

Abstract
Until a few years ago, celiac disease (CD) was thought to be a rare food intolerance that was confined to childhood and characterized by severe malabsorption and flat intestinal mucosa. Currently, CD is regarded as an autoimmune disorder that is common in the general population (affecting 1 in 100 individuals), with possible onset at any age and with many possible presentations. The identification of CD is challenging because it can begin not only with diarrhea and weight loss but also with atypical gastrointestinal (constipation and recurrent abdominal pain) and extra-intestinal symptoms (anemia, raised transaminases, osteoporosis, recurrent miscarriages, aphthous stomatitis and associated autoimmune disorders), or it could be completely symptomless. Over the last 20 years, the diagnostic accuracy of serology for CD has progressively increased with the development of highly reliable tests, such as the detection of IgA tissue transglutaminase and antiendomysial and IgG antideamidated gliadin peptide antibodies. The routine use of antibody markers has allowed researchers to discover a very high number of ‘borderline' cases, characterized by positive serology and mild intestinal lesions or normal small intestine architecture, which can be classified as potential CD. Therefore, it is evident that the ‘old celiac disease' with flat mucosa is only a part of the spectrum of CD. It is possible that serology could identify CD in its early stages, before the appearance of severe intestinal damage. In cases with a positive serology but with mild or absent intestinal lesions, the detection of HLA-DQ2 and HLA-DQ8 can help reinforce or exclude the diagnosis of gluten sensitivity.
Keywords: celiac disease; clinical presentation; diagnostic criteria; histology, genetics; serology
Introduction
In recent years, our understanding of celiac disease (CD) has been rapidly growing because of significant advances in knowledge about its pathogenic, epidemiological, clinical and diagnostic aspects.1, 2 CD, also known as celiac sprue or gluten-sensitive enteropathy, can be defined as a permanent intolerance to wheat gliadins and other cereal prolamins in the small bowel mucosa in genetically susceptible individuals. The main expression of the disorder is characteristic, though not specific, small intestine lesions that impair nutrient absorption and improve upon withdrawal of the responsible cereals. It is generally acknowledged that wheat (gliadins), rye (secalins) and barley prolamins (hordeins) are toxic for the intestinal mucosa of celiac patients due to their high glutamine (>30%) and proline (>15%) content, whereas prolamins of rice and maize are nontoxic due to a lower content of these two amino acids.3 Other studies have shown that oat prolamins (avenins), which have an intermediate glutamine and proline composition, are non-toxic, and only ingestion of a large amount of this cereal can provoke damage in the small bowel of celiac patients.4, 5
The genetic susceptibility to CD is confirmed by its high familial incidence (about 10% of first degree relatives of celiac patients are affected by the disease) and by its strict linkage with some human leukocyte antigen (HLA) class 2 alleles (up to 95% celiacs are HLA-DQ2 positive with the typical heterodimer DQA1*0501/DQB1*0201, whereas the remaining 5% are HLA-DQ8 positive HLA-DQB1*0302).6, 7 Because about 20–30% of healthy people in Western countries display the same HLA pattern, the presence of HLA-DQ2 and/or HLA-DQ8 must be considered an indispensable prerequisite for the disease but cannot alone justify the development of CD. Therefore, non-HLA genetic factors are likely to be required for the development of disease. An alteration in the terminal portion of chromosome 5 has been identified as a risk factor for both symptomatic and silent forms of CD, whereas a change in the terminal portion of the chromosome 11 possibly differentiates the two forms.8 Among the candidate genes, there is also a CD28/CTLA4 region on chromosome 2 that encodes receptors regulating T-lymphocyte activation.9 Recently, multiple genetic loci have been implicated in CD pathogenesis, but their impact on the development of the disease seems to be limited as compared to that of the HLA system.10
T-cell-mediated immune response plays a crucial role in the pathogenesis of the disease.11 There is strong evidence that the mucosal lesions of gluten-sensitive enteropathy are initiated within the lamina propria and are due to major histocompatibility complex class 2-expressing activated macrophages that present gliadin peptides to α/β T-cell receptor CD4+ lymphocytes. This leads to increased production of cytokines (e.g., interleukin-15, interferon-γ and tumor necrosis factor-α), which recruit nonspecific effector cells responsible for initial tissue damage. The identification of tissue transglutaminase (tTG or TG2) as the predominant autoantigen of CD has allowed researchers to gain new insights into the pathogenesis of this disorder;12 tTG belongs to a family of cytoplasmic calcium-dependent enzymes found in endothelial cells, erythrocytes, hepatocytes, lamina propria and small intestine epithelial cells. Damage to, or hyperpermeability of, the small intestine mucosa, caused by either toxic gluten fractions or other irritants, triggers the abundant extracellular release of cytosolic tTG. The following step is the crosslinking between the released tTG and gliadin, an excellent substrate for tTG, resulting in gliadin-tTG complexes and the creation of antigenic neoepitopes. Moreover, tTG selectively deamidates gliadin peptides, leading to a strongly enhanced T-cell-stimulatory activity.13
Epidemiological studies, performed by accurate serological screening in the general population, have radically changed our knowledge about CD prevalence, showing that the disease occurs worldwide much more frequently than previously thought (Table 1).14, 15, 16, 17, 18, 19, 20, 21 The highest reported prevalence is in Europe: 1 in 99 in Finland, 1 in 122 in Northern Ireland and 1 in 175 people in Italy.16, 20, 21 Until a few years ago, gluten-sensitive enteropathy was erroneously considered uncommon in the United States, but serological screening in healthy blood donors has revealed a prevalence approaching that of Europe (1 in 250).22 Whereas CD is well documented in Asians from India and Pakistan,23 it is rare among native Japanese and Chinese. The amazingly high CD prevalence in children of the Sahara (1 in 18) can be partially explained by the high amount of cereal (couscous) in the diet and by the particular genetic background of this population (i.e., high prevalence of HLA-DQ2/DQ8).24
Table 1. Epidemiology of celiac disease: serological screening in the general population confirmed by duodenal histology.
| Reference | Antibody testing | No. of cases | Age (years) | Celiac disease prevalence |
|---|---|---|---|---|
| Pittschieler KActa Paediatr 1996 | AGA | 4615 | 18–82 | 1∶513(1.95‰) |
| Corazza GRScand J Gastroenterol 1997 | EmA | 2237 | 20–87 | 1∶559(1.8‰) |
| Johnston SDLancet 1997 | EmA | 1823 | NR | 1∶122(8.0‰) |
| Kolho KLScand J Gastroenterol 1998 | EmA | 1070 | NR | 1∶134(7.5‰) |
| Ivarsson AJ Intern Med 1999 | EmA | 1894 | 25–74 | 1∶189(5.3‰) |
| Riestra SScand J Gastroenterol 2000 | EmA | 1170 | 2–89 | 1∶389(2.6‰) |
| Volta UDig Dis Sci 2001 | EmA | 3483 | 12–65 | 1∶175(0.57%) |
| Maki MN Engl J Med 2003 | tTGA | 3654 | 7–16 | 1∶99a(1%) |
Abbreviations: AGA, antigliadin antibodies; EmA: antiendomysial antibodies; NR, not reported; tTGA, antibodies to tissue transglutaminase.
1∶67 with positive serology, but without serological confirmation.
CD can manifest in any age group, from infants to the elderly. Retrospective analysis of clinical data shows that most adult celiacs had no sign of the disease during their childhood, thereby confirming that CD can develop in adulthood.25, 26, 27, 28 About 20% of diagnoses occur in people over 60 years of age.29 CD prevalence is higher in females than in males worldwide, with a mean F/M ratio of 2∶1.30
The clinical presentation of CD can be misleading because the symptoms vary tremendously from patient to patient and appear to depend largely on the length and the severity of small intestine damage (Table 2).31 Because of the heterogeneity among clinical signs and the lack of specificity of many presenting symptoms, the clinical diagnosis of CD is a challenge even for experts. Depending on the clinical, histopathological and immunological features, CD can be classified into the following forms: classical (typical), subclinical (atypical or mono-symptomatic), silent (asymptomatic) and potential/latent.32
Table 2. Presenting symptoms of celiac disease.
| General | Weakness, lassitude, malaise, weight loss, short stature |
|---|---|
| Gastrointestinal | Diarrhea, anorexia, nausea and vomiting, flatulence and abdominal distension, abdominal pain, constipation, motility disturbance, glossitis/aphthous ulcers |
| Metabolic | Anemia features, bleeding tendency, edema, cramps/tetany, dental enamel hypoplasia |
| Musculoskeletal | Bone pain and fractures, myopathy |
| Neuropsychiatric | Depression, anxiety, paraesthesia, peripheral neuropathy, cerebrospinal degeneration |
| Reproductive | Menstrual irregularities, recurrent miscarriages, abnormalities of sperm morphology and motility |
| Skin | Variety of rashes, petechiae |
The classical form is characterized by the typical malabsorption syndrome with diarrhea, flatulence, weight loss, fatigue, vomiting, abdominal pain and expression of severe intestinal damage affecting a large tract of small bowel. Thanks to the increased frequency of early CD diagnosis, this form is less and less often observed; however, some patients can still present with wasting cachexia characterized by muscle atrophy and cramps, severe hypoalbuminemia and electrolyte and metabolic disturbances, leading to hypocalcemic tetany and spontaneous bone fractures from marked osteoporosis.27, 28, 33
In the subclinical form, gastrointestinal manifestations can be mild or absent and are often completely overshadowed by extra-intestinal symptoms, especially in those patients with a mild mucosal lesion confined to the proximal small intestine. Patients in this group have an isolated iron-deficient anemia,34 unexplained folic acid deficiency and a hemorrhagic syndrome caused by vitamin K deficiency.35 Failure to thrive is an important sign of CD in childhood, and short stature is a possible sign of CD in adults.36 Dental enamel defects and aphthous stomatitis can suggest subclinical CD.37, 38 Sometimes, CD patients have a gluten-sensitive dysmotility involving the whole gastrointestinal tract, and the clinical expression may be dyspepsia, reflux, dysphagia or, more frequently, severe constipation.39 Irritable bowel syndrome, common in the general population, can hide a condition of gluten-sensitive enteropathy.40 Atypical CD can also present with only psychiatric and neurological symptoms. In the former group, depression and anxiety have been reported,41 whereas in the latter there are a number of idiopathic neurological disorders, such as epilepsy with and without parieto-occipital calcifications, cerebellar ataxia, intellectual deterioration with attention/memory impairment, brain atrophy, peripheral neuropathy, multiplex myoclonus (Friedreich's disease) and multiple sclerosis.42, 43, 44, 45, 46 Reproductive system dysfunction may be a sign of CD in both sexes. In females, there is a trend to late menarche, amenorrhea, early menopause and infertility, which respond positively to gluten withdrawal.47 Moreover, there is also a greater frequency of recurrent miscarriages (mainly in the first 3 months of pregnancy), stillbirth and neonatal death.48 In males, impotence, decreased sexual activity, infertility, abnormalities of sperm morphology and motility may be expressions of CD.49, 50 Another presentation of atypical CD is so-called celiac hepatitis, characterized by a gluten-dependent increase in serum transaminases without abnormality of any other liver functions.51 Both the serum transaminases and mild liver histological damage revert to normal after removal of gluten. Several disorders are associated with gluten-sensitive enteropathy.52, 53 The most well-known association is with dermatitis herpetiformis, a skin disease characterized by intensely pruritic papulovescicular lesions that occur symmetrically on the extensor surface of arms and legs and on the buttocks, trunk, neck and scalp.54 All patients with dermatitis herpetiformis show gluten-dependent intestinal damage, which is indistinguishable from that of CD because it can present with mild clinical CD symptoms or with no symptoms at all. The withdrawal of gluten reverses not only the intestinal, but also the skin lesions in most dermatitis herpetiformis patients. Many other pathological conditions, such as autoimmune disorders (insulin-dependent diabetes mellitus type 1, thyroid disorders, autoimmune hepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, alopecia, vitiligo, Addison's disease, Sjögren's syndrome, IgA nephropathy and IgA deficiency),55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65 idiopathic disorders (primitive dilated cardiomyopathy, atopy and inflammatory bowel disease)66, 67, 68 and chromosome disorders (Down, Turner and Williams syndromes),69, 70, 71 display a strong association with CD. The importance of diagnosing the subclinical form of CD associated with these disorders is twofold because a gluten-free diet not only prevents the clinical manifestations and complications of CD but also sometimes improves clinical symptoms of the associated disorders (Table 3).
Table 3. Disorders associated with celiac disease.
| Autoimmune | Idiopathic | Chromosomal | Miscellaneous |
|---|---|---|---|
| T1DM | Dilated cardiomyopathy | Down syndrome | Female and male infertility |
| Hashimoto thyroiditis | Epilepsy with or without occipital calcifications | Turner syndrome | Depression |
| Graves' disease | Cerebellar ataxia | Williams syndrome | Psychiatric diseases |
| Autoimmune hepatitis | Peripheral neuropathy | Aphthous stomatitis | |
| Primary biliary cirrhosis | Multiple myoclonus | ||
| Primary sclerosing cholangitis | Multiple sclerosis | ||
| Alopecia | Brain atrophy | ||
| Vitiligo | Inflammatory bowel disease | ||
| Psoriasis | Sarcoidosis | ||
| Dermatitis herpetiformis | Atopy | ||
| IgA deficiency | |||
| Autoimmune atrophic gastritis | |||
| Autoimmune hemolytic disease | |||
| Sjögren's syndrome | |||
| Myasthenia gravis | |||
| Addison's disease | |||
| IgA nephropathy |
Abbreviation: T1DM, diabetes mellitus type 1.
The silent form is diagnosed in patients who do not complain of any symptoms. Examples of patients with this condition include first-degree relatives of celiac patients72 and subjects of the general population who are identified through serological screening.20 It is common to find mild histological lesions usually confined to the proximal tract of the small intestine mucosa in these patients.
The potential form is a more and more frequently observed condition characterized by a normal villous architecture, but with some histological and immunologic features suggestive of the future development of CD. These features include an increased number of intraepithelial lymphocytes (IELs) (with an overexpression of γ/δ T-cell receptor lymphocytes); the presence of IgA and IgM gliadin antibodies (AGA) in the intestinal juices; and the presence of serum IgA antiendomysial antibodies (EmA), usually at low titer (<1∶40).73, 74 The potential form of the disease is considered the first step toward the flat mucosa characteristic of CD, and when the disease develops with the typical atrophy of small intestine mucosa, the condition is renamed ‘latent CD'. Potential celiacs are frequently asymptomatic or may suffer from mild intestinal symptoms, such as abdominal pain, resembling those of irritable bowel syndrome.
Diagnostic criteria
Small intestine biopsy
A long time ago, when CD was recognized as a new disease, its diagnosis was exclusively based on the finding of villous atrophy during a small intestine biopsy.75 The most relevant feature of the disease was histological change, and histology became the gold standard for diagnosis. Despite substantial changes in the mode of presentation and the availability of new diagnostic tools, small bowel mucosal biopsy has remained the gold standard for CD diagnosis until now. In recent years, a progressive decline in the use of this diagnostic tool has been evident. First, standardized technical procedures, often neglected in many medical centers, have been identified; with regard to small intestine histology, researchers have learned that when diagnosing CD, patchy and irregular mucosal lesions are just as relevant as continuous lesions.76 Therefore, at least four biopsy samples, i.e., two from the duodenal bulb and two from the second third of the duodenum, should be taken. It is also essential that biopsy samples are correctly oriented to avoid tangential artifacts and spurious shortening or absence of villi.77 Orientation is important not only for the evaluation of atrophic lesions but also for correct interpretation of minimal changes in small intestine mucosa. A well-oriented biopsy allows for a good evaluation of villi/crypt ratio (≥3∶1 in mucosa with normal architecture) and, above all, an accurate count of IELs, which is difficult to obtain with transverse sections and/or convoluted villi. Provided that the orientation is correct, the pathological interpretation of an intestinal biopsy is a major pitfall in CD diagnosis, particularly in the presence of mild intestinal lesions.78 Agreement between six different pathologists has been reported for the most extreme cases, such as normal mucosa and subtotal villous atrophy, whereas agreement was poor for cases of mild intestinal lesions (increased IELs and crypt hyperplasia), confirming the difficulty of getting a reproducible evaluation of non-atrophic lesions.79 When an increase in IELs is suspected as the sole marker of intestinal mucosal damage, CD3 staining by immunohistochemistry is a mandatory adjunctive technique needed to count them.80
It must also be emphasized that non-atrophic lesions of small intestine mucosa (Fig. 1a–b), characterized by an isolated increase in the number of IELs with or without crypt hyperplasia, indicate CD in only 10% of cases because many other conditions can be responsible for an increased number of IELs in the intestinal mucosa, including the following: food allergy, gastrointestinal infection (including Helicobacter pylori infection), Crohn's disease, ulcerative colitis, common variable immunodeficiency and autoimmune disorders.81 Therefore, pathologists should avoid overdiagnosing CD based only on an increased number of IELs. Immunohistochemical characterization of lymphocyte populations in the intraepithelial compartment using frozen sections of a duodenal biopsy may be useful for identifying gluten-sensitive patients among those with nonatrophic lesions. A high density of T cells with γ/δ receptors in the surface epithelium is a characteristic feature of gluten sensitivity. The mean proportion of γ/δ T-cells to IELs in gluten-dependent non-atrophic lesions ranges from 20% to 30%, but when non-atrophic lesions are not dependent on gluten, the proportion of γ/δ+T cells is about 2–3%. However, this technique has limited diagnostic utility due to the unavailability of formalin-fixed, paraffin-embedded tissue.82
Figure 1.

Different grades of small intestinal damage in coeliac disease patients: a-b normal villi and pathological increase of intraepithelial lymphocytes (IELs) (a- haematoxilin/eosin, b CD3 staining), c-d mild/moderate atrophy of villi and pathological increase of IELs (c- haematoxilin/eosin, d staining), e-f total villous atrophy and pathological increase of IELs (e- haematoxilin/eosin, f CD3 staining). Magnification x20.
The predictive value of histology is much greater in the presence of villous atrophy (mild, partial or subtotal) (Figure 1c–d–e–f). However, it should be remembered that other disorders, including common variable immunodeficiency, autoimmune enteropathy, Whipple disease and eosinophilic gastroenteritis, can cause nongluten-dependent villous atrophy.83
With its low degree of specificity, histology fails to be the gold standard of CD diagnosis, and it is time to recognize that for the majority of gluten-sensitive cases, histology alone cannot provide the diagnosis. Histology remains an important element in CD diagnosis, but pathological findings must be evaluated in the context of other relevant components, including clinical signs, serological markers and HLA haplotypes.84
Serological tests
Ideas about CD have progressed, and currently there is a general consensus that it is a heterogeneous autoimmune disorder, the diagnosis of which relies not only on histological findings but also on increasingly important serological and genetic tests.
Serology has become increasingly relevant to CD diagnosis.85 The availability of immunologic tests that confirm symptoms to be strictly related to food intolerance has definitely changed the diagnostic algorithm for CD. CD-related antibodies have allowed researchers to confirm a great number of borderline cases in patients with mild intestinal lesions and positive serology, diagnosing the so-called potential CD. It is likely that serology could identify CD in its early stages, before the appearance of a severe intestinal damage. At present, the most widely used approach is to perform an intestinal biopsy, independent of serology results, only in patients with severe malabsorption; in individuals at risk for CD (those with first-degree relatives, iron-deficient anemics, and patients with unexplained osteoporosis, cryptogenic hypertransaminasemia, CD-associated autoimmune disorders, etc.), gluten-sensitive enteropathy can be excluded by the absence of CD-related antibodies, and only those with a positive serology test should undergo intestinal biopsy.
There is a general agreement that the best strategy for CD serological screening is the detection of IgA tissue transglutaminase antibodies (tTGA).86 These antibodies are the most sensitive test for CD (up to 97%), whereas IgA EmA are employed as a confirmatory test in tTGA-positive cases due to their higher specificity (about 100% versus 91% of tTGA) (Table 4). ‘False positives' for tTGA have been observed in patients with inflammatory bowel disease, food allergy, irritable bowel syndrome, giardiasis, other intestinal infections and autoimmune disorders. These ‘false positives' cannot always be resolved by EmA because the results of EmA testing are reliable only in laboratories skilled in immunofluorescent assays.87 The recognition of an EmA-positive pattern is sometimes difficult due to their resemblance to antismooth muscle antibodies.
Table 4. Performance of serological markers for CD diagnosis.
| Sensitivity(%) | Specificity(%) | PPV(%) | NPV(%) | Diagnostic accuracy(%) | |
|---|---|---|---|---|---|
| IgA tTGA | 97 | 91 | 91 | 97 | 98 |
| IgA EmA | 94 | 100 | 100 | 94 | 97 |
| IgA AGA | 73 | 87 | 84 | 77 | 80 |
| IgG AGA | 73 | 77 | 75 | 75 | 75 |
| IgA DGP-AGA | 84 | 90 | 89 | 85 | 87 |
| IgG DGP-AGA | 84 | 99 | 98 | 87 | 92 |
Abbreviations: AGA, antigliadin antibodies; CD, celiac disease; DGP-AGA, antibodies to deamidated gliadin peptides; EmA, antiendomysial antibodies; NPV, negative predictive value; PPV, positive predictive value; tTGA, antibodies to tissue transglutaminase.
IgA AGA is an obsolete test with low sensitivity and specificity; therefore, the search for these antibodies should be abandoned, except in very young children (under 2 years of age).88 IgG tTGA should be used only for detecting CD in patients with IgA deficiency, a condition strictly related to CD.89
A new antibody test has been introduced into the serological work-up of CD. This test consists of antibodies binding to deamidated gliadin peptides (DGP-AGA) (Table 4). Both IgG and IgA DGP-AGA show a lower sensitivity for CD than IgA tTGA, but IgG DGP-AGA displays a very high specificity for CD (higher than tTGA and similar to EmA); moreover, these antibodies allow for the identification of all CD cases in IgA-deficient patients with a very high sensitivity in young children (aged less than 2 years). In light of these promising results, it is possible to hypothesize a new serological protocol for CD that uses only two tests—IgA tTGA and IgG DGP-AGA—instead of the current four tests—IgA tTGA, IgA EmA, IgA AGA and IgG tTGA. This new procedure would reduce the number of tests required, thus providing an obvious advantage in terms of cost-efficiency (Figure 2).90 Recent data have shown that IgA tTGA can be present in the intestine before the serum, thus predicting a forthcoming CD.91 These antibodies, detected by direct immunofluorescence on frozen sections of duodenal biopsies, display a high sensitivity for potential CD (78–100%), but their specificity is low because they are also present in a high proportion of patients with autoimmune disorders (diabetes mellitus type 1) or inflammatory bowel disease and in some controls.92 After improving their specificity, intestinal tTGA of the IgA class could be the best option for classifying non-atrophic intestinal lesions as gluten-dependent.
Figure 2.
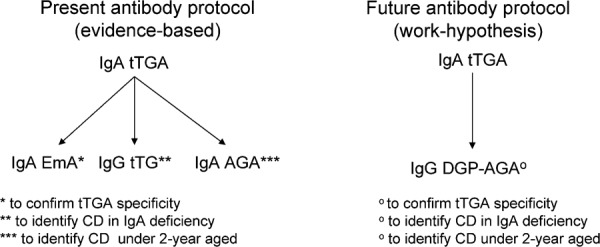
Comparison between the present and the new serological strategy for coeliac disease diagnosis. The combined search for IgG DGP-AGA to IgA tTGA in the diagnostic work-up of coeliac disease makes unuseful the detection of IgA EmA, IgG tTGA and IgA AGA. Abbreviations: tTGA, antibodies to tissue transglutaminase, EmA, antiendomysial antibodies, DGP-AGA, antideamidated gliadin peptide antibodies, AGA, antigliadin antibodies, CD, celiac disease.
Genetics
The puzzle of diagnosing gluten sensitivity includes another relevant element, genetic testing. As generally acknowledged, CD is closely related to a well-defined HLA pattern, characterized by the presence of HLA-DQ2 and/or HLA-DQ8.6, 7, 8 HLA testing should not be routinely performed in all CD cases, but it is strongly indicated when CD diagnosis is controversial (because of a discrepancy between histology and serology results) and in first-degree relatives of CD patients. A positive test alone (finding HLA-DQ2-DQA1*05, DQB1*02- and HLA-DQ8-DQB1*0302) is never diagnostic for CD because about 30% of the general population displays the same HLA pattern as CD patients. The most important clinical impact of the test is when the result is negative, because the absence of HLA-DQ2, HLA-DQ8 and DQB1*02 excludes a diagnosis of CD (negative predictive value 100%). In patients with potential CD (positive serology with mild or absent histological lesions), HLA genotyping is useful to reinforce (when positive) or discount (when negative) the presence of the condition. In patients with villous atrophy and negative serology, HLA negativity should alert us to search for another cause of flat mucosa (common variable immunodeficiency, autoimmune enteropathy, Whipple disease, etc.). In patients with positivity for IgA tTGA at low titer without IgA EmA and IgG DGP-AGA a negative HLA test gives evidence that IgA tTGA readings are ‘false positives'.
Conclusions
There is no doubt that our understanding of CD has changed in its clinical, histological and autoimmune features, so that it seems to be a new disease as compared to that described in medical books from only 10 or 20 years ago. Therefore, it is time to change the historical dogma that uses histology as the gold standard for the detection of CD. Today, in light of the current knowledge and emerging complex clinical problems, we propose that the true gold standard for the final diagnosis of CD is the decision made by the clinician. The clinician is the only one who knows the patient and establishes the tests and their timing and is, consequently, the only one who can correctly interpret the panel of available data (clinical, serological, histological and genetic). The role of the pathologist is not diminished because he is always an important contributor in CD diagnosis. An accurate assessment of the morphology of the duodenal mucosa, while avoiding any clinical conclusions (which are often misleading), remains crucial for the final diagnosis of CD. A multidisciplinary team coordinated by the clinician, including specialists, pathologists and laboratory technicians, can pave the way for improving the quality of CD diagnosis by compiling all of the pieces needed to solve the CD puzzle.
A small intestine biopsy is still necessary for CD diagnosis, but it must be emphasized that with the improved diagnostic accuracy of serology and genetics, there is a trend toward non-invasive diagnosis of CD. The European Society of Paediatric Gastroenterology Hepatology and Nutrition has recently proposed new criteria for CD diagnosis, suggesting that in children with severe malabsorption, CD can be diagnosed on the basis of a positive serological test and the presence of a typical genetic pattern (DQ2 and/or DQ8), without the need for a small intestine biopsy. This is an early attempt to avoid duodenal biopsy for CD diagnosis, indicating that the diagnostic criteria for CD might change radically in the next few years.
References
- Di Sabatino A, Corazza GR. Coeliac disease. Lancet. 2009;373:1480–1493. doi: 10.1016/S0140-6736(09)60254-3. [DOI] [PubMed] [Google Scholar]
- Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234–242. doi: 10.1053/gast.2000.8521. [DOI] [PubMed] [Google Scholar]
- Shewry PR, Tatham AS, Kasarda DD.Cereal proteins and coeliac diseaseIn: Marsh MN (ed.)Coeliac Disease. Oxford; Blackwell Scientific; 1992305–348. [Google Scholar]
- Janatuinen EK, Pikkarainen PH, Kemppainen TA, Kosma VM, Jarvinen RM, Uusitupa MI, et al. A comparison of diets with and without oats in adults with coeliac disease. N Engl J Med. 1995;333:1033–1037. doi: 10.1056/NEJM199510193331602. [DOI] [PubMed] [Google Scholar]
- Hardman CM, Garioch JJ, Leonard JN, Thomas HJ, Walker MM, Lortan JE, et al. Absence of toxicity of oats in patients with dermatitis herpetiformis. N Engl J Med. 1997;337:1884–1887. doi: 10.1056/NEJM199712253372604. [DOI] [PubMed] [Google Scholar]
- Sollid L, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105:910–922. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- Partanen J.Major histocompatibility complex and coeliac diseaseIn: Maki M, Collin P, Visakorpi JK (eds.)Coeliac Disease. Tampere; University of Tampere; 1997253–264. [Google Scholar]
- Greco L, Corazza G, Babron MC, Clot F, Fulchignoni-Lataud MC, Percopo S, et al. Genome search in celiac disease. Hum Genet. 1998;62:669–675. doi: 10.1086/301754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holopainen P, Arvas M, Sistonen P, Mustalahti K, Collin P, Maki M, et al. CD28/CTLA4 gene region on chromosome 2q33 confers genetic susceptibility to celiac disease. A linkage and family-based association study. Tissue Antigens. 1999;53:470–475. doi: 10.1034/j.1399-0039.1999.530503.x. [DOI] [PubMed] [Google Scholar]
- Dubois CA, Trynka G, Franke L, Hunt CA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–303. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godkin A, Jewell D. The pathogenesis of celiac disease. Gastroenterology. 1998;115:206–210. doi: 10.1016/s0016-5085(98)70382-8. [DOI] [PubMed] [Google Scholar]
- Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken E, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- Molberg O, McAdam SM, Korner M, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- Pittschieler K, Ladinser B. Coeliac disease screened by a new strategy. Acta Paediatr Suppl. 1996;412:42–45. doi: 10.1111/j.1651-2227.1996.tb14247.x. [DOI] [PubMed] [Google Scholar]
- Corazza GR, Andreani ML, Biagi F, Corrao G, Pretolani S, Giulianelli G, et al. The smaller size of the ‘coeliac iceberg' in adults. Scand J Gastroenterol. 1997;32:917–919. doi: 10.3109/00365529709011202. [DOI] [PubMed] [Google Scholar]
- Johnston SD, Watson RG, McMillan SA, Sloan J, Love AH. Prevalence of coeliac disease in Northern Ireland. Lancet. 1997;350:1370. doi: 10.1016/s0140-6736(05)65142-2. [DOI] [PubMed] [Google Scholar]
- Kolho KL, Farkkila MA, Savilahti E. Undiagnosed coeliac disease is common in Finnish adults. Scand J Gastroenterol. 1998;33:1280–1283. doi: 10.1080/00365529850172368. [DOI] [PubMed] [Google Scholar]
- Ivarsson A, Persson LA, Juto P, Peltonen M, Suhr O, Hernell O. High prevalence of undiagnosed coeliac disease in adults: a Swedish population-based study. J Intern Med. 1999;245:63–68. doi: 10.1046/j.1365-2796.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- Riestra S, Fernandez E, Rodrigo L, Garcia S, Ocio G. Prevalence of coeliac disease in the general population of Northern Spain. Scand J Gastroenterol. 2000;35:398–402. doi: 10.1080/003655200750023967. [DOI] [PubMed] [Google Scholar]
- Volta U, Bellentani S, Bianchi FB, Brandi G, de Franceschi L, Miglioli L, et al. High prevalence of celiac disease in Italian general population. Dig Dis Sci. 2001;46:1500–1505. doi: 10.1023/a:1010648122797. [DOI] [PubMed] [Google Scholar]
- Mäki M, Mustalahti K, Kokkonen J, Kulmala P, Haapalahti M, Karttunen T, et al. Prevalence of celiac disease among children in Finland. N Engl J Med. 2003;348:2517–2524. doi: 10.1056/NEJMoa021687. [DOI] [PubMed] [Google Scholar]
- Not T, Horvath K, Hill ID, Partanen J, Hammed A, Magazzu G, et al. Coeliac disease risk in USA: high prevalence of antiendomysium antibodies in healthy blood donors. Scand J Gastroenterol. 1998;33:494–498. doi: 10.1080/00365529850172052. [DOI] [PubMed] [Google Scholar]
- Gupta R, Reddy DN, Makharia GK, Sood A, Ramakrishna BS, Yachha SK, et al. Indian task force for celiac disease: current status. World J Gastroenterol. 2009;15:6028–6033. doi: 10.3748/wjg.15.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catassi C, Ratsch IM, Gandolfi L, Pratesi R, Fabiani E, El Asmar R, et al. Why is coeliac disease endemic in the people of the Sahara. Lancet. 1999;354:647–648. doi: 10.1016/s0140-6736(99)02609-4. [DOI] [PubMed] [Google Scholar]
- Maki M, Collin P. Coeliac disease. Lancet. 1997;349:1755–1759. doi: 10.1016/S0140-6736(96)70237-4. [DOI] [PubMed] [Google Scholar]
- Fasano A, Catassi C. Current approaches to diagnosis and treatment of celiac disease: an evolving spectrum. Gastroenterology. 2001;120:636–651. doi: 10.1053/gast.2001.22123. [DOI] [PubMed] [Google Scholar]
- Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346:180–188. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- Howdle PD, Losowsky MS.Coeliac disease in adultsIn: Marsh MN (ed.)Coeliac Disease. Oxford; Blackwell Scientific; 199249–80. [Google Scholar]
- Gasbarrini G, Ciccocioppo R, de Vitis I. Corazza GR; Club del Tenue Study Group Coeliac disease in the elderly. A multicentre Italian study. Gerontology. 2001;47:306–310. doi: 10.1159/000052819. [DOI] [PubMed] [Google Scholar]
- Ciclitira PJ. AGA technical review on celiac sprue. Gastroenterology. 2001;120:1526–1540. doi: 10.1053/gast.2001.24056. [DOI] [PubMed] [Google Scholar]
- Trier JS. Diagnosis of celiac sprue. Gastroenterology. 1998;115:211–216. doi: 10.1016/s0016-5085(98)70383-x. [DOI] [PubMed] [Google Scholar]
- Volta U. Coeliac disease: recent advances in pathogenesis, diagnosis and clinical signs. Rec Prog Med. 1999;1:37–44. [PubMed] [Google Scholar]
- McFarlane XA, Bhalla AK, Reeves DE, Morgan LM, Robertson DA. Osteoporosis in treated adult coeliac disease. Gut. 1995;36:710–714. doi: 10.1136/gut.36.5.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazza GR, Valentini RA, Andreani ML, D'Anchino M, Leva MT, Ginaldi L, et al. Subclinical coeliac disease is a frequent cause of iron-deficiency anaemia. Scand J Gastroenterol. 1995;30:153–156. doi: 10.3109/00365529509093254. [DOI] [PubMed] [Google Scholar]
- Volta U.Dallo screening alla diagnosiIn: Corazza GR (ed.)Malattia Celiaca, Educazione Permanente in Malattie Digestive. Roma; Il Pensiero Scientifico Editore; 1996529–35. [Google Scholar]
- Cacciari E, Salardi S, Volta U, Biasco G, Lazzari R, Corazza GR, et al. Can antigliadin antibody detect symptomless coeliac disease in children with short stature. Lancet. 1985;1:1469–1471. doi: 10.1016/s0140-6736(85)92251-2. [DOI] [PubMed] [Google Scholar]
- Aine L, Maki M, Collin P, Keyrilainen O. Dental enamel defects in celiac disease. J Oral Path Med. 1990;19:241–245. doi: 10.1111/j.1600-0714.1990.tb00834.x. [DOI] [PubMed] [Google Scholar]
- Ferguson R, Basu MK, Asquith P, Cooke WT. Jejunal mucosal abnormalities in patients with recurrent aphthous ulceration. BMJ. 1975;1:11–13. doi: 10.1136/bmj.1.6000.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti GM, Tursi A, Brandimarte G, Rubino C, Gasbarrini G. Dysmotility-like dyspeptic symptoms in coeliac patients: role of gluten and Helicobacter pylori infection. Dig Liv Dis. 2000;32:73–74. doi: 10.1016/s1590-8658(00)80051-1. [DOI] [PubMed] [Google Scholar]
- Sanders DS, Carter MJ, Hurlstone DP, Pearce A, Ward AM, McAlindon ME, et al. Association of adult coeliac disease with irritable bowel syndrome: a case–control study in patients fulfilling ROME II criteria referred to secondary care. Lancet. 2001;358:1504–1508. doi: 10.1016/S0140-6736(01)06581-3. [DOI] [PubMed] [Google Scholar]
- Hallert C, Derefeldt T. Psychi disturbances in adult coeliac disease. Clinical observations. Scand J Gastroenterol. 1982;17:17–19. doi: 10.3109/00365528209181037. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bouquet F, Greco L, Lambertini A, Tassinari CA, Ventura A, et al. Coeliac disease, epilepsy, and cerebral calcifications. Lancet. 1992;340:439–443. doi: 10.1016/0140-6736(92)91766-2. [DOI] [PubMed] [Google Scholar]
- Hadjivassiliou M, Gibson A, Davies-Jones GA, Lobo AJ, Stephenson TJ, Milford-Ward A. Does cryptic gluten sensitivity play a part in neurological illness. Lancet. 1996;347:369–371. doi: 10.1016/s0140-6736(96)90540-1. [DOI] [PubMed] [Google Scholar]
- Luorostainen L, Pirttila T, Collin P. Coeliac disease presenting with neurological disorders. Eur Neurol. 1999;42:132–135. doi: 10.1159/000008086. [DOI] [PubMed] [Google Scholar]
- Fantelli FJ, Mitsumoto H, Sebek BA. Multiple sclerosis and malabsorption. Lancet. 1978;311:1039–1040. doi: 10.1016/s0140-6736(78)90758-4. [DOI] [PubMed] [Google Scholar]
- Volta U, de Giorgio R. Gluten sensitivity: an emerging issue behind neurological impairment. Lancet Neurol. 2010;9:233–235. doi: 10.1016/S1474-4422(09)70357-6. [DOI] [PubMed] [Google Scholar]
- Ferguson R, Holmes GK, Cooke WT. Coeliac disease, fertility and pregnancy. Scand J Gastroenterol. 1982;17:65–68. doi: 10.3109/00365528209181045. [DOI] [PubMed] [Google Scholar]
- Sher KS, Mayberry JF. Female fertility, obstetric and gynecological history in coeliac disease: a case control study. Digestion. 1994;55:243–246. doi: 10.1159/000201155. [DOI] [PubMed] [Google Scholar]
- Farthing MJ, Edwards CR, Rees LH, Dawson AM. Male gonadal function in coeliac disease: 1 sexual dysfunction, infertility and semen quality. Gut. 1982;23:608–614. doi: 10.1136/gut.23.7.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farthing MJ, Rees LH, Edwards CR, Dawson AM. Male gonadal function in coeliac disease: 2 sexual dysfunction, infertility and semen quality. Gut. 1982;24:127–135. doi: 10.1136/gut.23.7.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta U, de Franceschi L, Lari F, Molinaro N, Zoli M, Bianchi FB. Coeliac disease hidden by cryptogenic hypertransaminasemia. Lancet. 1998;352:26–29. doi: 10.1016/s0140-6736(97)11222-3. [DOI] [PubMed] [Google Scholar]
- Cooper BT, Holmes GK, Cooke WT. Coeliac disease and immunological disorders. BMJ. 1978;1:537–539. doi: 10.1136/bmj.1.6112.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Reunala T, Pukkala E, Laippala P, Keyrilainen O, Pasternack A. Coeliac disease, associated disorders and survival. Gut. 1994;35:1215–1218. doi: 10.1136/gut.35.9.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reunala T, Collin P. Diseases associated with dermatitis herpetiformis. Br J Dermatol. 1997;136:315–318. [PubMed] [Google Scholar]
- Cronin CC, Shanahan F. Insulin-dependent diabetes mellitus and coeliac disease. Lancet. 1997;349:1096–1097. doi: 10.1016/S0140-6736(96)09153-2. [DOI] [PubMed] [Google Scholar]
- Counsell CE, Taha A, Ruddell WS. Coeliac disease and autoimmune thyroid disease. Gut. 1994;35:844–846. doi: 10.1136/gut.35.6.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta U, de Franceschi L, Molinaro N, Cassani F, Muratori L, Lenzi M, et al. Frequency and significance of anti-gliadin and anti-endomysial antibodies in autoimmune hepatitis. Dig Dis Sci. 1998;43:2190–2195. doi: 10.1023/a:1026650118759. [DOI] [PubMed] [Google Scholar]
- Volta U, Rodrigo L, Granito L, Petrolini L, Muratori P, Muratori P, et al. Celiac disease in autoimmune cholestatic liver disorders. Am J Gastroenterol. 2002;97:2609–2613. doi: 10.1111/j.1572-0241.2002.06031.x. [DOI] [PubMed] [Google Scholar]
- Volta U, Bardazzi F, Zauli D, de Franceschi L, Tosti A, Molinaro N, et al. Serological screening for coeliac disease in vitiligo and alopecia areata. Br J Dermatol. 1997;136:801–802. doi: 10.1111/j.1365-2133.1997.tb03684.x. [DOI] [PubMed] [Google Scholar]
- O'Leary C, Walsh CH, Wieneke P, O'Regan P, Buckley B, O'Halloran DJ, et al. Coeliac disease and autoimmune Addison's disease: a clinical pitfall. QJM. 2002;95:79–82. doi: 10.1093/qjmed/95.2.79. [DOI] [PubMed] [Google Scholar]
- Henriksson KG, Hallert C, Walan A. Gluten-sensitive polymyositis and enteropathy. Lancet. 1976;2:317. doi: 10.1016/s0140-6736(76)90772-8. [DOI] [PubMed] [Google Scholar]
- Teppo AM, Maury CP. Antibodies to gliadin, gluten and reticulin glycoprotein in rheumatic diseases: elevated levels in Sjogren's disease. Clin Exp Immunol. 1984;57:73–78. [PMC free article] [PubMed] [Google Scholar]
- O'Farrelly C, Marten M, Melcher D, McDougall B, Price R, Goldstein AJ, et al. Association between villous atrophy in rheumatoid arthritis and a rheumatoid factor and gliadin-specific IgG. Lancet. 1988;2:819–822. doi: 10.1016/s0140-6736(88)92784-5. [DOI] [PubMed] [Google Scholar]
- Fornasieri A, Sinco RA, Maldifassi P, Bernasconi P, Vegni M, D'Amico G. IgA-antigliadin antibodies in IgA mesangial nephropathy (Berger's disease) BMJ. 1987;295:78–80. doi: 10.1136/bmj.295.6590.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo F, Marino V, Ventura A, Bottaro G, Corazza G. Prevalence and clinical features of selective immunoglobulin A deficiency in coeliac disease: an Italian multicentre study. Gut. 1998;42:362–365. doi: 10.1136/gut.42.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curione M, Barbato M, de Biase L, Viola F, Lo Russo L, Cardi E. Prevalence of coeliac disease in idiopathic dilated cardiomiopathy. Lancet. 1999;354:222–223. doi: 10.1016/s0140-6736(99)01501-9. [DOI] [PubMed] [Google Scholar]
- Zauli D, Grassi A, Granito A, Foderaro S, de Franceschi L, Ballardini G, et al. Prevalence of silent coeliac disease in atopics. Dig Dis Sci. 2000;32:775–779. doi: 10.1016/s1590-8658(00)80354-0. [DOI] [PubMed] [Google Scholar]
- Kitis G, Holmes GK, Cooper BT, Thompson H, Allan RN. Association of coeliac disease and inflammatory bowel disease. Gut. 1980;21:636–641. doi: 10.1136/gut.21.7.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale L, Wimalaratna H, Brotodiharjo A, Duggan JM. Down's syndrome is strongly associated with coeliac disease. Gut. 1997;40:492–496. doi: 10.1136/gut.40.4.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonamico M. Celiac disease and Turner syndrome. J Pediatr Gastroenterol. 1999;29:107–108. doi: 10.1097/00005176-199907000-00029. [DOI] [PubMed] [Google Scholar]
- Giannotti A, Tiberio G, Castro M, Virgilii F, Colistro F, Ferretti F, et al. Coeliac disease in Williams syndrome. J Med Genet. 2001;38:767–768. doi: 10.1136/jmg.38.11.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazza GR, Valentini RA, Frisoni M, Volta U, Corrao G, Bianchi FB, et al. Gliadin immune reactivity is associated with overt and latent enteropathy in relatives of celiac patients. Gastroenterology. 1992;103:1517–1522. doi: 10.1016/0016-5085(92)91172-z. [DOI] [PubMed] [Google Scholar]
- Maki M, Holm K, Collin P, Savilahti E. Increase in γ/δ T cell receptor bearing lymphocytes in normal small bowel mucosa in latent coeliac disease. Gut. 1991;32:1412–1414. doi: 10.1136/gut.32.11.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz E, Bode J, Kingstone K, Ferguson A. Intestinal antibody pattern of coeliac disease: association with γ/δ T cell receptor expression by intraepithelial lymphocytes, and other indices of potential coeliac disease. Gut. 1994;35:476–482. doi: 10.1136/gut.35.4.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiner M. Duodenal biopsy. Lancet. 1956;1:17–19. doi: 10.1016/s0140-6736(56)91854-2. [DOI] [PubMed] [Google Scholar]
- Upton MP. ‘Give us this day our daily bread'. Evolving concepts in celiac sprue. Arch Pathol Lab Med. 2008;132:1594–1599. doi: 10.5858/2008-132-1594-GUTDOD. [DOI] [PubMed] [Google Scholar]
- Villanacci V. The problem of biopsies in the diagnosis of celiac disease. Gastrointest Endosc. 2009;69:983–984. doi: 10.1016/j.gie.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Brown I, Mino-Keduson M, Deshpande V, Lauwers GY. Intraepithelial lymphocytosis in architecturally preserved proximal small intestinal mucosa. Arch Pathol Lab Med. 2006;130:1020–1025. doi: 10.5858/2006-130-1020-ILIAPP. [DOI] [PubMed] [Google Scholar]
- Corazza GR, Villanacci V, Zambelli C, Milione M, Luinetti O, Vindigni C, et al. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastroenterol Hepatol. 2007;5:838–843. doi: 10.1016/j.cgh.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Corazza GR, Villanacci V. Coeliac disease. J Clin Pathol. 2005;58:573–574. doi: 10.1136/jcp.2004.023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar S, Nehra V, Murray JA, Dayharsh GA, Burgart LJ. Significance of intraepithelial lymphocytosis in small bowel biopsy samples with normal mucosa architecture. Am J Gastroenterol. 2003;98:2027–2033. doi: 10.1111/j.1572-0241.2003.07631.x. [DOI] [PubMed] [Google Scholar]
- Paparo F, Petrone E, Tosco A, Maglio M, Borrelli M, Salvati VM, et al. Clinical, HLA and small boel immunohistochemical features of children with positive serum antiendomysium antibodies and architecturally normal small intestinal mucosa. Am J Gastroeneterol. 2005;100:2294–2298. doi: 10.1111/j.1572-0241.2005.41134.x. [DOI] [PubMed] [Google Scholar]
- Oberhuber G, Grandtisch G, Vogelsang H. The histopathology of celiac disease: time for standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999;11:1185–1194. doi: 10.1097/00042737-199910000-00019. [DOI] [PubMed] [Google Scholar]
- Villanacci V, Catassi C, Rostami K, Volta U.Celiac disease: changing dogma on historical diagnosis. GastroHep 2010 ; January 13. http://www.gastrohep.com/freespeech/freespeech.asp?id=128
- Dieterich W, Laag E, Schopper H, Volta U, Ferguson A, Gillet H, et al. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115:1317–1321. doi: 10.1016/s0016-5085(98)70007-1. [DOI] [PubMed] [Google Scholar]
- Volta U, Fabbri A, Parisi C, Piscaglia M, Caio G, Tovoli F, et al. Old and new serological tests for celiac disease screening. Expert Rev Gastroenterol Hepatol. 2010;4:31–35. doi: 10.1586/egh.09.66. [DOI] [PubMed] [Google Scholar]
- Stern M. Working Group on Serologic Screening for Celiac Disease Comparative evaluation of serologic tests for celiac disease: a European iniative toward standardization. J Pediatr Gastroenterol Nutr. 2000;31:513–519. doi: 10.1097/00005176-200011000-00012. [DOI] [PubMed] [Google Scholar]
- Lagerqvist C, Dahlbom I, Hansson T, Jidell E, Juto P, Olcén P, et al. Antigliadin immunoglobulin A best in finding celiac disease in children younger than 18 months of age. J Pediatr Gastroenterol Nutr. 2008;47:428–435. doi: 10.1097/MPG.0b013e31817d80f4. [DOI] [PubMed] [Google Scholar]
- Cataldo F, Marino V, Ventura A, Bottaro G, Corazza GR. Prevalence and clinical features of selective immunoglobulin A deficiency in coeliac disease: an Italian multicentre study. Gut. 1998;42:362–365. doi: 10.1136/gut.42.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta U, Granito A, Parisi C, Fabbri A, Fiorini E, Piscaglia M, et al. Deamidated gliadin peptide antibodies as a routine test for celiac disease: a prospective analysis. J Clin Gastroenterol. 2010;44:186–190. doi: 10.1097/MCG.0b013e3181c378f6. [DOI] [PubMed] [Google Scholar]
- Salmi TT, Collin P, Järvinen O, Haimila K, Partanen J, Laurila K, et al. Immunoglobulin A autoanttibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming celiac disease. Aliment Pharmacol Ther. 2006;24:541–552. doi: 10.1111/j.1365-2036.2006.02997.x. [DOI] [PubMed] [Google Scholar]
- Maglio M, Florian F, Vecchiet M, Auricchio R, Paparo F, Spadaro R, et al. Majority of children with type 1 diabetes produce and deposit anti-tissue transglutaminase antibodies in the small intestine. Diabetes. 2009;58:1578–1584. doi: 10.2337/db08-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]