

Abstract
Aim:
To evaluate a mixed micellar drug delivery system composed of sodium cholate and phospholipid for oral administration of silybin, a promising hepatoprotectants.
Methods:
The optimum formulation of sodium cholate/phospholipid-mixed micelles containing silybin was obtained based on the study of pseudo-ternary phase diagram. The dissolution of silybin-mixed micelles was investigated. The pharmacokinetic characteristics and bioavailability after oral administration of silybin-mixed micelles and silybin-N-methylglucamine were compared in dogs.
Results:
The mean particle size of prepared mixed micelles was 75.9±4.2 nm. The largest solubility of silybin was found to be 10.0±1.1 mg/mL in the optimum formulation of mixed micelles. The silybin-sodium cholate/phospholipid-mixed micelles showed a very slow release of silybin 17.5% (w/w) within 72 h in phosphate buffer (pH 7.4) and 15.6% (w/w) in HCl solution (pH 1.2). After oral administration to dogs, the relative bioavailability of mixed micelles versus silybin-N-methylglucamine in dogs was 252.0%.
Conclusion:
Sodium cholate/phospholipid-mixed micelles are promising carriers in orally delivery of silybin, considering their capability of enhancing bioavailability and large-scale production.
Keywords: sodium cholate, phospholipid, mixed micelles, drug delivery system, silybin, bioavailability
Introduction
The ability of bile salts to enhance the oral bioavailability of poorly water-soluble drugs has been recognized for many years1, 2, 3. In the small intestine, particularly during lipid digestion, bile salts are capable to form stable mixed micelles consisting of fatty acids such as palmitic acid and phospholipids such as lecithin. They normally enhance the transport of lipophilic drugs across biological membranes and thereby enhance oral bioavailability4, 5, 6. Moreover, such micellar systems are known to improve the solubility of extremely lipophilic drugs7, 8, 9. Therefore, bile salts micelles and derived mixed systems are intensively investigated as drug carrier systems10, 11, 12, 13, 14, 15, 16. In addition, from an economic or commercial perspective, this technique simplifies the manufacturing process and allows for large-scale production.
Silymarin is an antihepatotoxic polyphenolic substance isolated from the milk thistle plant, Silybum marianum. Derivatives of milk thistle have been used as herbal remedies for almost 200 years. Silybin, the major active component of silymarin has been widely used to maintain liver health and treat liver disorders. However, the commercial oral preparations are poorly absorbed with a low bioavailability owing to its low solubility and do not provide effective treatment17, 18. Many researches have been reported concerning preparations with the aim to enhance the bioavailability of silybin, such as lipid microspheres18, self-microemulsifying17, silybin–phospolipid complex19, nanostructured lipid carriers20 and so on. However, developing a economic, large-scale produced drug carrier for efficient solubilization and oral administration of silybin remains a challenge. As we know, there is no silybin-bile salts/phospholipids-mixed micelles have been reported.
The aim of this study was to incorporate silybin into mixed micelles containing a bile salt sodium cholate and phospholipid, and to assess its bioavailability compared with silybin-N-methylglucamine, the soluble chemical derivative of silybin.
Materials and methods
Materials
Phospholipid was purchased from Shanghai TaiWei Pharmaceutical Industry Co Ltd (China). Sodium cholate was purchased from Sinopharm Chemical Reagent Co Ltd (China). Both silybin and silybin-N-methylglucamine were purchased from Jiangsu Zhongxing Pharmaceutical Industry Co Ltd (China), purity ≥98%, α-Naphthol was purchased from Tianjin Chemical Reagent Factory (China). The other chemical reagents were of analytical grade or better.
Preparation of silybin-sodium cholate/phospholipid-mixed micelles
Silybin-loaded sodium cholate/phospholipid-mixed micelles were prepared by a film dispersion method. Silybin, sodium cholate and phospholipid were dissolved in absolute ethyl alcohol by ultrasonic method. A lipid film of drug/micelle material mixture was formed after evaporation of ethyl alcohol by rotary evaporator (Heidolph, Germany). The lipid film was then hydrated and dispersed in 20 mL phosphate buffer (0.01 mol/L, pH 7.4) to form drug-loaded sodium cholate-phospholipid-mixed micelles while stirring at room temperature. The resulting mixed micelles were stored at 4 °C. Powders were obtained by freeze-drying method.
To obtain an optimum formulation of the mixed micelles, pseudo-ternary phase diagram was constructed according to the following procedures. The phase diagram was determined at 25 °C by stepwise addition of one composition to the other compositions at fixed ratios. After each addition, the samples were stirred, homogenized and kept at 25 °C. The occurrence of phase separation was observed within a period of 48 h. The phase boundary was determined by observing the changes in the sample appearance from turbid to transparent or from transparent to turbid. All the ratios in this study were reported as weight-to-weight ratios (w/w). Through a pseudo-ternary phase diagram investigation, the optimal formulation was selected for in vitro and in vivo evaluation.
Particle size analysis
Particle size measurements were performed at 25 °C by photon correlation spectroscopy on Malvern Zetasizer Nano Instrument (Malvern Instruments, Malvern, UK). Particle size was analyzed by the dispersion technology software provided by Malvern Instruments.
In vitro release study
The in vitro release of silybin-sodium cholate/phospholipid-mixed micelles was investigated compared to raw material product of silybin-N-methylglucamine with a dialysis method in phosphate buffer (pH 7.4) and HCl solution (pH 1.2). The temperatures of the media were set at 37 °C and the rotational speed of paddle was set at 70 round per min. The dialysis bag containing the drug formulation was immersed in a 50 mL polypropylene tube containing 40 mL of the dissolution medium. After a certain interval, aliquots of 3 mL were drawn from the medium for measurement of the silybin release and placed again into the same vessel so that the liquid volume was kept constant. Silybin release was determined spectrophotometrically at 288 nm. The calibration tests made with UV absorption measurements of pure silybin at different pH values studied in this work showed no changes in the spectra of drug. Release percentages were calculated as the ratio of silybin released/total silybin. The release data were presented as means of three determinations.
Determination of silybin in dog plasma by HPLC
Silybin in dog plasma was determined by HPLC with UV detection. The HPLC system (Waters, USA) consisted of two 510 pumps, a 486 UV detector and the HT-230A column heater. The UV detection wavelength was set at 288 nm. A Nova-pak C18 reserved phase analytical column(5 μm, 3.9 mm×150 mm) was employed in all the HPLC analyses. The HPLC mobile phase contained methanol/0.05 mol/L KH2PO4 (9.9:11). The mobile phase was pumped at a flow rate of 1.0 mL/min. The column temperature was set at 38 °C.
Silybin in dog plasma was extracted by liquid-liquid extraction procedures. In a screw cap glass tube, 200 μL of plasma was added, followed by adding 20 μL of internal standard solution (α-naphthol, 10 μg/mL in methanol), 200 μL of KH2PO4 (0.2 mol/L, pH 5), 20 μL of methanol, and 5 mL of diethyl ether. The mixture was thoroughly vortex-mixed for 5 min and then centrifuged for 10 min at 3000 round per min (Clements GS 200 centrifuge). The organic layer was transferred to a clean glass culture tube, and evaporated to dryness under a nitrogen flow at 45 °C. The dried residues were reconstituted in 100 μL of mobile phase, and 20 μL were injected into the HPLC system. Because silybin is an isomeric compound, two peaks with the retention time 6.3 min and 7.2 min were detected. The sum of the area of the two peaks was calculated for analysis. The ratio of peak area of silybin over the internal standard was used for quantitative analysis. The calibration was linear over a range from 10 to 1000 ng/mL (R=0.0016C+0.028) with a correlation coefficient (r) 0.9984. The LOD of current assay based on S/N=3 was 3 ng/mL in plasma. The method recoveries were all above 80% at high, middle and low concentrations, the intra- and inter-day variation was below to 15.0% (Table 1).
Table 1. Intra-, inter-day variation, and recoveries of silybin in dog plasma. n=5.
| Concentration (μg/mL) | Intra-day RSD (%) | Inter-day RSD (%) | Recovery (mean±RSD %) |
|---|---|---|---|
| 0.5 | 3.9 | 4.3 | 98.43±2.17 |
| 2 | 1.9 | 3.4 | 102.8±3.9 |
| 20 | 4.2 | 4.3 | 101.8±2.8 |
The concentration of silybin-N-methylglucamine was determined based on silybin. The treatment of silybin-N-methylglucamine blood samples was same as silybin blood samples.
Bioavailability study
Bioavailability of silybin-sodium cholate/phospholipid-mixed micelles was compared with that of raw material of silybin-N-methylglucamine. Six male and female dogs ageing 2 years, weighing 10.1±1.9 kg were purchased from the Jiangsu University Experiment Animal Center and were randomly and equally divided into two groups. All animal experiments were evaluated and approved by the Animal Ethics Committee of Jiangsu University.
The dogs were housed individually and acclimated in the laboratory for at least 1 week prior to testing. Before experiments the animals were fasted. They had free access to food but were deprived of water for 24 h prior to drug administration. A dose of each formulation (90 mg, expressed as silybin equivalents) was administered orally to the dogs in a three-way crossover design. The washout period between two consecutive treatments was 2 weeks. After administration, blood samples (approximately 4 mL) were collected from the jugular vein into a heparinized tube at 0.25, 0.5, l, 2, 4, 6, 8, 10, 12, 16, and 24 h. Blood samples were immediately separated by centrifugation (3000 round per min for 10 min) and plasma samples were withdrawn and stored at -20 °C until analyzed.
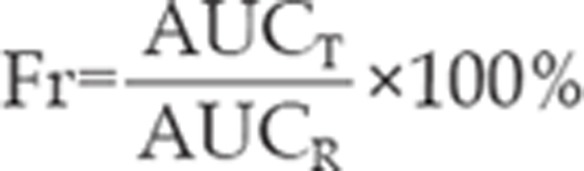 |
Peak concentration (Cmax) and peak time (Tmax) were derived directly from the experimental points. The other pharmacokinetic parameters were calculated using the program BAPP2.3 (supplied by the Center of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University) by compartmental analysis. The goodness of fit of the experimental data to the chosen compartmental model was determined by the Akaike Criterion (AIC) as well as by the correlation coefficient calculated by the program.
The relative bioavailability (Fr) is defined as ratio of AUC0–24 of silybin-sodium cholate/phospholipid-mixed micelles to the AUC0–24 of silybin-N-methylglucamine when same doses were administered and calculated using the formula, where T and R represent tested silybin-sodium cholate/phospholipid-mixed micelles and reference silybin-N-methylglucamine, respectively.
Statistical analysis
The Student's t-test was used to compare the pharmacokinetic parameters. The statistical difference was considered significant when P value<0.05.
Results
Preparation of silybin-sodium cholate/phospholipid-mixed micelles
A series of silybin-sodium cholate/phospholipid-mixed micelles were prepared and their properties were observed visually. Pseudo-ternary phase diagram was constructed to identify the drug-solubilized region and to optimize the concentration of sodium cholate, phospholipid and silybin as shown in Figure 1. The slashed areas indicated the clear drug-loaded micelles in the system. The optimal formulation of mixed micelles was selected regarding solubilization ability as described in Table 2.
Figure 1.

Pseudo-ternary phase diagram for silybin-sodium cholate/phospholipid-mixed micelles.
Table 2. Relative composition of the optimal silybin-sodium cholate/phospholipid-mixed micelles formulation.
| Drug/excipient | Composition (g) |
|---|---|
| Silybin | 0.2 |
| Sodium cholate | 0.3 |
| Phospholipid | 0.4 |
Particle size
The mean particle size of the best formulation of mixed micelles was 75.9±4.2 nm. It was found that the particle size distribution of mixed micelles was within a narrow range (Figure 2).
Figure 2.

Size and size distribution of silybin-sodium cholate/phospholipid-mixed micelles.
In vitro release
Figure 3 showed the dissolution profile of silybin from mixed micellar system and silybin-N-methylglucamine material in HCl (pH 1.2) and phosphate buffer saline (pH 7.4), respectively. Silybin release from mixed micellar system was always slower than the corresponding silybin-N-methylglucamine in both pH. In pH 7.4 phosphate buffer saline, silybin-N-methylglucamine escaped rapidly from the dialysis bag 36.3% within 12 h and the release was over 48% by 72 h. The profile of the silybin-sodium cholate/phospholipid-mixed micelles showed a very slow release of silybin 17.5% (w/w) in phosphate buffer saline and 15.6% (w/w) in HCl solution within 72 h.
Figure 3.

The dissolution profiles of silybin-sodium cholate/phospholipid-mixed micelles and reference silybin-N-methylglucamine in HCl (pH 1.2) and phosphate buffer saline (pH 7.4), respectively. (•) mixed micelles in HCl (pH 1.2); (*) mixed micelles in phosphate buffer saline (pH 7.4); (◊) silybin-N-methylglucamine in HCl (pH 1.2); (▴) silybin-N-methylglucamine in phosphate buffer saline (pH 7.4).
From Figure 3, we can know that the release profile of silybin-sodium cholate/phospholipid-mixed micelles in simulated gastric fluids (pH 1.2) was not significantly different from that in intestinal fluids (pH 7.4). But the release of silybin-N-methylglucamine was pH-dependent, the release accelerated with pH increasing.
Bioavailability
Oral bioavailability of silybin-sodium cholate/phospholipid-mixed micelles in dogs was compared with those of silybin-N-methylglucamine. The mean plasma concentration-time profiles of silybin after oral administration of silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine were shown in Figure 4. The pharmacokinetic parameters and the compartment model were analyzed by software program BAPP2.3. The results showed that both silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine plasma concentration–time curves can be fitted into the open one-compartment model. The relevant pharmacokinetic parameters for the compartmental analysis were listed in Table 3.
Figure 4.

The plasma concentration-time profiles of silybin after oral administration of silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine to dogs at a dose of 90 mg as silybin. (▴) silybin-sodium cholate/phospholipid-mixed micelles; (□) silybin-N-methylglucamine. Data are shown as mean±SE.
Table 3. The main pharmacokinetic parameters after oral administration silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine to dogs at a dose equivalent to 90 mg silybin.
| Parameters | Silybin-N-methyl-glucamine | Silybin-sodium cholate/phospholipid-mixed micelles |
|---|---|---|
| A (ng/mL) | 94.7 | 107.0 |
| Ke (h−1) | 0.189 | 0.088 |
| Ka (h−1) | 2.31 | 46.8 |
| t1/2 (h) | 3.66 | 7.85 |
| MRT (h) | 5.32 | 13.7 |
| Cmax (ng/mL) | 118.9 | 113.3 |
| Tmax (h) | 1 | 1 |
| Vc/F (mg·ng−1·mL-1) | 0.951 | 0.841 |
| AUC0–τ (h·ng/mL) | 439.7 | 1108.0 |
| AUC0–∞ (h·ng/mL) | 505.8 | 1372.4 |
Discussion
Silybin is poorly water-soluble (solubility <50 μg/mL) owing to its highly hydrophobic and nonionizable structure. A major problem in the development of an oral solid dosage form of this drug is the extremely poor aqueous solubility, possibly resulting in limited oral absorption21. In our study, silybin-mixed micelles were found to be readily soluble. The maximum solubility was found to be 10.0±1.1 mg/mL.
Normally, healthy kidneys are capable of filtering particles smaller than 10 nm (about 70 000 Da), and the liver can capture particles larger than 100 nm. Therefore, the ideal nanocarrier size is somewhere between 10 and 100 nm. The mean particle size of our prepared mixed micelles was 75.9±4.2 nm with a narrow distribution, suggesting it could be an ideal carrier from a biological point of view.
The dialysis method is the most suitable method that simulated the circumstance of drug in vivo. The release pattern of silybin-sodium cholate/phospholipid-mixed micelles in comparison with silybin-N-methylglucamine was investigated in not only simulated gastric fluids (pH 1.2), but also in intestinal fluids (pH 7.4). The drug release rate between mixed micelles and silybin-N-methylglucamine was significantly different at both pH ranges tested. The silybin-N-methylglucamine exhibited faster release than the silybin-sodium cholate/phospholipid-mixed micelles at all the pH ranges evaluated. The silybin-N-methylglucamine reached the 48% release within 72 h in pH 7.4 phosphate buffer saline. However, over eighty percent of the initially incorporated drug was still associated with the micelles even after 72 h incubation in pH 7.4 phosphate buffer saline. The silybin release behavior reflected the silybin incorporation stability in the mixed micelles. The drug incorporated into the inner core compartment stayed tightly inside the micelles. And no precipitation was observed during the dissolution tests. The results suggested that pH had little effect on the dissolution of silybin from the mixed micelles, while silybin-N-methylglucamine release markedly increased with increasing pH value. The reason may be that as an amine derivative, silybin-N-methylglucamine is unstable in low pH gastric condition, which may lead to a low bioavailability.
From Figure 4, it can be seen in dog plasma, silybin was measurable at 24 h post oral administration of silybin-sodium cholate/phospholipid-mixed micelles, but only up to 10 h after oral administration of silybin-N-methylglucamine; double peaks of maximum concentrations were observed when giving the mixed micelles, while only one peak was observed after giving silybin-N-methylglucamine. Wu et al also observed double peaks of silymarin plasma concentration-time profiles by adding bile salts to self-microemulsifying drug delivery system, this characteristic may be due to the effect of bile salt via the enterohepatic circulation22.
No significant difference was found in Cmax and Tmax values between silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine. The Cmax values were 118.9 and 113.3 ng/mL for silybin-sodium cholate/phospholipid-mixed micelles and silybin-N-methylglucamine with the same Tmax, respectively. However, their profiles were markedly different with mixed micelles exhibiting a very rapid absorption (Ka=46.8 h-1) compared to silybin-N-methylglucamine (Ka=2.31 h-1). Possible explanation for such a high absorption for silybin-sodium cholate/phospholipid-mixed micelles may be as follows: Firstly, bile salts and phospholipid in the mixed micelles may act as a enhancer for silybin transmembrane delivery, and therefore increase the absorption of silybin; Secondly, the bile salts, which has been shown to result in two peaks in blood postulated to be due to enterohepatic recycling, may be an important consideration for the interpretation of higher Ka. In addition, it is still difficult to find an appropriate pharmacokinetic model to describe the behavior of double-peak drug in vivo, the pharmacokinetic model we used in our study, may be not the best-fitting model, which caused another possibility of higher Ka values of silybin in mixed micelles.
The plasma AUC0–∞ for mixed micelles (1372.4 h·ng/mL) was approximately 2.7-fold greater than that for silybin-N-methylglucamine (505.8 h·ng/mL). The relative biolavailability of silybin-sodium cholate/phospholipid-mixed micelles was dramatically enhanced, which was 252.0% compared to silybin-N-methylglucamine. As is well known, in vitro dissolution testing is a powerful and useful method for determining product quality and sometimes to evaluate the clinical performance of dosage forms. However, in our experiments, the in vitro dissolution did not correlate well with in vivo bioavailability. Higher bioavailability but lower in vitro dissolution results were observed for silybin-sodium cholate/phospholipid-mixed micelles, while silybin-N-methylglucamine demonstrated lower bioavailability and higher in vitro dissolution. The reason for these may be that, water soluble characteristic of silybin-N-methylglucamine resulted in a faster drug release; the rapid and effective transmembrane transport of the sodium cholate and phospholipid in the mixed micelles leaded to a much higher bioavailability, on the contrary, silybin-N-methylglucamine without sodium cholate and phospholipids acting can not be absorbed effectively in vivo.
In conclusion, after oral administration to dogs, the silybin in sodium cholate/phospholipid-mixed micelles could provide the desired character for higher bioavailability and consequently lower dose. In addition, the mixed micelles should be particularly suitable for large-scale production due to its lower cost and easier scale-up. These results suggest that the mixed micelles may serve as suitable oral dosage form for poorly soluble drugs.
Author contribution
Jiang-nan YU and Xi-ming XU designed the research; Yuan ZHU, Min PENG, Shan-shan TONG, Xia CAO, and Hui QIU performed research; Xi-ming XU, Yuan ZHU, and Li WANG analyzed data; Li WANG wrote the paper.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (Grant No 30472098) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
References
- Sallee VL, Dietschy JM. Determinants of intestinal mucosal uptake of medium-chain fatty acids and alcohols. J Lipid Res. 1973;14:475–84. [PubMed] [Google Scholar]
- Westergaard H, Dietschy JM. The mechanism whereby bile acid micelles increase the rate of fatty acid and cholesterol uptake into the intestinal mucosal cell. J Clin Invest. 1976;58:97–108. doi: 10.1172/JCI108465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson FA. Intestinal transport of bile acids. Am J Physiol. 1981;241:G83–G92. doi: 10.1152/ajpgi.1981.241.2.G83. [DOI] [PubMed] [Google Scholar]
- O'Reilly JR, Corrigan OI, O'Driscoll CM. The effect of mixed micellar systems, bile acid/fatty acids, on the solubility and intestinal absorption of clofazimine (B663) in the anesthetized rat. Int J Pharm. 1994;109:147–94. [Google Scholar]
- Garidel P, Hildebrand A, Blume A. Membranolytic activity of bile salts: Influence of biological membrane properties and composition. Molecules. 2007;12:2292–326. doi: 10.3390/12102292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangi JS, Vyas SP, Dixit VK. Effect of various lipid-bile salt mixed micelles on transfer of amphotericin-B across the everted rat intestine. Drug Dev Ind Pharm. 1995;21:2021–7. doi: 10.3109/03639049809082364. [DOI] [PubMed] [Google Scholar]
- Wiedmann TS, Kamel L. Examination of the solubilization of drugs by bile salt micelles. J Pharm Sci. 2002;91:1743–64. doi: 10.1002/jps.10158. [DOI] [PubMed] [Google Scholar]
- Wiedmann TS, Liang W, Kamel L. Solubilization of drugs by physiological mixtures of bile salts. Pharm Res. 2002;19:1203–8. doi: 10.1023/a:1019858428449. [DOI] [PubMed] [Google Scholar]
- Hammad MA, Müller BW. Increasing drug solubility by means of bile salt-phosphatidylcholine-based mixed micelles. Eur J Pharm Biopharm. 1998;46:361–7. doi: 10.1016/s0939-6411(98)00037-x. [DOI] [PubMed] [Google Scholar]
- Dongowski G, Fritzsch B, Giessler J, Härtl A, Kuhlmann O, Neubert RHH. The influence of bile salts and mixed micelles on the pharmacokinetics of quinine in rabbits. Eur J Pharm Biopharm. 2005;60:147–51. doi: 10.1016/j.ejpb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Hammad MA, Müller BW. Solubility and stability of lorazepam in bile salt/soya phosphatidylcholine-mixed micelles. Drug Dev Ind Pharm. 1999;25:409–17. doi: 10.1081/ddc-100102190. [DOI] [PubMed] [Google Scholar]
- Guo J, Wu T, Ping Q, Chen Y, Shen J, Jiang G. Solubilization and pharmacokinetic behaviors of sodium cholate/lecithin-mixed micelles containing cyclosporine A. Drug Deliv. 2005;12:35–9. doi: 10.1080/10717540590889691. [DOI] [PubMed] [Google Scholar]
- Risovic V, Boyd M, Choo E, Wasan KM. Effects of lipid-based oral formulations on plasma and tissue amphotericin B concentrations and renal toxicity in male rats. Antimicrob Agents Chemother. 2008;47:3339–42. doi: 10.1128/AAC.47.10.3339-3342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamulitrat W, Burhenne J, Stremmel W. Cytoprotective functions of a bile salt-phospholipid conjugate lysophosphatidyl-ethanolamino-ursodeoxycholate: implications for treatment of liver diseases. J Hepatol. 2007;46:S287. [Google Scholar]
- Chamulitrat W, Burhenne J, Rehlen T, Pathil-Warth A, Stremmel W. A novel bile salt-phospholipid conjugate ursodeoxycholyl-lysophosphatidylethanolamide as a hepatic cytoprotective agent. J Hepatol. 2008;48:S59. doi: 10.1002/hep.22955. [DOI] [PubMed] [Google Scholar]
- Chamulitrat W, Burhenne J, Rehlen T, Pathil A, Stremmel W. Bile salt-phospholipid conjugate ursodeoxycholyl lysophosphatidylethanolamide as a hepatoprotective agent. Hepatology. 2009;50:143–54. doi: 10.1002/hep.22955. [DOI] [PubMed] [Google Scholar]
- Li X, Pei Y, Huang Y, Zhou Y, Zhang Y, Liu Y. In vitro and in vivo evaluation of a selfmicroemulsifying drug delivery system for silybin. J Chin Pharm Sci. 2009;18:342–7. [Google Scholar]
- Liu X, Sun Y, Zhang Y, Tang X. Preparation and in vitro-in vivo evaluation of silybin lipid microspheres. Asian J Pharm Sci. 2007;2:204–10. [Google Scholar]
- Xiao Y, Song Y, Chen Z, Ping Q. The preparation of silybin-phospholipid complex and the study on its pharmacokinetics in rats. Int J Pharm. 2006;307:77–82. doi: 10.1016/j.ijpharm.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Jia LJ, Zhang DR, Li ZY, Feng FF, Wang YC, Dai WT, et al. Preparation and characterization of silybin-loaded nanostructured lipid carriers. Drug Deliv. 2010;17:11–8. doi: 10.3109/10717540903431586. [DOI] [PubMed] [Google Scholar]
- Li F, Hu J, Jiang Y. Preparation and characterization of solid dispersions of silymarin with polyethylene glycol 6000. J Chin Pharm Sci. 2003;12:76–81. [Google Scholar]
- Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63:288–94. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]