) are two major NS1 mutations that are highly correlated with enhanced virulence. To investigate the effect of these mutations in H5N1 virulence, three H5N1-NS1 variants were constructed: NS51 (lacking 5 aa at position 80–84), NS51(I) (carrying a 5-aa insertion at position 80–84) and NS51(IM) (carrying both the 5-aa insertion and the D92E mutation). We examined the effects of these mutations on interferon (IFN) induction, tumor-necrosis factor (TNF)α response, p53 activity and apoptosis. We found that the D92E mutation eliminated NS1's repressive effect on IFN induction, while the 5-aa deletion resulted in enhanced resistance to TNFα responses. We also observed that all three variants exhibited a similar suppressive effect on p53 transcriptional activity, although none of them significantly influenced apoptosis of host cells. Our findings shed new light on the role of NS1 in the pathogenicity of H5N1 virus.
Keywords: H5N1 influenza virus, interferon, NS1 variants, p53, TNFα
Introduction
Non-structural protein 1 (NS1) is a virulence factor of the influenza virus and plays multiple roles in thwarting innate and adaptive immune responses and promoting the effective replication of the virus. NS1 primarily exerts its biological activity via the following strategies: (i) inhibition of cellular pre-mRNA splicing and polyadenylation; (ii) enhancement of nuclear-export machinery and efficiency of viral mRNA translation; (iii) activation of the PI3K/Akt signaling pathway; (iv) repression of antiviral immune response; and (v) involvement in apoptosis of host cells.1
The NS1 proteins of most influenza A virus strains have an average length of 230 amino acids (aa). Nevertheless, spontaneous deletion of amino-acid residues in NS1 protein is common in many influenza A strains, especially the H5N1 subtype. For example, the deletion of five residues at position 80–84 has been reported in several studies.2, 3, 4, 5 This deletion is not found in H5N1 strains isolated prior to 1999, but has been detected in the majority of H5N1 isolates since 2000. Other NS1 mutations, such as a deletion at position 191–1956 and a C-terminal truncation at position 221–230,7, 8 are also common. The biological significance of these deletions is not fully understood; however, recent findings have linked them with the pathogenicity or transmissibility of influenza A virus. For instance, a five-residue deletion at position 191–195 of NS1 is reportedly involved in the attenuation of this virus strain,6 while a deletion at position 80–84 within NS1 has been implicated in enhancing the virulence of H5N1 in chickens and mice.9
Other common variations of NS1 are amino-acid substitutions at particular sites (e.g., S42P, S42G, D92E, L103F, I106M and A149V), some of which are responsible for alterations in virus pathogenicity.10, 11, 12, 13, 14 Among them, the D92E substitution has drawn a great deal of attention due to its association with a potent resistance to the antiviral effects of cytokines and a high fatality rate.13, 15
Strain-specific amino-acid sequence diversity of NS1 is also an important factor that affects its function, as well as the overall virulence of the virus. For example, NS1 proteins from the A/VN/1203/04 (H5N1) and A/Hongkong/483/97(H5N1) strains exhibit differential interactions with the host cell CPSF30 (30KD subunit of cleavage and polyadenylation specificity factor).14 In addition, the NS1 proteins of the A/Brevig Mission/1/18(H1N1) and A/Mallard/Netherlands/12/2000(H7N3) strains, but not the A/Udorn/72(H3N2) strain, can interact with cellular Crk/CrkL (v-crk homolog-like).16
Therefore, different NS1 proteins may possess different biological properties and exert varying influences on host cells. In the present study, we attempted to elucidate the effects of NS1 on the virulence of influenza A virus by examining host responses (cytokine response, p53 activity and apoptosis) in the presence of overexpressed NS1 variants (with/without deletion at position 80–84 or alteration at position 92).
Materials and methods
Cell lines, viruses and reagents
Madin–Darby canine kidney (MDCK) cells, human lung carcinoma cells (A549), human cervix epithelial cells (Hela), human embryonic kidney cells (293T) and a human osteosarcoma cell line (Saos-2) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with antibiotics and 10% fetal calf serum. Influenza A virus A/Chicken /GD/1/05(H5N1) was passaged in MDCK cells.
QIAamp viral RNA mini kits were purchased from Qiagen (Hilden, Germany). DNase I, AMV reverse transcriptase and PrimeSTAR HS DNA polymerase were from TaKaRa (Dalian, China). Lipofectamine 2000, Trizol reagent and SYBR green I were from Invitrogen (Carlsbad, CA, USA). Poly I∶C was from Sigma (St Louis, MO, USA). interferon (IFN)α-2b and tumor-necrosis factor (TNF)α were purchased from Shanghai Genomics (Shanghai, China). Dual-luciferase reporter assay kits were from Promega (Madison, WI, USA). Human annexin V-FITC kits were from Bender Medsystems (Burlingame, CA, USA) and cell death detection ELISAPLUS kits were from Roche Diagnostics (Mannheim, Germany). Rabbit anti-p53 antibody and phospho-p53 antibody sampler kits were purchased from Cell Signaling Technology (Danvers, MA, USA), rabbit anti-Flag antibody, mouse anti-Flag antibody, mouse anti-β-actin antibody and peroxidase-conjugated goat anti-mouse antibody were purchased from Sigma (St Louis, MO, USA), Cy3-conjugated goat anti-rabbit antibody and peroxidase-conjugated goat anti-rabbit antibody were from Beyotime Biotechnology (Haimen, China) and Alexa Fluor 488-labeled goat anti-mouse antibody was from Molecular Probes (Eugene, OR, USA).
Construction of recombinant plasmids
To construct the PNF-NS51 plasmid, viral RNA from A/Chicken/GD/1/05(H5N1) was extracted, and the NS1 coding sequence was amplified by reverse transcription (RT)-PCR using the following primers: NS51-S (5′-TATGGATCCATGGATTCCAACACT GTG-3′) and NS51-A (5′-GACGGA TCCTCAAACTTTTGACTCAATTG-3′). PCR products were digested with BamHI and cloned into PNF vector (a modified pcDNA3 vector with N-terminal Flag tag).
Plasmid PNF-NS51(I), encoding a H5N1-NS1 protein with an insertion of five residues immediately next to position 79, was generated as follows. First, the N-terminal coding sequence of NS1 was amplified from the PNF-NS51 plasmid using the upstream NS51-S primer and the downstream NS51-IR primer: (5′-AGCTGGCACTGAAGCGATAGTCATTTTAAGTGCCTCATC-3′). Second, the C-terminal sequence of NS1 was amplified by PCR using primers NS51-IF (5′-AAA ATGACTATCGCTTCAGTGCCAGCTTCACGCTACCTA-3′) and NS51-A based on the PNF-NS51 plasmid. Third, the PCR products of the above N- and C-terminal coding sequences of NS1 were annealed and extended for 10 cycles followed by the addition of the primer sets NS51-S and NS51-A for an additional 30 cycles of amplification. Finally, the PCR product was cloned into the PNF vector's BamHI site to generate plasmid PNF-NS51(I).
To produce the PNF-NS51(IM) plasmid, a T to A point mutation was introduced into plasmid PNF-NS51(I) using the primers NS51-MF (5′-CGCTACCTAACTGAA ATGACTCTCGAGG-3′) and NS51-MR (5′-CCTCGAGAGTCATTTCAGTTAGGT AGCG-3′) following the protocol of the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA).
For the reporter assays, p21 and MDM2 gene promoter regions were amplified from human genomic DNA using two pairs of primers: p21F (5′-CAGAGATCTTCCCTCC ATCCCTATGCT-3′) and p21R (5′-GCTAAGCTTCTTCGGCAGCTGCTCACA-3′) for p21; MDMF (5′-GTCAGATCT AAAACCCCGGATGGTGAG-3′) and MDMR (5′-CGCAAGCTTTCTTGTTCCGAAGCTGGA-3′) for MDM2. P21-Luc and MDM-Luc reporter plasmids were subsequently generated by fusing the p21 and MDM2 gene promoters into the pGL3-basic vector using BglII and HindIII.
To construct the pcDNA3-p53 plasmid, a human p53 encoding sequence was generated by RT-PCR using primers p53F (5′-ATTGGATCCATGGAGGAGCCGCAGTCA-3′) and p53R (5′-CGAGAATTCTCAGTCTGAGTCAGGCCC-3′) and ligated into pcDNA3 via BamHI and EcoRI.
The human IFNβ promoter was cloned using the following primer pair: IFNB-F (5′-AGTAGATCTGAATTCTCAGGTCGTTTG-3′) and IFNB-R (5′-CTTAAGCTTAAA GGTTGCAGTTAGAATG-3′) and was subsequently inserted via BglII/HindIII into the pGL3-basic vector to create the pIFNβ-luc plasmid. pISRE-luc and pNF-κB-luc were purchased from Beyotime Biotechnology.
Immunofluorescence microscopy
Immunofluorescence analysis was performed as described previously.17 Briefly, Hela or A549 cells grown on glass coverslips were transfected with recombinant plasmids. After 24 h, cell monolayers were washed once with phosphate-buffered saline (PBS) and fixed for 10 min with 4% paraformaldehyde at room temperature. The cells were then permeabilized with 0.2% Triton X-100 for 10 min and blocked for 30 min in PBS containing 5% bovine serum albumin (BSA), followed by incubation with rabbit anti-Flag antibody (1∶700) alone or with a mixture of mouse anti-Flag antibody (1∶700) and rabbit anti-p53 (1∶600) antibody for 2 h. After three washes with PBS, the coverslips were incubated with Cy3-conjugated goat anti-rabbit antibody or reacted with Alexa Fluor 488-labeled goat anti-mouse antibody (1∶300) and Cy3-conjugated goat anti-rabbit antibody (1∶250) for 1 h and then stained with Hoechst 33258 for 5 min. Finally, the coverslips were washed three times with PBS and observed with an Olympus IX70 fluorescence microscope.
Reporter assays
Confluent Hela or Saos-2 cells in 24-well plates were transfected with different reporter plasmids (pIFNβ-luc, pISRE-luc, pNF-κB-luc, P21-Luc or MDM-Luc) and H5N1-NS1 effector plasmids along with an internal control plasmid, pRL-CMV, in the presence or absence of poly I∶C, IFNα-2b, TNFα, or overexpressed p53. In addition, pcDNA3 was supplemented to normalize the total amount of DNA in each transfection. After 36 h of transfection, the cells were washed once with PBS, lysed with 100 µl passive lysis buffer (Promega), and the luciferase activity was measured using the Dual-Luciferase Assay System (Promega) and a TD-20/20 luminometer (Turner Biosystems, Sunnyvale, CA, USA) according to the manufacturer's instructions. All experiments were repeated independently, and the results are expressed as the average values of firefly luciferase activity normalized to Renilla luciferase activity.
RT-PCR
Hela cells were transfected with plasmids encoding different NS1 variants or an empty vector for 24 h and subjected to further transfection with poly I∶C for 6 h, followed by a 6 h incubation with Dulbecco's modified Eagle's medium containing 40 µg/ml of poly I∶C. Total RNA was isolated using Trizol reagent (Invitrogen) according to the manufacturer's instructions and treated with DNase I for 1 h. RT-PCR was performed using the indicated primer pairs for human IFNβ (IFNB-S: 5′-GACGCCGCATTGACCATCTA-3′ and IFNB-A: 5′-AGTTCCTTAGGATTTCC ACTCT-3′) or β-actin (Actin-S: 5′-CCAAGGCCAACCGCGAGAAGATGAC-3′ and Actin-A: 5′-AGGGTACATGGTGGTGCCGCCAGAC-3′). RT-PCR products were separated by 2% agarose electrophoresis, stained with SYBR green I and visualized under ultraviolet light (UV). The relative abundance of IFNβ mRNA was calculated after normalization with β-actin.
Western blotting
293T cells (80% confluence) were transfected with plasmids expressing NS1 variants or an empty vector for 24 h. Laemmli sample buffer was added, and samples were collected and boiled for 5 min. After a brief sonication, the lysate was subjected to 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The membrane was blocked for 2 h in Tris-buffered saline with Tween 20 (TBST) containing 5% non-fat milk and incubated overnight at 4 °C with specific antibodies against total p53 and phosphorylated p53 at different sites (Ser6, Ser9, Ser15, Ser20, Ser37, Ser46 or Ser392). β-actin antibody (1∶3000) was also used as a control. After three washes with PBS, membranes were exposed to peroxidase-conjugated secondary antibody (1∶3000) for 2 h. Immunoblots were developed using the enhanced chemiluminescence substrate ECL (Pierce, Rockford, IL, USA) or diaminobenzidine (DAB).
Cell fractionation study
Transfected 293T cells were washed with PBS and suspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml of leupeptin and 2 µg/ml aprotinin). After 15 min incubation on ice, 1% NP40 was added and the samples were vortexed for 10 s. The lysate was centrifuged at low-speed (1300g, 5 min, 4 °C), and the supernatant (cytoplasmic fraction) was collected and further centrifuged at high-speed (20 000g, 15 min, 4 °C) to remove insoluble aggregates. Nuclei in the pellet were washed once in buffer A and lysed for 30 min in buffer B (20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, 25% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml leupeptin and 2 µg/ml aprotinin). Soluble components (nuclear fraction) were collected by centrifugation (12 000g, 5 min, 4 °C). The resulting cytoplasmic and nuclear fractions were mixed with an equal volume of 2× SDS sample buffer, boiled for 10 min, and briefly sonicated for 30 s. The sample was finally subjected to SDS–polyacrylamide gel electrophoresis followed by western blot analysis using p53 antibody, as described earlier (DAB).
Annexin V/propidium iodide (PI) double staining
Hela cells were transfected with different H5N1-NS1-expressing plasmids. Two days after transfection, the cells were harvested, washed once with PBS, and incubated with FITC-conjugated annexin V and PI in binding buffer for 10 min at room temperature in the dark. Cells were further analyzed by flow cytometry immediately following incubation.
Analysis of apoptosis by ELISA
The extent of apoptosis induced by different H5N1-NS1 variants at 48 h post-transfection was assessed in Hela cells using a Cell Death Detection ELISAPLUS kit (Roche Diagnostics) as per the manufacturer's instructions. This kit allows the quantitative measurement of mono- and oligonucleosomes released into the cytoplasm from apoptotic cells utilizing two different antibodies against DNA and histones. Briefly, the cell lysate (20 µl/well) was placed into a streptavidin-coated microplate, and 80 µl of the immunoreagent containing a mixture of anti-histone-biotin and anti-DNA-POD antibody was added. After incubation for 2 h at 25 °C with gentle shaking at 300 r.p.m., the unbound antibodies were washed away with incubation buffer and 2,2'-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS), the substrate of POD, was added. The absorption values were measured at 405 nm with a reference wavelength of 490 nm. The relative nucleosome abundance is expressed as a ratio of A405 to A490.
Results
H5N1-NS1 variants and their subcellular localization
We constructed three H5N1-NS1 recombinant plasmids: PNF-NS51 carrying a coding sequence for H5N1-NS1 with a natural deletion of 5 aa at position 80–84; PNF-NS51(I) carrying a coding sequence for H5N1-NS1 with the missing 5 aa inserted; PNF-NS51(IM) carrying a coding sequence for H5N1-NS1 with both a 5-aa insert and a D92E point mutation. All constructs contained an artificially introduced Flag tag at the N terminus of NS1 (Figure 1a).
Figure 1.
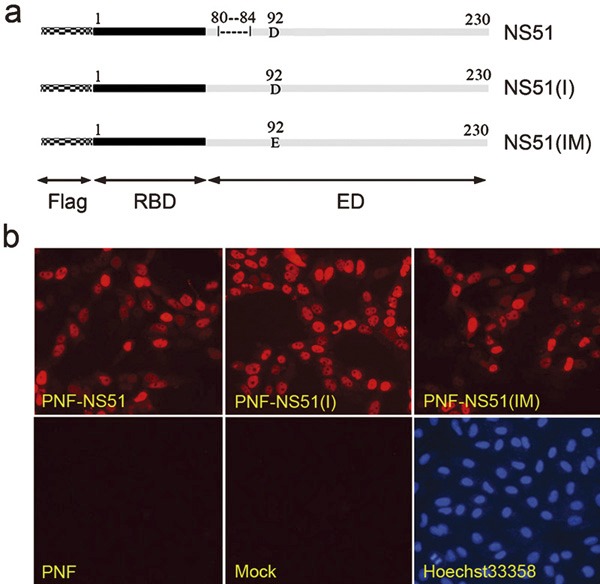
Three H5N1-NS1 variants and their subcellular localization. (a) Schematic diagram of H5N1-NS1 variants. The mosaic bars and Flag represent the Flag tag at the N terminus of NS1. The solid black bars and RBD indicate the RNA-binding domain mapped between residues 1 and 73. The solid gray bars and ED indicate the effector domain at 74–230 residues. NS51: H5N1-NS1 variant with a 5-aa deletion at position 80–84; NS51(I): H5N1-NS1 variant with a 5-aa insertion at the corresponding position; NS51(IM): H5N1-NS1 variant carrying both the 5-aa insertion and a D92E mutation. (b) Intracellular distribution of H5N1-NS1 variants in Hela cells. Cells were transfected with indicated plasmids or empty PNF vector. At 24 h post-transfection, cells were fixed, permeabilized and stained with rabbit anti-Flag antibody and Cy3-conjugated goat anti-rabbit secondary antibody followed by nuclear staining with Hoechst 33258. aa, amino acid; NS1, non-structural protein 1.
NS1 is known to preferentially localize in the nuclei of infected cells. As shown in Figure 1b, all H5N1-NS1 variants were distributed mainly within the nuclei and minimally in the cytoplasm of transfected cells. An N-terminal Flag tag in all three H5N1-NS1 constructs had no obvious influence on NS1 localization. In addition, the similar localization patterns of all three variants indicated that the two NS1 mutations did not interfere with the normal distribution of the H5N1-NS1 protein.
Differential influence of H5N1-NS1 variants on IFN production
The potent ability of NS1 to antagonize the IFN system has proven to be an important factor contributing to elevated replication and virulence of influenza A virus. To identify the potential impact of a 5-aa deletion and D92E mutation on the induction and action of IFN, two reporter constructs, pIFNβ-luc and pISRE-luc, were selected for subsequent reporter assays in the presence or absence of different H5N1-NS1 variants. In empty vector-treated Hela cells, the transcriptional activity of the IFNβ promoter following poly I∶C stimulation was about four times higher than in untreated controls. However, when different H5N1-NS1 variants were introduced into cells individually, the relative luciferase activity induced by IFN-β promoter exhibited apparent variation. Expression of NS51 and NS51(I) showed comparable repressive effects on IFNβ promoter activity induced by poly I∶C (approximately 56 and 63% decrease relative to the control) (Figure 2a). However, expression of NS51(IM) failed to inhibit poly I∶C-stimulated transcription of the IFNβ reporter gene. Indeed, NS51(IM) treatment slightly enhanced reporter activity (Figure 2a). Tests on the ISRE-luc construct showed that all three H5N1-NS1 variants had no significant effect on IFN-responsive promoter activity induced by IFNα-2b (Figure 2b). To further confirm the effect of H5N1-NS1 variants on IFN induction, we performed semiquantitative RT-PCR experiments to determine the level of IFNβ and β-actin mRNA expression in Hela cells following exogenous expression of NS1 variants. Consistent with the results of our reporter assays, NS51 and NS51(I) downregulated the transcription of IFNβ to a similar degree, while NS51(IM) did not substantially affect the mRNA level of IFNβ (Figure 2c).
Figure 2.
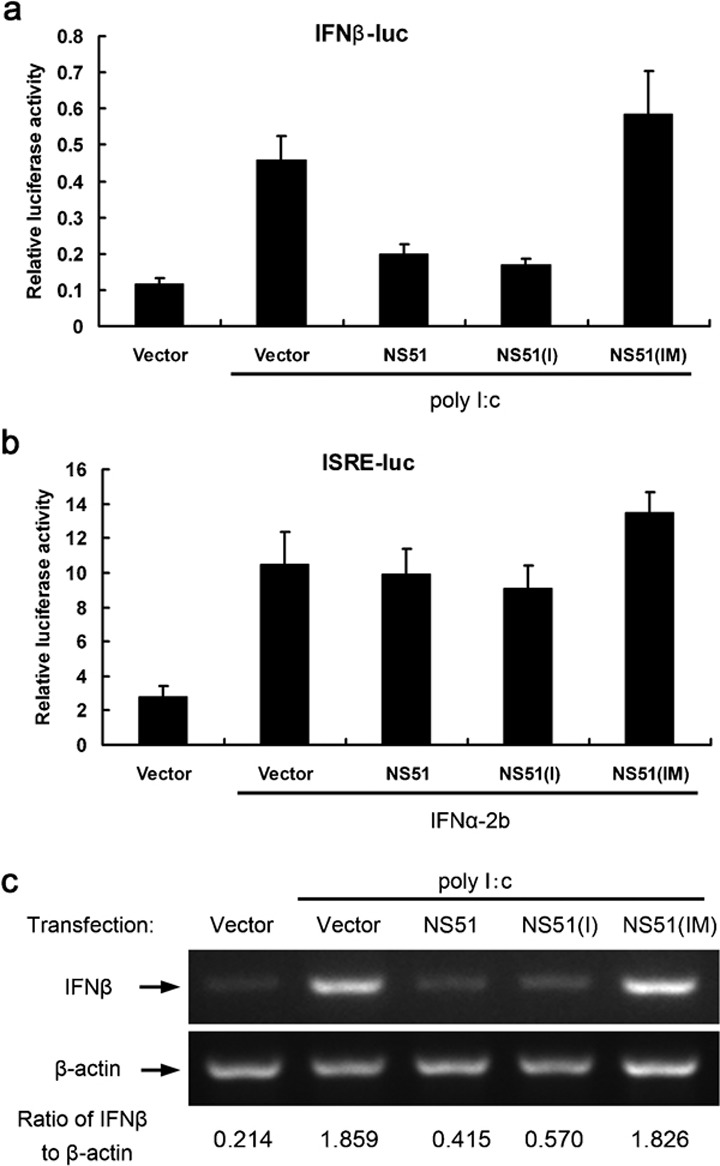
Effects of H5N1-NS1 variants on the production and action of IFN. The IFNβ promoter- (a) or ISRE promoter-containing reporter construct (b) and pRL-CMV plasmids were transfected into Hela cells along with the indicated NS1 expression vectors for 24 h with or without subsequent stimulation with poly I∶C (a) or IFNα-2b (b) for 12 h. Relative luciferase activities were plotted as means±SD from three independent experiments. (c) Influence of H5N1-NS1 variants on IFNβ mRNA levels. Hela cells were transfected with the indicated plasmids for 24 h with or without 12 h poly I∶C stimulation. Total RNA was extracted and subjected to semi-quantitative RT-PCR analysis using specific primer sets for IFNβ and β-actin. The ratio of IFNβ to β-actin is shown at the bottom of the graph following quantification with the Quantity One program. IFN, interferon; NS1, non-structural protein 1.
H5N1-NS1 variant-mediated inhibition of TNFα-mediated activation of NF-κB promoter activity
NS1 has been shown to circumvent the action of TNFα, which is a powerful suppressor of influenza A replication.13, 15 TNFα can activate NF-κB, which is a key target of the TNFα signaling transduction pathway. To address whether three H5N1-NS1 variants can act as TNFα antagonists, an NF-κB promoter-containing plasmid was used in reporter assays. All H5N1-NS1 variants inhibited NF-κB promoter activity in response to TNFα (Figure 3). Compared to the empty vector, relative luciferase activity was reduced by 74.6, 53.3 and 55.2% upon the expression of NS51, NS51(I) and NS51(IM), respectively. The suppressive effect of NS51 was about 40% higher than that of NS51(I) and 35.2% higher than that of NS51(IM) .
Figure 3.
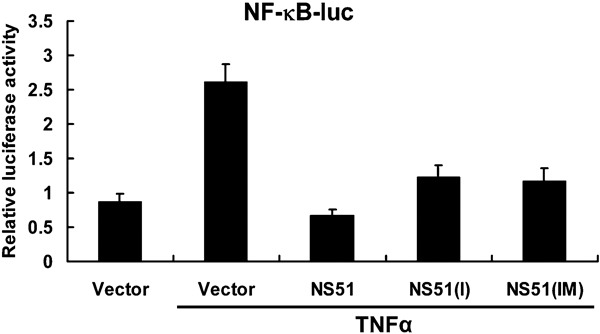
Suppressive effects of H5N1-NS1 variants on TNFα-mediated NF-κB promoter activity. Hela cells were transfected with pNF-κB-luc, pRL-CMV and NS1 variant constructs. After 30 h, cells were incubated with TNFα for 6 h and luciferase activity was measured. The data are presented as means±SD of three independent experiments. NS1, non-structural protein 1; NF-κB, nuclear factor-kappaB; TNF, tumor-necrosis factor.
H5N1-NS1 variants did not alter the phosphorylation status and intracellular distribution pattern of p53
p53 has been recently shown to be able to defend against a subset of viruses, including influenza A virus.18 The expression level and activity of p53 can be substantially increased by treatment with IFN or TNFα.19, 20, 21 Based on the observed effects of H5N1-NS1 variants on IFN production and TNFα action, we investigated the influence of H5N1-NS1 variants on p53 activity. Since the activation of p53 is closely associated with its phosphorylation status and nuclear accumulation, we first examined the effects of H5N1-NS1 variants on the phosphorylation state of p53 at seven serine sites (Ser6, 9, 15, 20, 37, 46 and 392) and found no significant changes (Figure 4a). Moreover, the total expression level of p53 was constant in both NS1-transfected cells and controls (Figure 4a).
Figure 4.
Phosphorylation and intracellular distribution of p53 following expression of H5N1-NS1 variants. (a) Western blot analysis of the phosphorylation state of p53 at seven serine sites. 293T cells were transfected with NS1-coding plasmids or empty vector. After 24 h, cellular lysates were subjected to western blot analysis using specific antibodies as described in ‘Material and methods'. (b) Cellular fractionation assay for the cytoplasmic and nuclear localization pattern of p53 in 293T cells. C, cytoplasm; N, nucleus. (c) Immunofluorescent detection of p53 within A549 cells transfected with H5N1-NS1 variants. Localization of NS1 protein (green), p53 (red), overlapped images of NS1 and p53 (yellow), and nuclei (blue) stained with Hoechst 33258 were viewed using an Olympus IX70 fluorescence microscope. The percentage of bright red nuclei (p53-positive nuclei) in 10 random fields relative to the overall number of nuclei in the same fields was calculated. NS1, non-structural protein 1.
Next, we performed cell fractionation analysis and immunofluorescence staining to investigate the influence of different H5N1-NS1 variants on the intracellular distribution of p53. As shown in Figure 4b, there was no significant difference in the abundance of p53 within the cytoplasm or nucleus in 293T cells, including empty vector-transfected cells. In addition, although colocalization of each H5N1-NS1 variant and endogenous p53 in nuclei was observed in a proportion of A549 cells (Figure 4c, yellow arrows), the existence of H5N1-NS1 variants in other A549 cells did not lead to marked nuclear accumulation of p53 (Figure 4c, white arrows). Meanwhile, the percentage of p53-positive nuclei among all experimental groups, including controls, remained almost unchanged. These results demonstrated that all three H5N1-NS1 variants did not affect the normal distribution of p53 in host cells regardless of the cell types used.
H5N1-NS1 variants reduced the transcriptional activity of p53
Based on the partial colocalization of H5N1-NS1 variants and p53 observed in our immunofluorescence experiments, we speculated that H5N1-NS1 variants may influence the function of p53. Active p53 is linked with a variety of physical or pathological processes, such as DNA repair, cell-cycle arrest, apoptosis and viral resistance. Most of these functions are associated with transcriptional activity of p53. Therefore, reporter assays were carried out to determine the effect of H5N1-NS1 variants on p53 transcriptional activity. Saos-2, a p53-defective cell line, and two reporter plasmids, p21-luc and MDM-luc with respective p21 and MDM2 promoters, were selected because p21 and MDM2 are two biological p53 target genes.
When the pcDNA3-p53 plasmid was co-transfected with the reporter plasmids, the relative luciferase activities of p21-luc and MDM2-luc increased approximately 35-fold and 265-fold respectively (Figure 5a and b). Nevertheless, coexpression of NS51, NS51(I) or NS51(IM) with p53 reduced p21-luc activity to 41.4, 35.7 and 44.8% of that of the vector-transfected group (Figure 5a). A similar inhibitory effect was also observed in the MDM-luc reporter assay, albeit to a lesser extent. The relative values of MDM-luc activity accounted for 69.0, 70.2 and 78.1% of the control group (Figure 5b).
Figure 5.
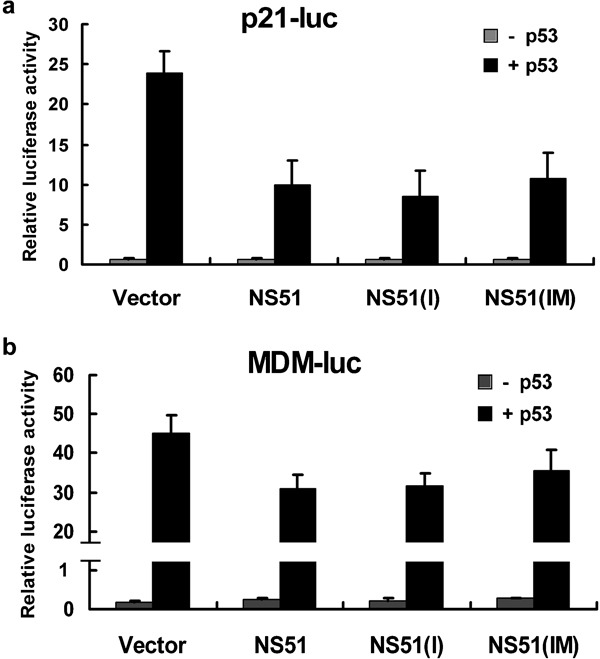
Effects of H5N1-NS1 variants on p53 transcriptional activity. Saos-2 cells were transfected with p21-luc (a) or MDM-luc (b), pRL-CMV and NS1 plasmids with or without pcDNA3-p53. At 36 h post-transfection, cells were harvested and analyzed for luciferase expression. Relative activities are shown as means±SD from three independent experiments. NS1: non-structural protein 1.
H5N1-NS1 variants had no significant influence on the apoptosis of Hela cells
Given that the relationship between influenza A virus, NS1 and apoptosis has been described in a number of studies,22, 23, 24 we compared the effects of different H5N1-NS1 variants on the apoptotic response of Hela cells. As seen in Figure 6a, although the percentage of both early-apoptotic cells (which possess a single-positive staining pattern detected by FITC-labeled annexin V) and late-apoptotic/necrotic cells (annexin V/PI double-positive staining pattern) were mildly elevated upon expression H5N1-NS1 variants relative to the controls, release of mono- or oligonucleosomes from apoptotic cells was unchanged (Figure 6b).
Figure 6.
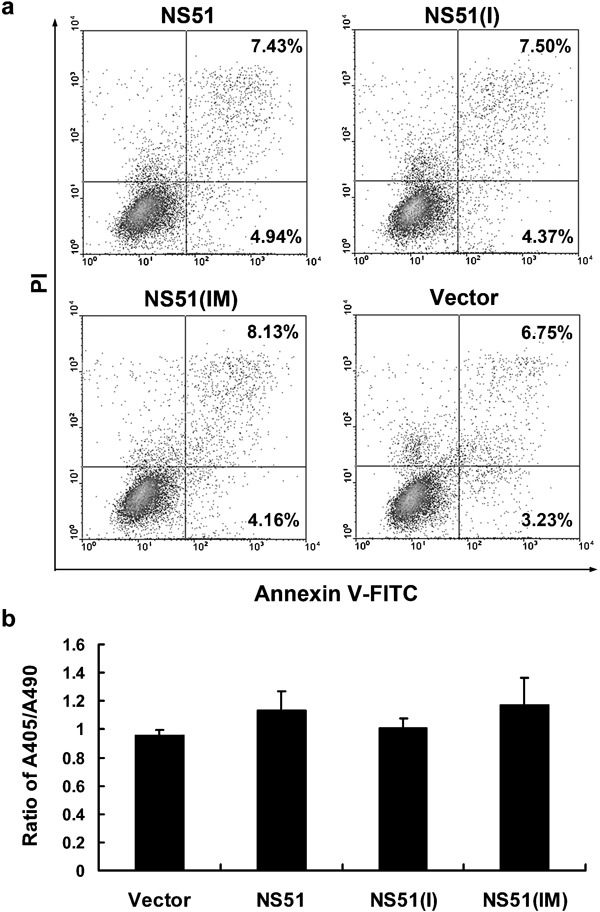
Effect of H5N1-NS1 variants on apoptosis. Hela cells were transfected with the indicated plasmids. After 48 h, double-staining was performed using annexin V-FITC/PI (a). Oligonucleosomal DNA fragmentation was measured by ELISA and expressed as the ratio of A405/A490 (b). The bars represent means±SD from three experiments. NS1, non-structural protein 1; PI, propidium iodide.
Discussion
The NS1 protein of the highly pathogenic H5N1 virus has undergone continuous evolution in birds and some mammals since the virus was isolated from fatal cases in Hong Kong in 1997. One important determinant contributing to the high virulence of H5N1/1997 is a D92E mutation in the NS1 protein.13, 15 Surprisingly, the D92E mutation rate in NS1 of H5N1 virus has decreased gradually since 1997. In 2000, a new strain of H5N1 virus with a 5-aa deletion at position 80–84 of NS1 emerged;2, 3 this deletion can be observed in almost all H5N1 influenza virus strains isolated during 2001–2009.
To evaluate the potential influence of NS1 variants on the pathogenicity of H5N1 virus, three H5N1-NS1 variants with diverse amino-acid compositions at position 80–84 or 92, named NS51, NS51(I) and NS51(IM), were constructed. All H5N1-NS1 variants exhibited similar nuclear distribution patterns, implying that the mutation at position 80–84 or 92 did not disturb the normal localization of NS1.
The capacity of NS1 protein to antagonize cytokine responses (especially to IFN and TNFα) has long been considered a major factor promoting effective viral replication. NS1 may exert its antagonistic effects on the IFN system through three primary strategies: limiting IFN production, blocking IFN-induced gene expression or suppression of the effector molecule involved in the IFN signaling pathway (e.g., PKR).1 Examination of the effects of three H5N1-NS1 variants on the induction and action of IFN demonstrated that NS51 and NS51(I) significantly downregulated IFNβ production (Figure 2a and c), despite the fact that all H5N1-NS1 variants failed to affect IFN-induced reporter gene expression governed by an ISRE-containing promoter (Figure 2b). Additionally, one study reported that PR8/NS1 did not reduce the IFN-induced gene expression from ISRE but efficiently hindered IFNβ induction, corroborating our results.25 The above data suggest that the IFN resistance effect induced by NS51 and NS51(I) variants is primarily mediated by inhibition of IFN production rather than by interference with the IFN signaling pathway.
The presence of glutamic acid at position 92 (E92) in NS1 has been shown to be essential for countering antiviral cytokine responses.13, 15 However, the exact mechanism responsible for the enhanced virulence conferred by this mutation has not been fully established. Recently, Lipatov and colleagues reported that a reassortant virus harboring a D92E mutation in its H5N1/NS1 gene provokes significantly higher levels of inflammatory cytokines (including IFN) than viruses whose NS1 protein contains D92.4 Consistent with their findings, our data indicated that the D92E mutation entirely abrogated the repressive effect of the NS51(I) variant on IFNβ production. IFNβ is capable of promoting expression of a variety of cytokines and chemokines, including IL10, RANTES(CCL5), MCP-1(CCL2), MCP-3(CCL7), MCP-2(CCL8), MIG(CXCL9) and IP-10(CXCL10).26, 27, 28 Because an elevated expression of cytokines and chemokines has been proposed to contribute to the unusual severity and mortality of H5N1 infection,29, 30 our results provide evidence in support of the hypothesis that the high pathogenicity of H5N1 virus may be associated with a reduced repression on IFN production conferred by the D92E mutation.
Recently, Long and co-workers reported that a 5-aa deletion at position 80–84 of NS1 contributes to increased virulence of H5N1 in chickens and mice.9 Moreover, crystallographic analysis of the NS1 protein from a highly pathogenic H5N1 virus (A/Vietnam/1203/2004) revealed that this 5-aa deletion results in a more pronounced twisting of the effector domain of NS1.7 However, the mechanism by which this deletion contributes to H5N1 virulence remains unknown. Thus, we examined the effects of NS51 and another two variants (NS51(I) and NS51(IM)) on NF-κB activity. NS51 exhibited a more powerful repressive effect on TNFα-induced NF-κB promoter activity than NS51(I) and NS51(IM) (Figure 3). Because TNFα can exert potent antiviral functions during influenza A infection,31 the enhanced resistance to TNFα conferred by the 5-aa deletion could be partly responsible for the increased pathogenicity of the H5N1 virus.
In a previous study, p53 has been demonstrated to be an important mediator of host defense against influenza A.18 Additionally, the expression level, phosphorylation state, nuclear accumulation and transcriptional activity of p53 are substantially increased during influenza A infection.18, 32 Together with observations by Zhirnov, who described decreased serine phosphorylation (Ser15 and Ser46) and nuclear localization of p53 upon delNS1 virus infection,33 it appears that both influenza A infection and the NS1 protein are correlated with the regulation of p53 activity. Activation of p53 occurs primarily through post-transcriptional mechanisms, such as protein stabilization by phosphorylation, increased nuclear localization and enhanced DNA-binding via conformational change. In our study, neither the phosphorylation level nor the nuclear accumulation of p53 was altered in cells transfected with H5N1-NS1 variants (Figure 4). However, we observed partial colocalization of endogenous p53 and H5N1-NS1 variants (Figure 4c). Furthermore, we found that p53-induced activities of p21-luc and MDM-luc were reduced to similar levels by three H5N1-NS1 variants (Figure 5). The above results suggest an inherent association of NS1 and p53, although the exact relationship between these two proteins needs to be further examined. The discrepancies between our results and Zhirnov's observation may be attributed to the specific experimental systems used: we detected the influence of NS1 on p53 via exogenous transfection of NS1-encoding plasmids, whereas Zhirnov used wild-type and NS1-deficient viruses to examine the effect of NS1. The enhancement of p53 activity in the presence of NS1 observed by Zhirnov may be due to the elevated DNA damage induced by wild-type influenza A virus. DNA breakage or damage is a common phenomenon during influenza A infection34 and is the most important trigger of p53 activation.35 Compared with wild-type influenza A virus, delNS1 virus always displays a reduced replication capability in host cells,36 which may induce a comparatively mild level of DNA damage, leading to reduced elevation of p53 activity.
Apoptosis is a complicated process and involves numerous extracellular signals and intracellular molecules, including IFN, TNFα and p53. In addition, an association between NS1 and apoptosis has been observed by many studies. However, the exact role of NS1 in the apoptotic response has not been fully understood, given that NS1 has been found to exhibit either pro- or antiapoptotic function in different experimental systems. It has been shown that the overexpression of H5N9 NS1 in MDCK or Hela cells is sufficient to trigger apoptosis.22 Transfection of NS1 from the A/HK/483/97(H5N1) strain into human airway epithelial cells can also induce apoptosis.23 However, in another report, the ectopic expression of PR8/NS1 protein was shown to produce an antiapoptotic response in MDCK cells through the activation of the PI3K/Akt signaling pathway.24 In our study, although H5N1-NS1 variants affected IFN induction, the TNFα response and p53 activity, they did not significantly promote or inhibit the apoptosis of Hela cells. Furthermore, none of these H5N1-NS1 variants markedly influenced the apoptotic process in MDCK cells (data not shown). Therefore, the role of NS1 in apoptosis is currently far from clear. A possible explanation for these discrepancies is that different NS1 proteins or cells were used in these studies.
In conclusion, our results indicate that three H5N1-NS1 variants differentially affected the production of IFN, inhibited the TNFα response and exerted similar inhibitory effects on the transcriptional activity of p53, although they did not markedly influence apoptosis of host cells. Our findings might partly explain the elevated virulence of H5N1 virus.
Acknowledgments
We are grateful to Dr William Ba-Thein for helpful discussion and editing of the manuscript. We thank Dr Xu Liyan for the use of a TD20/20 luminometer. This work was supported by the National Natural Science Foundation of China (No. 30771988and No. 30972766), Specialized Research Fund for the Doctoral Program of Higher Education (No. 20094402110004), Guangdong Natural Science Foundation (No. 8151503102000022 and 9451503102003499), Outstanding Young Scientists Foundation of Guangdong Province Education Department (No. LYM08056), State Key Lab of Agriculture Microbiology Open Foundation (No. AML200910) and Shantou University Medical College Research Foundation.
References
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Li KS, Guan Y, Wang J, Smith GJ, Xu KM, Duan L, et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- Guan Y, Poon LL, Cheung CY, Ellis TM, Lim W, Lipatov AS, et al. H5N1 influenza: a protean pandemic threat. Proc Natl Acad Sci USA. 2004;101:8156–8161. doi: 10.1073/pnas.0402443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatov AS, Andreansky S, Webby RJ, Hulse DJ, Rehg JE, Krauss S, et al. Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: the role of cytokines and B- and T-cell responses. J Gen Virol. 2005;86:1121–1130. doi: 10.1099/vir.0.80663-0. [DOI] [PubMed] [Google Scholar]
- Zhou H, Jin M, Chen H, Huag Q, Yu Z. Genome-sequence analysis of the pathogenic H5N1 avian influenza A virus isolated in China in 2004. Virus Genes. 2006;32:85–95. doi: 10.1007/s11262-005-5849-9. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Yang H, Chen W, Cao W, Zhong G, Jiao P, et al. A naturally occurring deletion in its NS gene contributes to the attenuation of an H5N1 swine influenza virus in chickens. J Virol. 2008;82:220–228. doi: 10.1128/JVI.00978-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Prasad BV. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature. 2008;456:985–988. doi: 10.1038/nature07444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JX, Peng DX, Liu YL, Wu YT, Liu XF. Virulence of H5N1 avian influenza virus enhanced by a 15-nucleotide deletion in the viral nonstructural gene. Virus Genes. 2008;36:471–478. doi: 10.1007/s11262-007-0187-8. [DOI] [PubMed] [Google Scholar]
- Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, et al. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82:1146–1154. doi: 10.1128/JVI.01698-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang Y, Jiao P, Wang A, Zhao F, Tian G, et al. The NS1 gene contributes to the virulence of H5N1 avian influenza viruses. J Virol. 2006;80:11115–11123. doi: 10.1128/JVI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan NR, Basler CF, Garcia-Sastre A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol. 2003;77:13257–13266. doi: 10.1128/JVI.77.24.13257-13266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SH, Hoffmann E, Webster RG. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat Med. 2002;8:950–954. doi: 10.1038/nm757. [DOI] [PubMed] [Google Scholar]
- Twu KY, Kuo RL, Marklund J, Krug RM. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J Virol. 2007;81:8112–8121. doi: 10.1128/JVI.00006-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SH, Hoffmann E, Webster RG. The NS1 gene of H5N1 influenza viruses circumvents the host anti-viral cytokine responses. Virus Res. 2004;103:107–113. doi: 10.1016/j.virusres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Heikkinen LS, Kazlauskas A, Melén K, Wagner R, Ziegler T, Julkunen I, et al. Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J Biol Chem. 2008;283:5719–5727. doi: 10.1074/jbc.M707195200. [DOI] [PubMed] [Google Scholar]
- Li W, Wang G, Zhang H, Zhang D, Zeng J, Chen X, et al. Differential suppressive effect of promyelocytic leukemia protein on the replication of different subtypes/strains of influenza A virus. Biochem Biophys Res Commun. 2009;389:84–89. doi: 10.1016/j.bbrc.2009.08.091. [DOI] [PubMed] [Google Scholar]
- Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz-Cherry S. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol. 2005;79:8802–8811. doi: 10.1128/JVI.79.14.8802-8811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- Yeung MC, Lau AS. Tumor suppressor p53 as a component of the tumor necrosis factor-induced, protein kinase PKR-mediated apoptotic pathway in human promonocytic U937 cells. J Biol Chem. 1998;273:25198–25202. doi: 10.1074/jbc.273.39.25198. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Qiu F, Tashiro S, Onodera S, Ikejima T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem Biophys Res Commun. 2008;376:483–488. doi: 10.1016/j.bbrc.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Schultz-Cherry S, Dybdahl-Sissoko N, Neumann G, Kawaoka Y, Hinshaw VS. Influenza virus ns1 protein induces apoptosis in cultured cells. J Virol. 2001;75:7875–7881. doi: 10.1128/JVI.75.17.7875-7881.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WY, Tang JW, Yeung AC, Chiu LC, Sung JJ, Chan PK. Avian influenza virus A/HK/483/97(H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J Virol. 2008;82:2741–2751. doi: 10.1128/JVI.01712-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C, Wolff T, Pleschka S, Planz O, Beermann W, Bode JG, et al. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol. 2007;81:3058–3067. doi: 10.1128/JVI.02082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman A, Comely S, Lackenby A, Hartgroves LC, Goodbourn S, McCauley JW, et al. NS1 proteins of avian influenza A viruses can act as antagonists of the human alpha/beta interferon response. J Virol. 2007;81:2318–2327. doi: 10.1128/JVI.01856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MO, Suh HS, Brosnan CF, Lee SC. Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-beta. J Neurochem. 2004;90:297–308. doi: 10.1111/j.1471-4159.2004.02487.x. [DOI] [PubMed] [Google Scholar]
- Rani MR, Shrock J, Appachi S, Rudick RA, Williams BR, Ransohoff RM. Novel interferon-beta-induced gene expression in peripheral blood cells. J Leukoc Biol. 2007;82:1353–1360. doi: 10.1189/jlb.0507273. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Ransohoff RM, Peppler R, VanderBrug Medendorp S, Lehmann P, Alam J. Interferon beta induces interleukin-10 expression: relevance to multiple sclerosis. Ann Neurol. 1996;40:618–627. doi: 10.1002/ana.410400412. [DOI] [PubMed] [Google Scholar]
- Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease. Lancet. 2002;360:1831–1837. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhang J, Li W, Xin G, Su Y, Gao Y, et al. Apoptosis and proinflammatory cytokine responses of primary mouse microglia and astrocytes induced by human H1N1 and avian H5N1 influenza viruses. Cell Mol Immunol. 2008;5:113–120. doi: 10.1038/cmi.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SH, Webster RG. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol. 2002;76:1071–1076. doi: 10.1128/JVI.76.3.1071-1076.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Wang X, Guo L, Qiu Y, Li X, Yu H, et al. Influenza A virus induces p53 accumulation in a biphasic pattern. Biochem Biophys Res Commun. 2009;382:331–335. doi: 10.1016/j.bbrc.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Zhirnov OP, Klenk HD. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis. 2007;12:1419–1432. doi: 10.1007/s10495-007-0071-y. [DOI] [PubMed] [Google Scholar]
- Vijaya Lakshmi AN, Ramana MV, Vijayashree B, Ahuja YR, Sharma G. Detection of influenza virus induced DNA damage by comet assay. Mutat Res. 1999;442:53–58. doi: 10.1016/s1383-5718(99)00058-3. [DOI] [PubMed] [Google Scholar]
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Talon J, Salvatore M, O'Neill RE, Nakaya Y, Zheng H, Muster T, et al. Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci USA. 2000;97:4309–4314. doi: 10.1073/pnas.070525997. [DOI] [PMC free article] [PubMed] [Google Scholar]