

Abstract
Innate responses combine with adaptive immunity to generate the most effective form of anti-Aspergillus immune resistance. Whereas the pivotal role of dendritic cells in determining the balance between immunopathology and protective immunity to the fungus is well established, we determined that epithelial cells (ECs) also contributes to this balance. Mechanistically, EC-mediated protection occurred through a Toll-like receptor 3/Toll/IL-1 receptor domain-containing adaptor-inducing interferon (TLR3/TRIF)-dependent pathway converging on indoleamine 2,3-dioxygenase (IDO) via non-canonical nuclear factor-κB activation. Consistent with the high susceptibility of TRIF-deficient mice to pulmonary aspergillosis, bone marrow chimeric mice with TRIF unresponsive ECs exhibited higher fungal burdens and inflammatory pathology than control mice, underexpressed the IDO-dependent T helper 1/regulatory T cell (Th1/Treg) pathway and overexpressed the Th17 pathway with massive neutrophilic inflammation in the lungs. Further studies with interferon (IFN)-γ, IDO or IL-17R unresponsive cells confirmed the dependency of immune tolerance to the fungus on the IFN-γ/IDO/Treg pathway and of immune resistance on the MyD88 pathway controlling the fungal growth. Thus, distinct immune pathways contribute to resistance and tolerance to the fungus, to which the hematopoietic/non-hematopoietic compartments contribute through distinct, yet complementary, roles.
Keywords: aspergillosis, epithelial cells, IDO, Th17/Treg, transplantation tolerance
Introduction
Dendritic cells (DCs) orchestrate the adaptive immune responses to Aspergillus fumigatus.1, 2, 3 The capacity of DCs to initiate different adaptive immune responses to the fungus largely relied upon specialization and cooperation between distinct DC subsets and the discriminative recognition of fungal morphotypes by distinct innate recognition receptors.4, 5, 6 Because of their heterogeneity and plasticity, DCs are central in the early decision-making mechanisms that results in a given type of immune response to the fungus and determine the balance between immunopathology and protective immunity generated by the host–fungus interaction.1 However, infectious or inflammatory conditions can profoundly alter the functions of steady-state DC subsets and recruit inflammatory-type DCs to the lung. This might be important for clearing the pathogenic stimulus, but could at the same time lead to immune pathology.7 In this regard, DCs expressing the enzyme indoleamine 2,3-dioxygenase (IDO), by means of activating regulatory T cells (Tregs), help to tame overzealous and exaggerated inflammatory responses in infection and allergy to the fungus8, 9 and confer protective tolerance in hematopoietic transplantation.6 As a matter of fact, IL-23 produced by inflammatory DCs and the T helper 17 (Th17) pathway, which downregulate tryptophan catabolism, favored pathology and served to accommodate the seemingly paradoxical association of chronic inflammation with fungal persistence.10, 11, 12
No longer considered innocent bystanders,13 airway epithelial cells (ECs) are central participants in innate and adaptive immune responses as well as mucosal inflammation and allergy.14 Through the activation of Toll-like receptors (TLRs) by endogenous and exogenous ligands, ECs may play a central role in determining the balance between a state of ‘mucosal homeostasis', as is required for optimal organ function, and ‘mucosal injury', leading to mucosal inflammation and barrier breakdown.13 Recent evidence indicated that ECs may also contribute to immunity to respiratory pathogens. ECs provided protection against Mycobacterium tuberculosis via an interferon (IFN)-γ/IDO axis culminating in the inhibition of Th17 cell responses.15 A. fumigatus conidia are internalized by ECs and, once inside, some conidia traffic to late endosomes/lysosomes, where they can germinate. This has lent to the credence that ECs may serve as reservoirs for immune evasion.16, 17 However, respiratory ECs also sense germinating conidia through MyD88-dependent and -independent pathways.18 Of interest, IDO overexpression from the airway ECs was found to restrain CD4+ T-cell activation to the fungus, an activity that was nevertheless dispensable in the presence of IDO-expressing DCs.19
In the present study, we assess the contribution of ECs to protective immunity to Aspergillus and the possible mechanisms underlying this activity in experimental models of pulmonary aspergillosis. We found that ECs contribute to immunity to the fungus by providing protective tolerance through a TLR3/Toll/IL-1 receptor domain-containing adaptor-inducing IFN (TRIF)-dependent pathway converging on IDO, a key regulator of the Th1/Treg versus Th17 pathway balance. Because the MyD88 pathway mainly contributed to antifungal resistance by restraining the fungal growth, our findings suggest that distinct immune pathways account for resistance and tolerance to the fungus, to which the hematopoietic/non-hematopoietic compartments contribute through distinct, yet complementary, roles.
Material and methods
Mice
Female C57BL/6 and BALB/c mice, 8–10 weeks old, were purchased from Charles River (Calco, Italy). Homozygous Tlr3−/−, Tlr4−/−, Myd88−/−, Trif−/−, Indo−/−(Indotm 1 Alm/J), Il17ra−/−, Ifng−/− , Ifnra1−/−, Il1r1−/− or Casp1−/− mice on a C57BL/6 background were bred under specific pathogen-free conditions at the Animal Facility of Perugia University (Perugia, Italy). Experiments were performed according to the Italian Approved Animal Welfare Assurance A-3143-01.
Fungal strains, infections and treatments
The strain of A. fumigatus was obtained from a fatal case of pulmonary aspergillosis at the Infectious Diseases Institute of the University of Perugia. Viable conidia (>95%) were obtained by growth on Sabouraud dextrose agar (Difco Laboratories, Detroit, MI, USA) supplemented with chloramphenicol for 4 days at room temperature. Swollen conidia were obtained as described.1 For infection, mice were anesthetized by intraperitoneal (i.p.) injection of 2.5% avertin (Sigma-Aldrich, St Louis, MO, USA) before instillation of a suspension of 2×107 conidia/20 µl saline intranasally (i.n.). Fungi were suspended in endotoxin-free (Detoxi-gel; Pierce, Rockford, IL, USA) solutions (<1.0 EU/ml, as determined by the Limulus amebocyte lysate method). Mice were monitored for fungal growth (colony-forming unit (CFU)/organ, mean±SE) and histopathology (periodic acid-Schiff (PAS) staining of lung tissue sections). Bronchoalveolar lavage (BAL) was performed by cannulating the trachea and washing the airways with 3 ml of phosphate-buffered saline (PBS) to collect the BAL fluid. Total and differential cell counts were done by staining BAL smears with May–Grünwald Giemsa reagents (Sigma-Aldrich) before analysis. At least 200 cells per cytospin preparation were counted and the absolute number of each cell type was calculated. Photographs were taken using a high-resolution Olympus DP71 microscope (Olympus, Milan, Italy). Mice were treated i.p. daily, starting 3 days before and up to 3 days after the infection with PBS or 20 µg/kg of a mixture of L-kynurenine, 3-hydorxykynurenine and 3-hydroxyanthranilic acid (Sigma-Aldrich). Treatment with 200 µg/mouse of IL-17-neutralizing monoclonal antibodies (clone TC11-18H10; PharMingen, San Diego, CA, USA)10 or isotype control antibody (rat IgG; Sigma-Aldrich) was done i.p. the day of and a day after the infection.
Generation of bone marrow chimeras and hematopoietic stem cell transplantation (HSCT)
Donor wild-type (WT) and knockout (KO) mice were euthanized with CO2 asphyxiation and cervical dislocation, and femurs and tibias were removed aseptically. Bone marrow was flushed with cold Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum and 2 mM L-glutamine (Invitrogen Srl, Milan, Italy). Cells were washed twice with PBS without calcium and magnesium supplemented with 1% fetal calf serum. T-cell depletion was obtained upon incubation of the cells with MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) conjugated to rat antimouse CD5 monoclonal antibodies (Ly-1; clone 53–7.3) for 15 min at 4 °C followed by magnetic sorting in a magnetic cell separator (CliniMACS cell separation system; Miltenyi Biotec). Viable cells were counted in a hemacytometer with a trypan blue exclusion assay. Recipient WT and KO mice were irradiated with 9 Gy and reconstituted no later than 6 h after the last irradiation with 1×106 T-repleted (chimeras) or T-depleted (HSCT) cells by intravenous injection. Mice were given sulfamethoxazole (150 mg/ml) and trimethoprim (30 mg/ml) in drinking water for the first 3 weeks of reconstitution. HSCT mice were infected i.n. a week later with Aspergillus conidia, while chimeras were used no earlier than 4 weeks after transplantation. Before use in experiments, all mice were bled from the retro-orbital plexus, and the peripheral blood lymphocytes were analyzed by reverse transcription-PCR for the expression of Myd88, Trif, Ifng, Indo and Il17ra to assess the degree of chimerism.
Cell isolation
For isolation of lung cells, lungs were aseptically removed and cut into small pieces in cold medium. The dissected tissue was then incubated in medium containing collagenase XI (0.7 mg/ml; Sigma-Aldrich) and type IV bovine pancreatic DNase (30 µg/ml; Sigma-Aldrich) for 30–45 min at 37 °C. The action of the enzymes was stopped by adding 10 ml of medium, and digested lungs were further disrupted by gently pushing the tissue through a nylon screen. The single-cell suspension was then washed and centrifuged at 200 g. Contaminating red blood cells were lysed, and cells were then washed with PBS containing 0.5% fetal calf serum, counted and incubated with fluorescein isothiocyanate (FITC)-labeled GL3 (hamster anti-γδ T-cell receptor; PharMingen) followed by anti-FITC MicroBeads (Miltenyi Biotech) or with CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotech) before magnetic cell sorting.10
EC purification, stimulation and treatment with small interfering RNA (siRNA)
Lung ECs, at ∼99% expressing cytokeratin, on pan–cytokeratin antibody staining of cytocentrifuge preparations, and >90% viable on trypan blue exclusion assay, were isolated as described.20 The average yield of tracheal cells was 1.7×105 cells/trachea (±0.58×105 (SD)). ECs (106) were stimulated with resting or swollen conidia (1∶1 ratio), 100 U IFN-γ 10 µg/ml Zymosan, 10 µg/ml Poli(I:C), 10 µg/ml ultrapure lipopolysaccharide from Salmonella minnesota Re 595 (all from Sigma-Aldrich), and 10 µg/ml ODN-CpG1 for 1 h (nuclear factor (NF)-κB and IFN regulatory factor 3) or 18 h (IDO and cytokines). ECs were seeded into 96-well plates and then siRNA was added in a final concentration of 1 µM for 72 h according to the manufacturer's instructions (Dharmacon RNAi Technologies, Chicago, IL, USA). The silencing effects of siRNAs were confirmed by western blotting. In vitro experiments were done in the presence of 2% fetal bovine serum.
Flow cytometry
Flow cytometry analyses involving surface expression of T-cell receptor-γδ, CD4 and CD25 molecules have previously been described.10 Stains were preblocked with FcBlock (antimouse CD16/CD32; PharMingen).
Western blotting
Protein phosphorylation was assessed on ECs stimulated as above. Blots of cells lysates were incubated with rabbit polyclonal antibodies recognizing the anti-phospho-IKKα (Ser180)/-IKKβ (Ser181) rabbit antibodies (Cell Signaling Technology, Danvers, MA, USA) were used for western blotting of phospho-IKKα and -IKKβ. Blots were developed with the Enhanced Chemiluminescence Detection Kit (Amersham Pharmacia Biotech, Milan, Italy).1 Immunoblotting for IDO was performed with rabbit polyclonal IDO-specific antibodies on ECs after 18-h stimulation. The positive control consisted of IDO-expressing MC24 transfectants and the negative control of mock-transfected MC22 cells. Scanning densitometry was done on a Scion Image apparatus (Scion Corp, Frederick, MD, USA). The pixel density of bands was normalized against total proteins or β-tubulin.
Kynurenine assay
Kynurenine concentrations in the supernatants of 18-h EC cultures were measured by high-performance liquid chromatography.10
ELISA, ELISPOT and real-time PCR
The level of cytokines in culture supernatants (24 h) of ECs was determined by Kit ELISA (R&D System, Milan, Italy). The detection limits of the assays were <10 for IL-12p70, <3 for IL-10, <30 for IL-23, and <10 for IL-17A and IL-17F. Real-time RT-PCR was performed using the iCycler iQ detection system (Bio-Rad, Hercules, CA, USA) and SYBR Green chemistry (Finnzymes Oy, Espoo, Finland). Cells were lysed and total RNA was extracted using RNeasy Mini Kit (Qiagen, Milan, Italy) and was reverse transcribed with Sensiscript Reverse Transcriptase (Qiagen) according to the manufacturer's directions. The PCR primers were as described.1, 21 Amplification efficiencies were validated and normalized against Gapdh. The thermal profile for SYBR Green real-time PCR was at 95 °C for 3 min, followed by 40 cycles of denaturation for 30 s at 95 °C and an annealing/extension step of 30 s at 60 °C. Each data point was examined for integrity by analysis of the amplification plot. The mRNA-normalized data were expressed as relative cytokine mRNA in stimulated cells compared to that of mock-infected cells. Cytokine-producing cells were enumerated by ELISPOT assay on purified CD4+ cells as described.21 Results were expressed as the mean number of cytokine-producing cells (±SE) per 104 cells, calculated using replicates of serial twofold dilutions of cells.
Statistical analysis
Data were analyzed by GraphPad Prism 4.03 program (GraphPad Software, San Diego, CA, USA). Student's t-test or analysis of variance and Bonferroni's test were used to determine the statistical significance (P) of differences in organ clearance and in vitro assays. The data reported are either from one representative experiment out of two to four independent experiments (western blotting and RT-PCR) or pooled from three to five experiments, otherwise. The in vivo groups consisted of 6–8 mice/group.
Results
Trif−/− mice are highly susceptible to pulmonary aspergillosis and develop pathogen-induced inflammation
We evaluated parameters of infection, inflammation and adaptive immunity in Trif−/− and mice with pulmonary aspergillosis. Despite the ability of both strains to restrain the fungal growth eventually, the initial fungal growth was higher in the lung and brain of KO than WT mice (Figure 1a). Inflammation and signs of parenchymal damage were also greatly exacerbated in Trif−/− mice, which failed to resolve and showed the presence of numerous PAS-positive cells within the epithelial layer. Confirming previous studies,4, 22, 23 Myd88−/− mice were not susceptible to infection and inflammation (Figure 1b). The number of polymorphonuclear cells increased and maintained elevated in the lung parenchyma and the BAL fluids (insets of Figure 1b also showing numerous hyphae, arrows) of Trif−/− as compared to WT or Myd88−/− mice. Gene expression analysis of the lung confirmed the higher and more persistent inflammatory response in Trif−/− than WT/Myd88−/− mice, as revealed by the higher mRNA expression of Cxcl1, Cxcl2 and Mpo genes as well as genes for inflammatory cytokines, such as Il1a (Figure 1c). As expected, the levels of Ifna and Ifnb1 were instead lower in Trif−/− than WT/Myd88−/− mice (Figure 1c). Tlr3−/− (manuscript in preparation) but not Tlr4−/− mice24 mimicked the patterns of susceptibility to infection and inflammation of Trif−/− mice, a finding revealing a previously undefined, crucial role for the TLR3/TRIF pathway in aspergillosis. The failure to resolve inflammation was not secondary to a deficient conidiocidal activity of lung cells either from naive or 3-day-infected mice (the % conidiocidal activity ranged from 23±2 to 50±7 (WT), 22±4 to 48±8 (Trif−/−) and 28±8 to 65±12 (Myd88−/−)). In vitro, the conidiocidal activity of purified neutrophils from each type of naive mice was not different (the % conidiocidal activity was 35±4 for WT, 32±6 for Trif−/−or 41±8 for Myd88−/− cells). Thus, the TLR3/TRIF pathway is not required for the conidiocidal activity of lung effector cells but unexpectedly protects from unintended inflammation.
Figure 1.
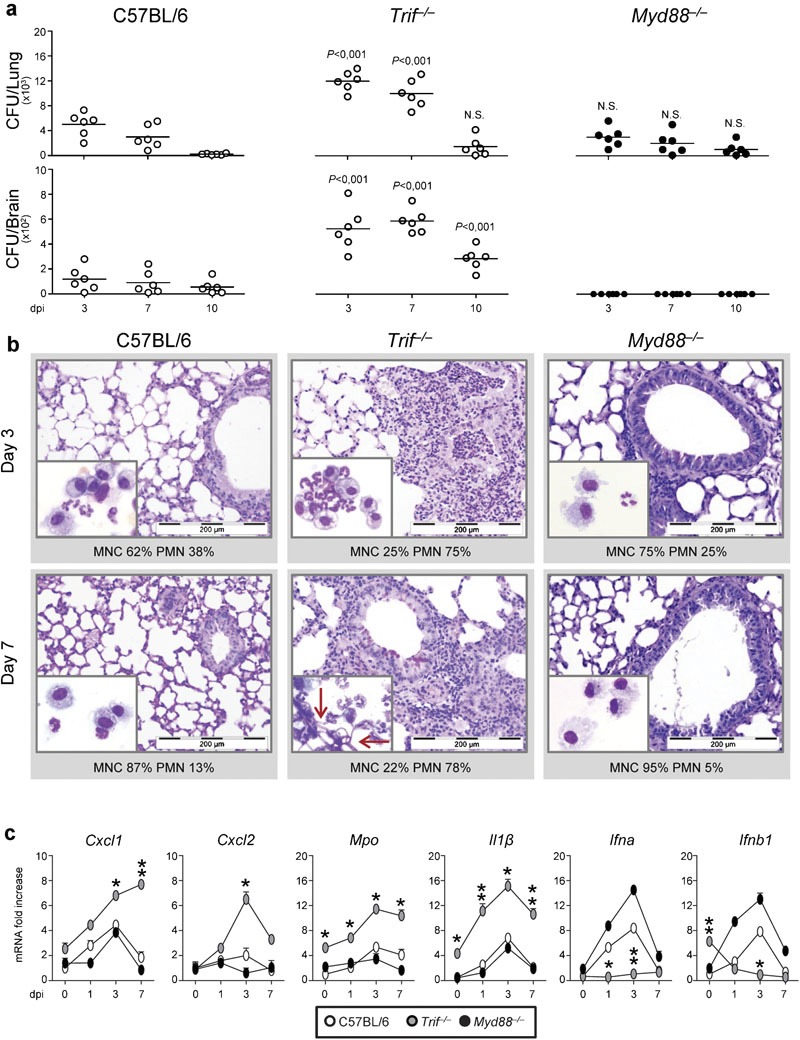
Trif−/− mice are highly susceptible to pulmonary aspergillosis and develop pathogen-induced inflammation. Mice were infected i.n. with live Aspergillus conidia. (a) Fungal growth (CFU±SE). (b) Lung histology (PAS staining) and BAL morphometry (% PMN or % MNC) at different dpi. P, Trif−/− versus WT mice. (c) Inflammatory chemokine and cytokine gene expression in the lung by real-time RT-PCR. Note the sustained inflammatory cell recruitment in lungs and BAL (May–Grünwald Giemsa staining in the inset) in Trif−/− mice. Bars indicated magnifications. Day 0 indicates uninfected animals. Data are pooled from four experiments or representative of two experiments (for histology). *P<0.05, **P<0.01, ***P<0.001, Trif−/− versus WT mice. BAL, bronchoalveolar lavage; CFU, colony-forming unit; dpi, days post-infection; i.n., intranasally; MNC, mononuclear cells; PAS, periodic acid-Schiff; PMN, polymorphonuclear cells; RT-PCR, PCR with reverse transcription; WT, wild-type.
The subverted innate inflammatory response to the fungus10 and the requirement for TLR3/TRIF in the activation of tolerogenic DCs in aspergillosis1 would predict altered adaptive Th/Treg cell responses in Trif−/− mice. This was indeed the case as shown by the results of Th/Treg cell activation in response to the fungus. Consistent with the DC cytokine profile in response to the fungus,1 the levels (gene expression and protein production) of IL-12p70 were lower and those of IL-23 higher in Trif−/− than WT/Myd88−/− mice (Figure 2a). Accordingly, the production of IFN-γ/IL-10 in the lung (Figure 2b) and the corresponding Tbet/Foxp3-specific transcripts in the thoracic lymph nodes (Figure 2c) were lower and those of IL-17A/IL-17F (Figure 2b) and the Rorc transcript (Figure 2c) higher in Trif−/− than WT/Myd88−/− mice.
Figure 2.
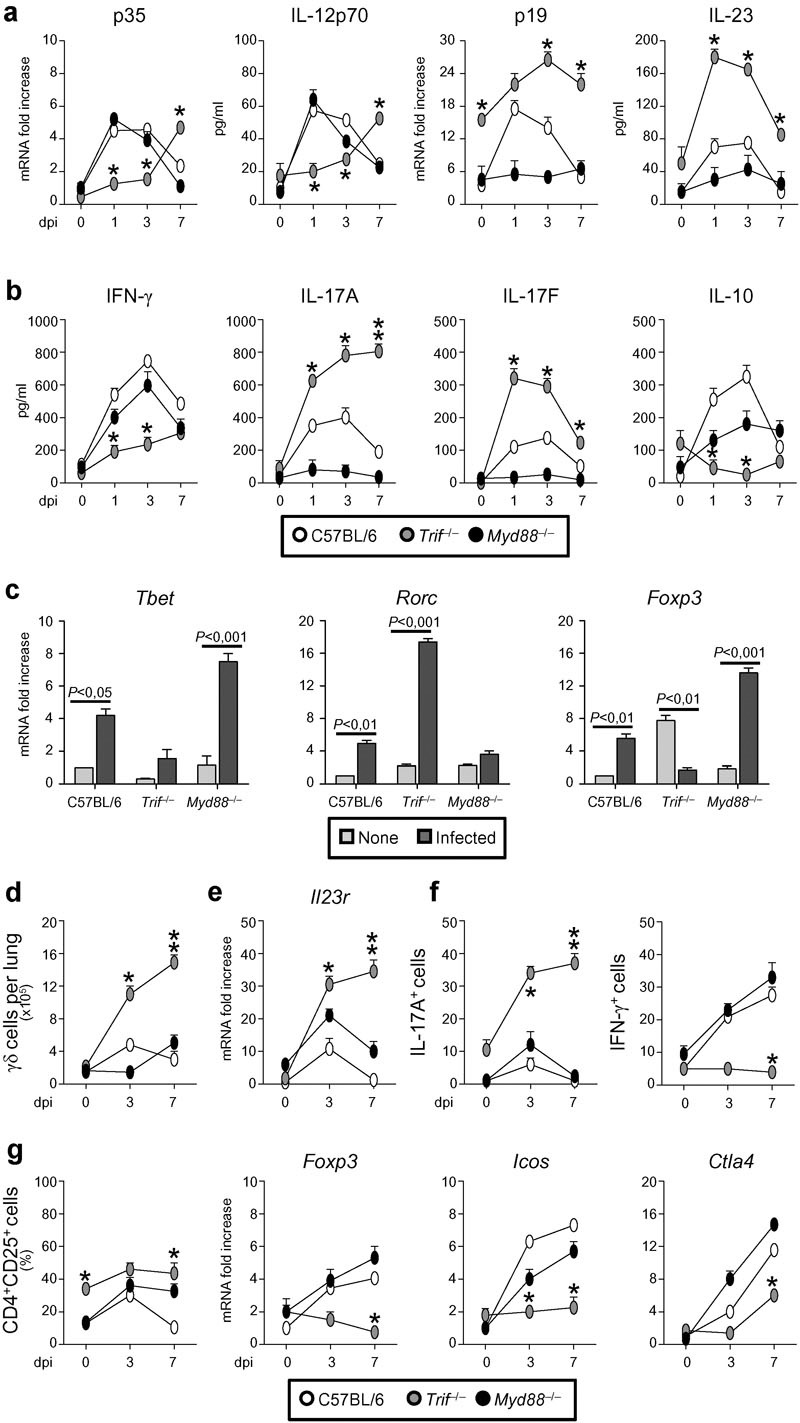
Trif−/− mice upregulate Th17 responses and downregulate Treg responses. Mice were infected i.n. with live Aspergillus conidia. (a, b) Cytokine gene expression in the lung at different dpi (RT-PCR). (c) Cytokine gene expression in CD4+ T cells from thoracic lymph nodes at 7 dpi (RT-PCR). (d) Absolute numbers of γδ TCR+ in the lung; (e) Il23r expression on purified γδ TCR+ cells by real-time RT-PCR; (f) number of γδ TCR+ cells producing IL-17A or IFN-γ by ELISPOT assay. (g) Percentage of CD4+CD25+ T cells in the lung at different dpi and Foxp3, Icos and Ctla4 gene expression by real-time RT-PCR. Data are pooled from three experiments. *P<0.05, **P<0.01, ***P<0.001, Trif−/− versus WT mice. dpi, days post-infection; IFN, interferon; i.n., intranasally; RT-PCR, PCR with reverse transcription; Th17, T helper 17; WT, wild-type.
Susceptibility to aspergillosis has been associated with the prevalent expansion of a subset of γδ TCR+ cells expressing IL-23R and producing IL-17A over that of CD4+CD25+Foxp3+ Tregs.10 We found that γδ+ cells producing IL-17A and expressing IL-23R were expanded in Trif−/− as compared to WT/Myd88−/− mice, whereas γδ+ cells producing IFN-γ were lower (Figure 2d–f). In contrast, despite the expansion of CD4+CD25+ T cells in Trif−/− mice, these cells failed to express the Foxp3, Icos and, partially, Ctla4 transcripts (Figure 2g). As predicted by the histological findings, Th2 (Gata3/Il4/Il13) cell responses were also higher in Trif−/− than WT mice and, accordingly, Trif−/− mice were more susceptible to fungal allergy than WT mice (Supplementary Figure 1). Thus, similar to what described in candidiasis,21 and consistent with the role of the TRIF pathway in promoting tolerogenic DCs in response to the fungus,1 TRIF deficiency is associated with the impairment of Th1/Treg protective responses and upregulation of inflammatory Th2/Th17 cell responses.
The TLR3/TRIF pathway activates IDO on ECs
We have already shown that the TLR3/TRIF pathway activates IDO in lung DCs through the non-canonical NF-κB pathway1 and that IDO+ DCs conferred antifungal protection upon adoptive transfer into hematopoietic transplantation.6 We assessed IDO protein expression and activation in the lung of WT, Trif−/− and Myd88−/− mice in infection as well as in ECs upon stimulation in vitro. Trif−/− mice failed to induce IDO as opposed to WT or Myd88−/− mice. However, IL-17A neutralization restored IDO expression in Trif−/− mice and increased it in WT mice (Figure 3a). These data point to the opposing activity of the TRIF and IL-17 pathways on IDO expression and confirm previous results obtained in experimental candidiasis21 and M. tuberculosis infection.15 Paralleling the in vivo data, IDO activation was observed in ECs from WT and Myd88−/− but not Trif−/− mice upon IFN-γ stimulation, as revealed by protein expression and kynurenine production (Figure 3b). The failure of resting conidia to activate IDO in ECs (Figure 3b), prompted us to search for microbial stimuli that would induce IDO in ECs. We found that swollen conidia and selected TLR ligands, Poli(I:C) and ODN-CpG, but not others, zymosan or lipopolysaccharide, promoted IDO protein expression in ECs (Figure 3c). Thus, IDO activation in ECs in response to Aspergillus occurs through a TLR3/TRIF pathway. Further studies showed that, similar to what observed in DCs,1 IDO activation in ECs occurred through non-canonical NF-κB, and not IFN regulatory factor 3, the two major pathways downstream TLR3 signaling.25 Despite the fact that both pathways were activated in response to swollen conidia or Poli(I:C), the inhibition of non-canonical NF-κB by siRNA prevented downstream IDO activation (Figure 3c). Thus, germinating conidia are sensed in infection by ECs through a TLR3/TRIF pathway culminating in IDO activation and production of type I IFNs and IL-10 (Figure 3d).
Figure 3.
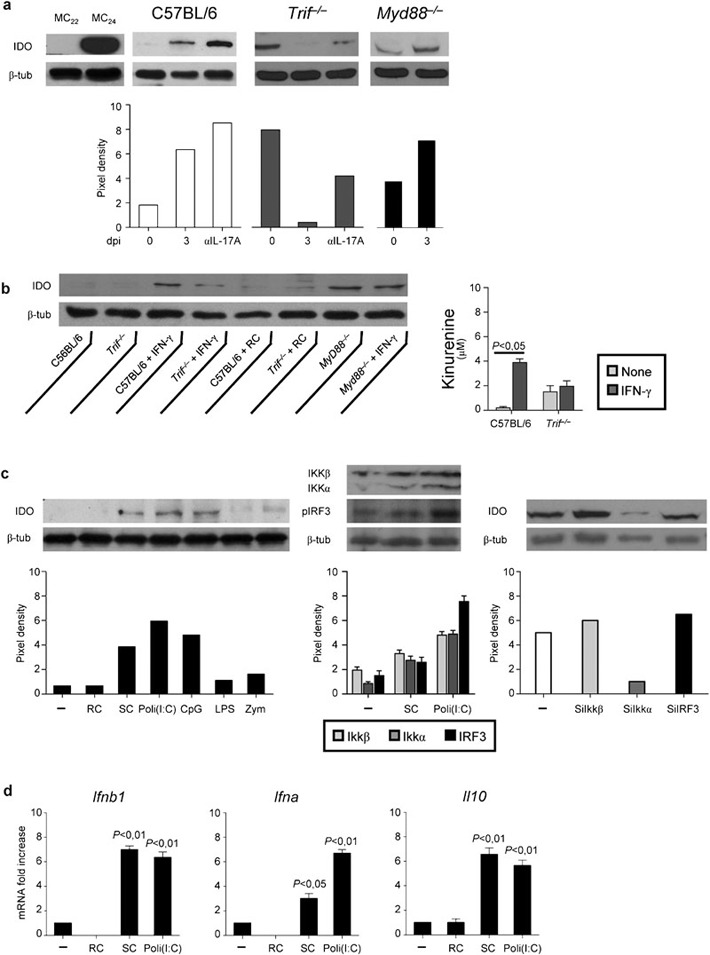
The TLR3/TRIF pathway activates IDO on ECs. Mice were infected i.n. with live Aspergillus conidia and treated with 200 µg IL-17A-neutralizing antibody or isotype control antibody the day of and a day after the infection. (a) IDO protein expression in the lung. IDO expression was analyzed by immunoblotting with rabbit polyclonal IDO-specific antibody on whole cell lysates. The positive control consisted of IDO-expressing MC24 transfectants and the negative control of mock-transfected MC22 cells. (b) IDO protein expression and kynurenines production in ECs purified from lungs of naive mice and stimulated in vitro with RC (1∶1 ratio) or 100 U IFN-γ for 18 h. (c) IDO expression (immunoblotting) in ECs from C57BL/6 mice stimulated with RC, SC or TLR ligands for 18 h. IKKα, IKKβ and IRF3 expressions by western blotting were performed after 1 h stimulation with Poli(I:C). For siRNA, cells were incubated with 1 µM siRNA for 72 h before 1 h stimulation with Poli(I:C). The silencing effects of siRNAs were confirmed by western blotting. (d) Cytokine gene expression by RT-PCR in Poli(I:C)-stimulated cells for 18 h. Scanning densitometry was done on a Scion Image apparatus. The pixel density of bands was normalized against total proteins or β-tubulin. EC, epithelial cell; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; i.n., intranasally; IRF3, IFN regulatory factor 3; RC, resting conidia; RT-PCR, PCR with reverse transcription; SC, swollen conidia; siRNA, small interfering RNA; TLR3/TRIF, Toll-like receptor 3/Toll/IL-1 receptor domain-containing adaptor-inducing IFN.
The IL-17/Th17 pathway worsens and kynurenine ameliorates the infection in Trif−/− mice
Because the type I IFN pathway constrains Th17 differentiation, Th17-mediated inflammation26 and prevents chronic lung damage in a respiratory fungal infection,27 and the inflammasome/IL-1β mediates antifungal host defense28 as well as pulmonary inflammation and fibrosis,29, 30 we assessed the contribution of the defective type I IFNs and the upregulated IL-1β production to susceptibility to aspergillosis by infecting mice lacking IFN-αβ receptor 1 (Ifnra1−/−), which are incapable of responding to type I IFNs, as well Il1r1−/− or Casp 1−/− mice, which fail to express the NLRP3 inflammasome-dependent antifungal immunity.31 We found that, despite variable levels of fungal growth in the lung, the tissue inflammatory pathology and cell infiltration were not different in the different types of mice as compared to WT mice (Figure 4a) and no evidence of upregulated Th17 responses was observed in the draining thoracic lymph nodes (data not shown). To explore whether, similar to what observed in hyperinflamed mice highly susceptible to aspergillosis,10 IL-17A blockade or exogenous supply of kynurenines would ameliorate the course of the infection, we subjected Trif−/− mice to treatment with either IL-17A neutralizing antibody or exogenous kynurenines. We found that the fungal growth was restrained, the neutrophil recruitment in BAL and the levels of IL-17A in the lung decreased and those of IL-10 increased upon IL-17A neutralization (Figure 4b). Similarly, treatment with kynurenines decreased both the fungal growth and the inflammatory response in Trif−/− mice and, consistent with the apoptotic activity on IL-17-producing γδ+ cells,10 lowered IL-17A production (Figure 4c). Together, these findings extend the therapeutic effects of kynurenines in aspergillosis and confirm the contribution of the upregulated IL-17/Th17 pathway in susceptibility to infection/inflammation in condition of TRIF deficiency to which neither the type I IFNs deficiency nor the inflammasome activation/IL-1 signaling obviously contributes.
Figure 4.
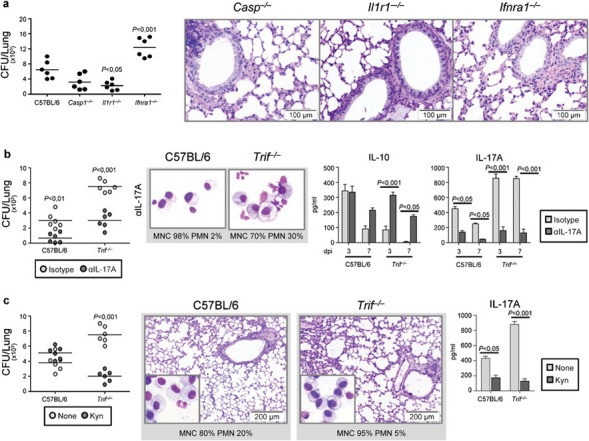
The IL-17/Th17 pathway worsens and kynurenines ameliorate the infection in Trif−/− mice. (a) Fungal growth (CFU±SE) and lung histology (PAS staining) in Ifnra1−/−, Il1r1−/− or Casp1−/− mice 4 days after the i.n. infection with live Aspergillus conidia. Bars indicated magnifications. (b) Fungal growth (CFU±SE), BAL morphometry (%PMN or % MNC) (at 3 days after the infection) and cytokine production in mice infected as above and treated i.p. with 200 µg IL-17A-neutralizing or isotype control antibody the day of and a day after the infection. (c) Fungal growth (CFU±SE), lung histology (PAS staining) and IL-17A production in infected mice treated daily, from 3 days before to 3 days after the infection, with PBS or 20 µg/kg of a mixture of L-kynurenine, 3-hydroxykynurenine and 3-hydroxyanthranilic acid. Assays were a day after the last kynurenins treatment. Bars indicated magnifications. Data are pooled from two experiments. P, KO versus WT mice and treated versus untreated mice. BAL, bronchoalveolar lavage; CFU, colony-forming unit; dpi, days post-infection; i.n., intranasally; i.p., intraperitoneal; KO, knockout; MNC, mononuclear cells; PAS, periodic acid-Schiff; PBS, phosphate-buffered saline; PMN, polymorphonuclear cells; Th17, T helper 17; WT, wild-type.
Mice with Trif−/− non-hematopoietic cells upregulate Th17 responses and downregulate Th1/Treg responses to A. fumigatus
The finding that both DCs1 and ECs activate IDO in infection, and IDO crucially regulates immunity and tolerance to the fungus,8, 9, 10 led us to investigate the relative contribution of IDO activity in the hematopoietic versus non-hematopoietic compartment in protective immunity to the fungus. To this purpose, chimeric mice with TRIF unresponsive hematopoietic or non-hematopoietic cells were assessed for susceptibility to infection and inflammation. We found that TRIF deficiency, particularly in ECs, greatly increased the susceptibility to aspergillosis, both in terms of fungal growth and inflammatory response in the lungs. These findings were not observed in mice with MyD88 unresponsive cells on both compartments, in which no inflammatory pathology were observed, despite evidence of fungal outgrowth (Figure 5). These results would predict a division of labor between the MyD88 and TRIF pathways in infection, namely, the control of fungal growth by the former and the ensuing inflammation by the latter. The results obtained in chimeric mice with cells of either compartment simultaneously unresponsive to TRIF or MyD88 confirmed the crucial contribution of the MyD88 pathway to the control of fungal growth and the contribution of the TRIF pathway, particularly on non-hematopoietic cells, to the control of the inflammatory response. The fungal growth and the inflammatory response were indeed higher in mice with TRIF unresponsive non-hematopoietic cells as compared to mice with TRIF unresponsive hematopoietic cells (Figure 5), a finding suggesting the ability of TRIF responsive non-hematopoietic cells to compensate for the lack of TRIF in hematopoietic cells.
Figure 5.
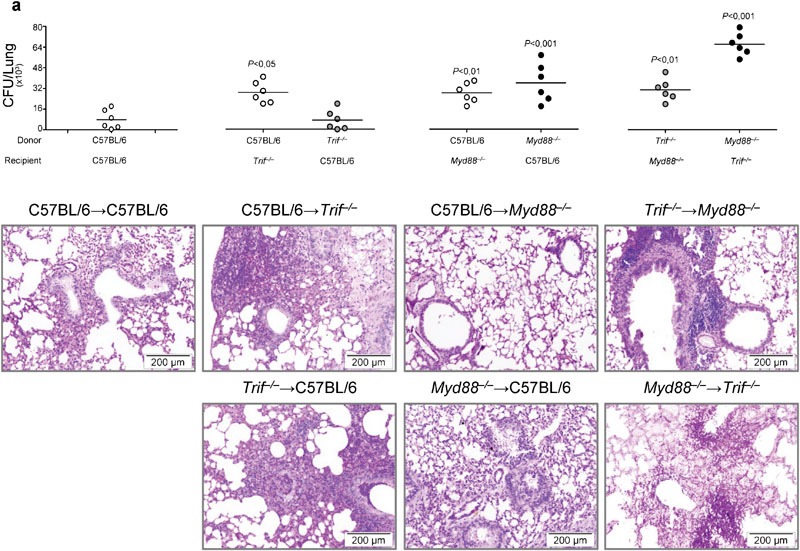
Bone marrow chimeric mice with Trif−/− non-hematopoietic cells are highly susceptible to Aspergillus fumigatus infection and inflammation. Mice were infected i.n. with live Aspergillus conidia. (a) Fungal growth (CFU±SE) and lung histology (PAS staining) in bone marrow chimeric WT and KO mice, at 3 dpi. Donor WT and KO mice received 1×106 viable bone marrow cells, 4 weeks before the infection. Note the increased susceptibility to aspergillosis, in terms of both fungal growth and the inflammatory response in the lung of chimeric mice with TRIF unresponsive non-hematopoietic cells as opposed to MyD88 unresponsive cells. Bars indicated magnifications. Data are representative of one experiment out of three. P, KO→WT, WT→KO, KO→KO versus WT→WT mice. CFU, colony-forming unit; dpi, days post-infection; i.n., intranasally; KO, knockout; PAS, periodic acid-Schiff; TRIF, Toll/IL-1 receptor domain-containing adaptor-inducing interferon; WT, wild-type.
Further experiments in an experimental model of allogeneic T cell-depleted hematopoietic transplantation (HSCT)2 confirmed that TRIF deficiency in either donor or recipient cells was associated with a worsening infection characterized by a limited fungal growth (Figure 6a) but an exaggerated inflammatory response (Figure 6b), increased IL-17A expression and decreased IFN-γ/IL-10 responses in the lung (Figure 6c). The finding that Foxp3 expression was lower in condition of both donor and recipient TRIF deficiencies (Figure 6c), further supports the contribution of the TRIF pathway to the activation of tolerogenic responses to the fungus. As a matter of fact, IDO protein expression was lower in the lung of mice, particularly in condition of recipient TRIF deficiency (data not shown). Confirming the findings of chimeric mice, the MyD88 pathway was mainly responsible for the control of fungal growth in both donor and recipient cells (Figure 6a) and, interestingly, of IL-17A production (Figure 6c).
Figure 6.
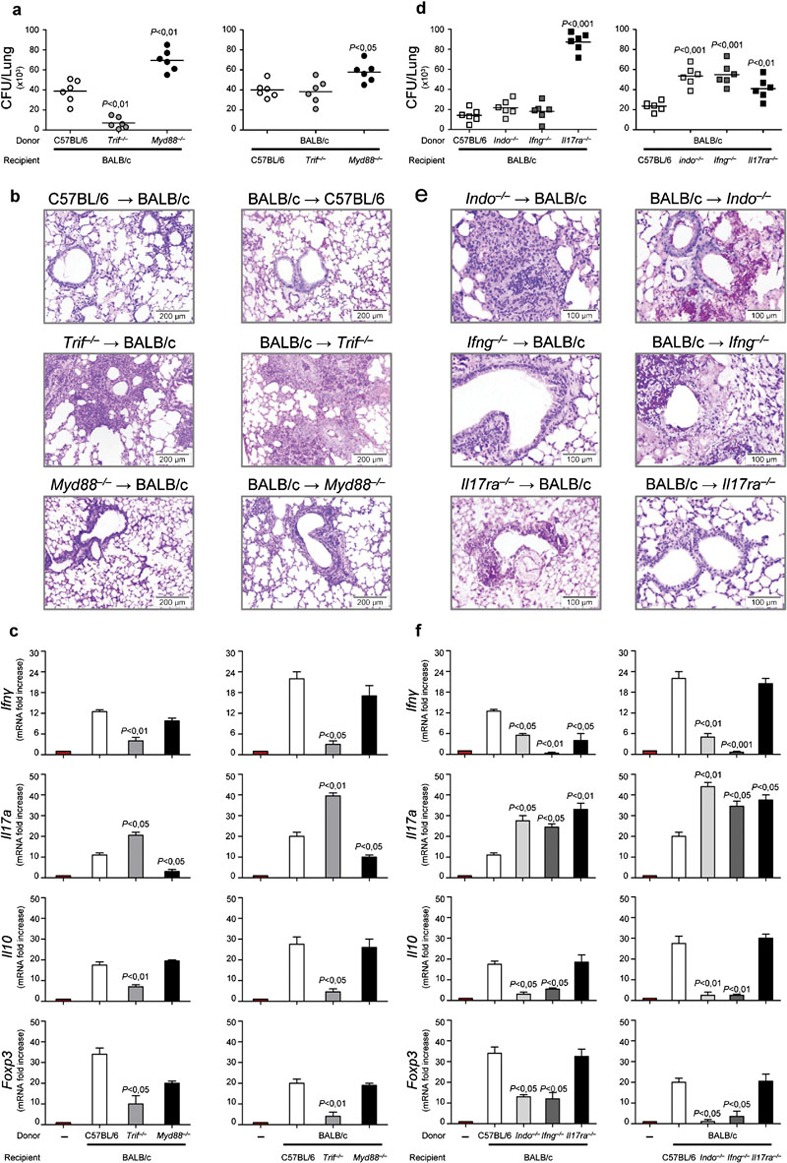
Trif−/−/Ifng−/−/Indo−/− non-hematopoietic cells upregulate Th17 responses and downregulate Th1/Treg responses to Aspergillus fumigatus in hematopoietic transplantation. HSCT mice were infected i.n. with Aspergillus live conidia a week later the infusion of 1×106 viable T-depleted bone marrow cells. (a, d) Fungal growth (CFU±SE), (b, e) lung histology (PAS staining) and (c, f) cytokine/Foxp3 gene expression (RT-PCR in lung and TLN, respectively) at 3 dpi. Note the unrestrained fungal growth in condition of MyD88 deficiency as well as the exaggerated inflammatory response, increased Il17a expression and decreased Ifng/Il10/Foxp3 expression in condition of TRIF, IFN-γ or IDO deficiency, particularly in recipient cells. Bars indicated magnifications. Representative of two experiments. P, KO→WT, WT→KO versus WT→WT mice. CFU, colony-forming unit; dpi, days post-infection; HSCT, hematopoietic stem cell transplantation; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; i.n., intranasally; KO, knockout; PAS, periodic acid-Schiff; RT-PCR, PCR with reverse transcription; Th, T helper; TLN, thoracic lymph nodes; Treg, regulatory T cell; TRIF, Toll/IL-1 receptor domain-containing adaptor-inducing IFN; WT, wild-type.
The IFN-γ/IDO and IL-17R pathways differently contribute to immune protection to A. fumigatus in hematopoietic transplantation
To confirm the pivotal contribution of IDO in infection, we evaluated parameters of infection, inflammation and Th cytokine production in condition in which either donor or recipient cells were deficient for IDO. Given the antagonistic activity of IFN-γ and IL-17 on IDO activation,10, 11 we also comparatively used donors or recipients deficient in IFN-γ or IL-17R. Deficiency of IDO greatly contributed to susceptibility to the infection in terms of increased fungal growth (Figure 6d), inflammatory pathology (Figure 6e) and decreased IFN-γ/IL-10 production (Figure 6f), particularly in recipient mice. Similar results were obtained with IFN-γ KO recipient mice (Figure 6d–f). In contrast, IL-17R deficiency, particularly in donor cells, was associated with increased fungal growth but not inflammatory response (Figure 6d and e). While the production of IL-10 was unaffected, a decreased IFN-γ production was observed in condition of IL-17R deficiency (Figure 6f), a finding consistent with the Th1-supporting role of the IL-17R signaling in fungal infections.32 Of interest, the finding that the fungal growth was also higher in IL-17R KO recipients as compared to controls indicates a role for recipient IL-17A in the control of fungal growth.
Discussion
This study has revealed a previously unappreciated role for ECs in aspergillosis, namely, the contribution of ECs to protective immunity to Aspergillus through a TLR3/TRIF-dependent pathway converging on IDO. We provide the direct evidence that ECs must be able to induce IDO for the proper control of the infection and the associated inflammatory response. Tryptophan metabolites and IDO represent not only effector host defense pathways, but also a means of balancing the generation of Th17 and Tregs.10, 11 Tregs and IL-17-producing T cells mediate opposing responses in aspergillosis, as clearly shown in mice highly susceptible to aspergillosis where the augmentation of IL-17-driven inflammation occurred concomitantly with the reduction of anti-inflammatory Treg responses, resulting in excessive inflammation.10 A reciprocal antagonistic relationship was found between IDO and the Th17 pathway, with IDO inhibiting Th17 responses11, 12 and IL-17 inhibiting IDO.12 Indeed, IDO+ DCs provided protective tolerance to the fungus in experimental HSCT, by crucially affecting the balance between immunity and tolerance in the lung.6 These data are consistent with the role of IDO in hematology,33 its requirement for the generation of Tregs34 and for protection against graft-versus-host disease.35, 36 However, allogeneic lung transplants expressing recombinant IDO in endothelial cells and ECs dramatically reduced acute cellular rejection37 and ECs provided protection against M. tuberculosis via an IFN-γ/IDO axis culminating in the inhibition of Th17 cell responses.15 We found here that ECs contribute to the immunoregulatory function of IDO in vivo and may compensate for the lack of IDO on hematopoietic cells. This may accommodate the recent finding by Paveglio et al.19 showing the superior role of IDO+DCs in contributing to allergic airway inflammation. ECs activate IDO via TLR3, which is abundantly expressed both intracellularly and on the cell surface of ECs,38 and IFN-γ to which ECs also respond.39 The failure to activate IDO likely accounted for the high levels of inflammation seen in Trif−/− mice, as proved by the immunorestorative effects of exogenously administered kynurenines, known to induce apoptosis of IL-17A+ γδ+ cells.10 Although ECs were found to induce Treg expansion,13, 14, 40 restrained CD4+ T-cell expansion in response to Aspergillus,19 and produce IL-10 (this study), the therapeutic effects of kynurenines will undoubtedly cover IDO deficiency also in DCs. As a matter of fact, the highly polarized Th2/Th17 responses seen in Trif−/− mice are immunological features compatible with the functional program activated in lung DCs by the TRIF pathway.1 Interestingly, Poli(I:C) also has a direct costimulatory effect on IFN-γ+ γδ+ cells,41 a finding suggesting that the protective effect of the TLR3/TRIF pathway in infection could be achieved through multiple mechanisms.
The TLR3/IDO axis has been reported to act as an innate antiviral defense.42 Irrespective of whether IDO itself may contribute to the antimicrobial innate function of ECs, our data indicate an important role for the MyD88 pathway in ECs in the control of fungal growth, a finding suggesting the possible antifungal effector activity of ECs. In this regard, despite the fact that airways ECs phagocytose Aspergillus conidia,16, 17 the ability of conidia to survive in ECs17 was taken to indicate airways ECs as a possible fungus reservoir. More recently, ECs were found to respond to Aspergillus conidia via MyD88-dependent and -independent pathways18 and by upregulating antimicrobial peptides43, 44 endowed with antifungal activity.45 Thus, the production of IL-17A, known to induce antimicrobial peptides,46 is of particular interest and worth of further investigation.
Overall, the data of the present study shed light on pathways of immune resistance and tolerance to the fungus that likely take place in a hematopoietic transplantation setting. It appears that protective tolerance to the fungus is achieved through a TLR3/TRIF-dependent pathway activating Th1/Tregs via IDO expressed on both the hematopoietic/non-hematopoietic compartments. The MyD88 pathway instead likely provides antifungal resistance, i.e., the ability to restrict the fungal growth, likely through defensins and other effector mechanisms. However, the ability of mice to clear the fungus in the relative absence of the MyD88 pathway,4, 22, 23 clearly indicates redundancies and hierarchy in antifungal mechanisms of resistance. Ultimately, the finding that both Candida albicans21 and A. fumigatus, two major human fungal pathogens, exploit the TRIF-dependent pathway at the interface with the mammalian hosts, indicate that the combinatorial configuration of antifungal resistance and tolerance is an advantageous option.
Several genetic polymorphisms in pattern recognition receptors, most remarkably TLRs, have been described to influence susceptibility to aspergillosis in distinct clinical settings.47, 48, 49 In addition to the mechanistic insights regarding the TRIF/IDO function in the hematopoietic/non-hematopoietic compartments, the present study suggests the utility of the TRIF/IDO genetic screening to identify patients at high risk for aspergillosis and, whenever possible, for adequate donor selection.
Acknowledgments
We thank Cristina Massi Benedetti for digital art and editing. This work was supported by the Specific Targeted Research Project ‘Sybaris' (LSHE-CT-2006), contract number 037899 (FP7) and by the Italian Projects PRIN 2007KLCKP8_004 (to LR) and 2007XYB9T9_001 (to SB). CC and AC were financially supported by fellowships from Fundação para a Ciência e Tecnologia, Portugal (contracts SFRH/BD/65962/2009 and SFRH/BPD/46292/2008, respectively).
Footnotes
Note: Supplementary information is available on the Cellular & Molecular Immunology website(http://www.nature.com/cmi/).
Supplementary Information
References
- Bonifazi P, D'Angelo C, Zagarella S, Zelante T, Bozza S, de Luca A, et al. Intranasally delivered siRNA targeting PI3K/Akt/mTOR inflammatory pathways protects from aspergillosis. Mucosal Immunol. 2010;3:193–205. doi: 10.1038/mi.2009.130. [DOI] [PubMed] [Google Scholar]
- Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E, et al. A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood. 2003;102:3807–3814. doi: 10.1182/blood-2003-03-0748. [DOI] [PubMed] [Google Scholar]
- Romani L.Dendritic cells in Aspergillus infection and allergyIn: Latgè JP, Steinbach WJ (eds). Aspergillus and Aspergillosis. Washington, DC; ASM Press; 2008247–261. [Google Scholar]
- Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, et al. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. . J Immunol. 2004;172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- Mezger M, Kneitz S, Wozniok I, Kurzai O, Einsele H, Loeffler J. Proinflammatory response of immature human dendritic cells is mediated by dectin-1 after exposure to Aspergillus fumigatus germ tubes. J Infect Dis. 2008;197:924–931. doi: 10.1086/528694. [DOI] [PubMed] [Google Scholar]
- Romani L, Bistoni F, Perruccio K, Montagnoli C, Gaziano R, Bozza S, et al. Thymosin alpha1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood. 2006;108:2265–2274. doi: 10.1182/blood-2006-02-004762. [DOI] [PubMed] [Google Scholar]
- Geurtsvan Kessel CH, Lambrecht BN. Division of labor between dendritic cell subsets of the lung. Mucosal Immunol. 2008;1:442–450. doi: 10.1038/mi.2008.39. [DOI] [PubMed] [Google Scholar]
- Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. 2007;13:579–586. doi: 10.1038/nm1563. [DOI] [PubMed] [Google Scholar]
- Montagnoli C, Fallarino F, Gaziano R, Bozza S, Bellocchio S, Zelante T, et al. Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J Immunol. 2006;176:1712–1723. doi: 10.4049/jimmunol.176.3.1712. [DOI] [PubMed] [Google Scholar]
- Romani L, Fallarino F, de Luca A, Montagnoli C, D'Angelo C, Zelante T, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- Romani L, Zelante T, de Luca A, Fallarino F, Puccetti P. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J Immunol. 2008;180:5157–5162. doi: 10.4049/jimmunol.180.8.5157. [DOI] [PubMed] [Google Scholar]
- Zelante T, de Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–2706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- Gribar SC, Richardson WM, Sodhi CP, Hackam DJ. No longer an innocent bystander: epithelial Toll-like receptor signaling in the development of mucosal inflammation. Mol Med. 2008;14:645–659. doi: 10.2119/2008-00035.Gribar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer AK, Bartz H, Fey F, Schmidt LM, Dalpke AH. Airway epithelial cells modify immune responses by inducing an anti-inflammatory microenvironment. Eur J Immunol. 2008;38:1689–1699. doi: 10.1002/eji.200737936. [DOI] [PubMed] [Google Scholar]
- Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. . Immunity. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylnka JA, Hissen AH, Wan AN, Moore MM. Intracellular and extracellular growth of Aspergillus fumigatus. . Med Mycol. 2005;43 Suppl 1:S27–S30. doi: 10.1080/13693780400029247. [DOI] [PubMed] [Google Scholar]
- Wasylnka JA, Moore MM. Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J Cell Sci. 2003;116:1579–1587. doi: 10.1242/jcs.00329. [DOI] [PubMed] [Google Scholar]
- Balloy V, Sallenave JM, Wu Y, Touqui L, Latge JP, Si-Tahar M, et al. Aspergillus fumigatus-induced interleukin-8 synthesis by respiratory epithelial cells is controlled by the phosphatidylinositol 3-kinase, p38 MAPK, and ERK1/2 pathways and not by the Toll-like receptor-MyD88 pathway. J Biol Chem. 2008;283:30513–30521. doi: 10.1074/jbc.M803149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paveglio SA, Allard J, Foster Hodgkins SR, Ather J, Bevelander M, Mayette Campbell J, et al. Airway epithelial indoleamine 2,3-dioxygenase inhibits CD4+ T cells during Aspergillus fumigatus antigen exposure Am J Respir Cell Mol Biol 2010. in press. [DOI] [PMC free article] [PubMed]
- D'Angelo C, de Luca A, Zelante T, Bonifazi P, Moretti S, Giovannini G, et al. Exogenous pentraxin 3 restores antifungal resistance and restrains inflammation in murine chronic granulomatous disease. J Immunol. 2009;183:4609–4618. doi: 10.4049/jimmunol.0900345. [DOI] [PubMed] [Google Scholar]
- de Luca A, Montagnoli C, Zelante T, Bonifazi P, Bozza S, Moretti S, et al. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J Immunol. 2007;179:5999–6008. doi: 10.4049/jimmunol.179.9.5999. [DOI] [PubMed] [Google Scholar]
- Bretz C, Gersuk G, Knoblaugh S, Chaudhary N, Randolph-Habecker J, Hackman RC, et al. MyD88 signaling contributes to early pulmonary responses to Aspergillus fumigatus. . Infect Immun. 2008;76:952–958. doi: 10.1128/IAI.00927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, Ro G, van Epps HL, Simpson T, Leiner I, Sant'Angelo DB, et al. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity. 2006;25:665–675. doi: 10.1016/j.immuni.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Bellocchio S, Moretti S, Perruccio K, Fallarino F, Bozza S, Montagnoli C, et al. TLRs govern neutrophil activity in aspergillosis. J Immunol. 2004;173:7406–7415. doi: 10.4049/jimmunol.173.12.7406. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci USA. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner N, Swain S, McInnerney K, Han S, Harmsen AG. Type-I IFN signaling suppresses an excessive IFN-gamma response and thus prevents lung damage and chronic inflammation during pneumocystis (PC) clearance in CD4 T cell-competent mice. Am J Pathol. 2010;176:2806–2818. doi: 10.2353/ajpath.2010.091158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci USA. 2010;107:3030–3033. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeck H, Ruland J. SYK kinase signaling and the NLRP3 inflammasome in antifungal immunity. J Mol Med. 2010;88:745–752. doi: 10.1007/s00109-010-0631-4. [DOI] [PubMed] [Google Scholar]
- de Luca A, Zelante T, D'Angelo C, Zagarella S, Fallarino F, Spreca A, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010;3:361–373. doi: 10.1038/mi.2010.22. [DOI] [PubMed] [Google Scholar]
- Curti A, Trabanelli S, Salvestrini V, Baccarani M, Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood. 2009;113:2394–2401. doi: 10.1182/blood-2008-07-144485. [DOI] [PubMed] [Google Scholar]
- Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol. 2008;181:5396–5404. doi: 10.4049/jimmunol.181.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood. 2009;114:5062–5070. doi: 10.1182/blood-2009-06-227587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood. 2008;111:3257–3265. doi: 10.1182/blood-2007-06-096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu L, Fletcher BS, Visner GA. Novel action of indoleamine 2,3-dioxygenase attenuating acute lung allograft injury. Am J Respir Crit Care Med. 2006;173:566–572. doi: 10.1164/rccm.200509-1413OC. [DOI] [PubMed] [Google Scholar]
- Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by Toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, et al. Interferon-gamma inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2004;31:573–582. doi: 10.1165/rcmb.2004-0195OC. [DOI] [PubMed] [Google Scholar]
- Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–1284. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesch D, Beetz S, Oberg HH, Marget M, Krengel K, Kabelitz D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J Immunol. 2006;176:1348–1354. doi: 10.4049/jimmunol.176.3.1348. [DOI] [PubMed] [Google Scholar]
- Suh HS, Zhao ML, Rivieccio M, Choi S, Connolly E, Zhao Y, et al. Astrocyte indoleamine 2,3-dioxygenase is induced by the TLR3 ligand poly(I:C): mechanism of induction and role in antiviral response. J Virol. 2007;81:9838–9850. doi: 10.1128/JVI.00792-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseeva L, Huet D, Femenia F, Mouyna I, Abdelouahab M, Cagna A, et al. Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol . 2009;9:33. doi: 10.1186/1471-2180-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SE, Scott BL, Clement CG, Larson DT, Kontoyiannis D, Lewis RE, et al. Stimulated innate resistance of lung epithelium protects mice broadly against bacteria and fungi. Am J Respir Cell Mol Biol. 2010;42:40–50. doi: 10.1165/rcmb.2008-0260OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smet K, Contreras R. Human antimicrobial peptides: defensins, cathelicidins and histatins. Biotechnol Lett. 2005;27:1337–1347. doi: 10.1007/s10529-005-0936-5. [DOI] [PubMed] [Google Scholar]
- Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochud PY, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, et al. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med. 2008;359:1766–1777. doi: 10.1056/NEJMoa0802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A, Cunha C, Carotti A, Aloisi T, Guarrera O, Di Ianni M, et al. Polymorphisms in Toll-like receptor genes and susceptibility to infections in allogeneic stem cell transplantation. Exp Hematol. 2009;37:1022–1029. doi: 10.1016/j.exphem.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Carvalho A, Pasqualotto AC, Pitzurra L, Romani L, Denning DW, Rodrigues F. Polymorphisms in Toll-like receptor genes and susceptibility to pulmonary aspergillosis. J Infect Dis. 2008;197:618–621. doi: 10.1086/526500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.