

Abstract
Celiac disease (CD) is one of the most common food intolerances in developed world. It affects genetically susceptible individuals and has severe consequences if it remains undiagnosed. A disease known for more than a century, it is still the focus for experts from various fields of research and development. Geneticists, pathologists, immunologists, food engineers and dieticians share their knowledge and expertise to improve the conditions of CD patients. With new insights in the pathomechanism of gluten processing and antigen presentation in CD, it was possible to improve the diagnostic antigen mimicking the primary epitope in CD. These celiac neo-epitopes are comprised of a complex of gliadin peptides crosslinked with transglutaminase (tTg). They are an early diagnostic marker for CD which occurs up to 6 months earlier than classical markers known to miss a certain amount of CD patients.
Keywords: celiac disease, tissue transglutaminase, celiac neo–epitope, gluten, diagnostic marker
Celiac disease
Celiac disease (CD) is a common inflammatory disease of the small intestine that is mainly triggered and maintained by the intake of wheat gluten and related cereals. It is characterized by an autoimmune response in genetically susceptible individuals resulting in small intestinal mucosal damage.
CD was first described in the first to second century by Aretaeus of Cappadochia. In 1887, Samuel Jones Gee described typical symptoms of CD, like diarrhea, faintness and growth retardation, and cure by means of a diet.1 In 1950, Willem-Karel Dicke, observed the role of gluten in CD and thus became the pioneer in gluten-free diet (GFD).2 Four years later, Paulley gave the first description of typical villous atrophy of the small intestine.3 A milestone in the pathology of CD was the introduction of the Marsh criteria in 1992,4 wherein the patterns of the celiac lesions were classified in four gradual stages. Seven years later, Oberhuber and colleagues further standardized the classification by introducing three subtypes of stage 3.5 In 1997, Schuppan and Dieterich identified tissue transglutaminase (tTg) as the one of the main autoantigens in CD.6 The most recent milestone was the identification of digestion resistant gliadin peptides by Shan in 2002.7
This review gives an overview of the latest trends in CD.
Epidemiology
With a currently estimated prevalence of 1% in Western population, CD is one of the most common inflammatory diseases of the small intestine.8 The statistical probability ranges between 0.5 and 1.26% in the general population of Europe and the United States.9 Recently, a finish study reported an increase of CD in elderly people wherein a prevalence of 1∶47 was found in randomly selected subjects older than 52 years of age.10
In general, CD can affect individuals from any age, but two peaks can be seen in the early childhood (below 6 years) and between the fourth and fifth decade of life. Whereas the prevalence in children ranges from 0.31 to 0.9%,9, 11 CD affects 1–2% of the adults in Europe12 and 0.4–0.95% of the adults in United States.9
CD occurs more often in women, with a female to male ratio of between 2∶1 and 3∶1.13 Patients older than 60 years of age in which CD is diagnosed are more frequently male.14
The prevalence of CD is increasing. This may be a result of improved screening methods, but also environmental factors are discussed7 such as the introduction of wheat in modern food industry worldwide7.
The development of CD is determined by both environmental and genetic factors. Environmental factors include infant feeding, infections and socioeconomic features. Onset of gluten introduction plays an important role in infant feeding. Thereby, it is crucial if the child is still breastfed or not during the first exposure.16, 17 Currently, the European Society for Paediatric Gastroenterology, Hepatology and Nutrition recommends the exposure to small amounts of gluten between 4 and 7 months of age during breastfeeding.18 But it still needs to be clarified whether breastfeeding is a permanent protection against CD or only delays clinical onset. Association of infection and the development of CD have been described for HCV infection.19 Recently, a prospective study was published that postulates frequent rotavirus infections as an independent risk factor.20 The effect of infections with other common intestinal microorganisms still has to be elucidated.
Besides environmental factors, genetic predisposition contributes to the development of CD. The most dominant genetic risk factors are the genotypes encoding the histocompatibility leukocyte Ag (HLA) class II molecules DQ2 and DQ8. About 90% of CD patients carry the DQ2 heterodimer whereas most of the remainder expresses DQ8. CD in patients who are negative for both DQ2 and DQ8 is extremely rare.21
The strong genetic association of CD is indicated by the high concordance between monozygotic twins (80%). In contrast, dizygotic twins have only a concordance of 11%, which is in accordance with the risk for first-degree relatives.9, 22
DQ2 and DQ8 are expressed in 30–35% of the Caucasian populations. Due to the fact that only 2–5% of gene carriers suffer from CD, there might be other non-HLA-linked genes that contribute to the development of CD.23
Pathomechanism
As CD is a complex inflammatory disorder, numerous genetic and environmental factors are involved in its pathomechanism.24, 25 One important example of such an environmental factor is gluten which elicits CD. Briefly, CD evolves as a consequence of an abnormal immune response to gluten.
Gluten proteins can be differentiated into ethanol-insoluble glutenins and ethanol-soluble gliadins. Gliadins are unusually rich in glutamine (eightfold normal content) and proline (threefold).26 These proline residues protect gluten peptides from proteolytic degradation by pancreatic and brush border proteases during digestion. Therefore, undigested gliadin fragments reach the lamina propria. The peptides are modified by tissue tTg by either deamidation or transamidation.27 Tissue tTg is an enzyme that is expressed in many organs, including the small intestine. In active lesions of celiac patients, it is located in the extracellular space of the subepithelial region, but also at the epithelial brush border and can deamidate gluten peptides there.28
In the deamidation reaction, a neutral glutamine residue is converted into a negatively charged glutamate. The negative charges of the peptide facilitate the binding to the anchor positions of HLA-DQ2 and HLA-DQ8 on antigen-presenting cells.29, 30, 31 In the transamidation reaction, the tissue tTg catalyzes the crosslinking of glutamine residues of the gliadin peptide and a lysine residue of an acceptor protein resulting in an isopeptide bond.32 A special form of transamidation of tTg was described in situ, wherein gliadins are covalently coupled to tTg, resulting in high molecular weight products. This process is termed autocatalysis.6, 33 Therefore, the uptake of these complexes by antigen-presenting cells and the subsequent intracellular processing may lead to the presentation of peptides from gliadin, tTg and crosslinked neo-epitopes.
The activation of gluten-specific T cells is a crucial step in the development of CD. This underlines the important role of HLA genes in the pathomechanism of CD.32
Disease symptoms
Since the degenerating microvilli of the intestinal mucosa are the main pathological hallmark of CD, the symptoms are naturally associated with the uptake and provision of nutrients. Classical symptoms are therefore related to malabsorption and malnutrition. It is important to stress that CD can occur during any period of a person's life. The main starting points, however, are in early childhood and between 40 and 50 years of age.34 Clinical manifestations vary according to the age group: in early childhood CD appears with classical symptoms whereas the spectrum of symptoms in adults is wider than in children. The classical signs were first observed in the young and may best be summarized as a direct result of the defects during nutrient absorption: diarrhea, growth deficits, abdominal bloating, steatorrhea and a wide range of other symptoms.35 In addition, there are a large proportion of patients who develop symptoms not considered typical for CD. As an example, the symptoms of anemia which are linked to the lack of iron are considered. Since the deficient absorption of nutrients is problematic for the development and metabolic maintenance, CD should be considered a systemic disease with a plethora of symptoms linked to the one or the other deficiency. The reasons for this wide range of symptoms are considered due to the differences in the age of onset, the immunological background, the gender and the genetic background.
Disease classification
CD can be classified in several subtypes. The classification is carried out according to several criteria: the clinical presentation, i.e. clinical symptoms, the degree of villous atrophy and the presence of antibodies against CD-specific antigens (Table 1). As classical CD, clinical symptoms such as diarrhea and abdominal bloating are present in conjunction with a high degree of villous atrophy and the presence of CD-specific antibodies. As stated earlier, CD can occur with atypical symptoms such as iron deficiency anemia or extraintestinal symptoms. Such cases usually show the presence of antibodies and a variable degree of villous atrophy. In addition to these overt cases of CD, a large group of patients develop types of CD which are classified as silent, potential or latent. Silent and potential CD is characterized by the lack of clinical symptoms and low or no villous atrophy. In both types, antibodies specific for CD are present. In the latent type, the remission after therapy is incomplete and even without symptoms, the villous atrophy and CD-specific antibodies persist. Finally, the presence of refractory CD is diagnosed when older patients show no or only transient response to therapy by GFD and the symptoms are persistent.36 This group comprises mainly adults over the age of 50 who, even after strict GFD, have no improved histology.34
Table 1. Celiac disease subtypes with symptoms, histological presentation and serology.
| CD subtype | Symptoms | Histology | Serology |
|---|---|---|---|
| Classic | Diarrhea malabsorption symptoms | Villous atrophy Marsh 3a-c | Positive |
| Silent | Inconspicuous | Normal | Positive |
| Potential | Inconspicuous | Normal, up to Marsh 1 | Positive |
| Latent | Atypical reduced under GFD | Marsh 1-2 | Positive |
| Refractory | Classic unresponsive to GFD | Classic unresponsive to GFD | Positive unresponsive to GFD |
Associated disease
A wide range of other conditions can be associated with the CD.35, 37 This group is mainly comprised of other autoimmune diseases. One example is type 1 diabetes where 2–5% of patients also suffer from CD. Other associated diseases are, among others, autoimmune thyroiditis, autoimmune hepatitis and osteoporosis.38 For type 1 diabetes, it has been shown that this condition is better treatable when CD is diagnosed and a GFD is followed.39 Similarly, osteoporosis treatment shows stronger improvement in CD patients following GFD.40 Other diseases which are associated with CD are Turner, Williams and Downs syndrome.41 Of specific importance is the association of CD with IgA deficiency. Since anti-tTg antibodies of IgA class are used as the main diagnostic tool, care has to be taken if patients present IgA deficiency.
Therapy
In most cases, a GFD is the appropriate therapy for CD. The patients respond well to this therapy. Furthermore, the GFD is a save and well-tolerated therapy which, very much in contrast with other autoimmune diseases, is free of obvious side effects. The main problems are associated with the compliance to the therapy. Many common food products contain at least traces of wheat or grain material. In order to have a good compliance, patients often have to rely on specifically labeled gluten-free products. This brings an added financial and social burden since these products are generally more expensive and not easily available.42 The GFD can be supported by the supplementation with relevant nutrients like iron, certain vitamins, calcium and vitamin D.
Several strategies are under way to ameliorate the symptoms of CD where patients are not able to adhere to strict GFD or where a transient exposure to gluten cannot be avoided. These strategies are still under development and aim to reduce the exposure to gluten in the gut. This can be achieved by the digestion of gliadin with specific enzymes, e.g. prolyl endopeptidases, which result in the breakdown of gliadin in the gut.43 However, these strategies are not yet finalized.
More problematic issues involve possible non-responders. Patients displaying refractory CD generally show no long-term response to a GFD. The treatment of refractory CD involves the use of corticosteroids or immunosuppressive drugs.34
Complications
Since CD is a disease where malabsorption of nutrients is a central feature, secondary conditions are the logical result. With the associating diseases already described above, the resulting symptoms are of relevance in untreated CD. After the initiation of a GFD, special care has to be taken to consider the associated conditions. The effect of CD therapy can be reflected by the progress in the therapy of the associated disease. For example, the changes of the bone mineral density should be monitored in osteoporosis-associated CD.40
Besides the complication of refractory CD, the risk for the development of enteropathy-associated T-cell lymphomas with an overall incidence is 0.5–1.0 cases per million in Western countries.44 The survival rate in these cases is poor, since the patients are mainly unresponsive to GFD and develop several life-threatening complications.
Diagnosis of CD
The diagnosis of CD can be separated in three fields: the histological, the serological and the genetical diagnosis. Whereas the former have a high positive predictive value, the latter has a high negative predictive value and is thus mainly used to exclude false positive cases. Currently, less than one in seven patients (celiac iceberg) is correctly diagnosed with CD and the diagnostic delay ranges from 5 to 11 years. Due to the complications which can arise from undiagnosed and thus untreated CD, it is important to have highly sensitive and specific diagnostic tests.
Histology
The histological findings are based on several small intestinal biopsies (4–6 well-oriented samples) and are to be interpreted by an experienced pathologist. The samples are classified in the modified Marsh criteria.5 It divides the mucosal damage into four stages: a normal duodenal mucosa (type 0), a mucosa with increased percentage of interepithelial lymphocytes (>40 per 100 epithelial cells; type 1), inflammatory infiltrates and crypt hyperplasia but still well preserved villi (type 2). Type 3 is further divided into three subclasses based on the level of villous atrophy: mild (3a), moderate (3b) and total (3c) and type 4 refers to irreversible total hypoplasia of the mucosa.
Most publications still consider duodenal biopsy as the ‘gold standard' for CD (no biopsy and no CD). However, some authors recently revised this rigid claim if very high levels of and/or several different serological antibodies and genetic susceptibility are present.45 Additionally, if serology (positive) and histology (negative) are inconsistent and CD is strongly suspected (e.g. family history, persisting symptoms, DQ2/8 positive), the biopsy samples should be re-examined by an expert pathologist.46
On the other hand, there are also reports of seronegative, biopsy-positive CD patients;47 this shows that the classical serological approach does not cover all the celiac patients.
Serology
Currently, several antigens are used in ELISA-based assays for the diagnosis of CD including α-gliadin, deamidated gliadin peptides (DGPs), tissue tTg, tTg mixed with deamidated gliadin and finally the novel neo-epitopes which consist of a complex of gliadin peptides modified and crosslinked with tissue tTg (Figure 1).48, 49, 50 In order to exclude IgA deficiency, a condition which occurs more frequent in CD patients (10- to 15-fold higher than the normal population), the total amount of IgA has to be determined.51 In this case, the serology has to rely on IgG-based assays only.52
Figure 1.
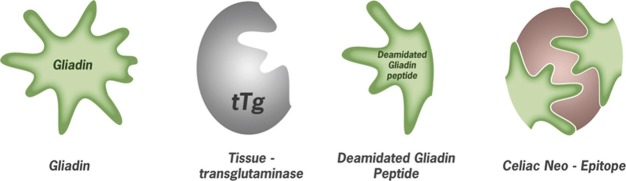
Relevant antigens used in CD diagnosis; from left to right: gliadin, the alcohol soluble fraction of gluten mainly used in pediatric diagnosis; tissue tTg discovered as the main autoantigen from endymosium immunofluorescence; deamidated gliadin peptides resistant to further digestion by pancreatic enzymes and modified by tTg and celiac neo-epitope: a complex resulting from transamidation of deamidated gliadin peptides with tTg. CD, celiac disease; tTg, transglutaminase.
As a separate serological test, the detection of endomysial antibodies (EMAs) is also used for the detection of CD. EMAs utilizing monkey esophagus as a substrate in immunofluorescence (EMA) lead in 1997 to the discovery of tissue tTg as one of the key auto-antigens in CD.6
Tissue tTg is an enzyme which plays a major role in the toxicity of gliadin peptides. Antibodies against tTg characterize CD as an autoimmune disease.53 The sensitivity of tTg-based assays ranges from 75.3 to 100% with a specificity from 91.8 to 100%, whereas EMA sensitivity ranges from 70.5 to 100% with a specificity from 89.8 to 100%. The sensitivity of tTg IgA is slightly higher in comparison to EMA IgA, whereas the specificity of EMA is slightly higher than that of tTg.54 Assays utilizing tTg are currently the most widely accepted serological test for CD.
Assays based on α-gliadin have been used for a long time especially in the diagnosis of pediatric patients. Their lower specificity and sensitivity was tolerated since it was the only antigen available for pediatric patients younger than 2 years. With the discovery of digestion-resistant gliadin peptides7 and their modification by tissue tTg,55, 56, 57 gliadins are more and more replaced by DGPs in pediatric samples.58, 59
One of the main motivations to replace gliadin with DGPs lies in their better specificity, whereas the sensitivity of DGPs is only slightly increased. Lately, the use of DGP IgG and tTg IgA as the main serological parameters is often recommended.58 Even though a meta-analysis of all available publications on DGPs at that time shows that the tTg assays still outperform DGP-based assays. This meta-analysis also stresses the fact that many of the analyzed studies are biased in the way that serologically prescreened serum samples have been used (in 9 of 11 studies) which thus may result in higher sensitivity values.50
Even though, the binding of DGPs to DQ2 molecules and its effects on T cells have been thoroughly examined and are still the focus in CD research,23 the complete mechanism how autoantibodies develop to tissue tTg still remains poorly understood. It has been postulated that the formation of complexes between gliadin peptides and tTg and its further processing in the antigen-presenting cells is supporting epitope spreading from neo-epitopes to gliadin peptides and tTg.24, 53, 60 The authors believe this to be the primary antigen for the induction of CD since it is completely non-self and unknown to the immune system (unpublished results).
The neo-epitope assays using a covalently crosslinked complex of gliadin peptides with tTg in contrast to those assays using only DGP/tTg mixtures53 are based on this hypothesis and show a good sensitivity in CD detection.49, 58, 61, 62, 63, 64 Several prospective studies elucidating the value of these neo-epitope antibodies as a predictive marker for CD are currently in progress showing promising results and indicate that these antibodies in some cases occur up to 6 months before antibodies to tTg, DGPs or mucosal damage.65 These results emphasize the importance to follow up supposedly false positive results in patients genetically predisposed to CD with persisting symptoms.
Genetic testing
The DQ2 and/or DQ8 HLA complex is present in 30–35% of the Caucasian population (genetically susceptible to CD) but only 2–5% of these develop CD.21 Nearly all (>99.5%) of CD patients share either the DQ2 or the DQ8 HLA genotype; therefore, a DQ2/DQ8 positive test is a necessary but not a sufficient condition for CD and can only be used to exclude false positive results and thus to confirm a CD diagnosis in case of positive serology and histology.66 But one still has to take into account that about 0.4% of CD patients are DQ2 and DQ8 negative.21
Decision making
Due to the heterogeneity of CD, diagnosis is a process which has to take into account several factors and should be done in communication with geneticist, gastroenterologist, pathologist, dietician and general physician. A current review tries to simplify the available diagnostic algorithms, also considering the most recent findings.67
They suggest a ‘4 out of 5' rule: the diagnosis of CD is confirmed when at last four of the following criteria are fulfilled: typical symptoms of CD; positivity of CD IgA class antibodies at high titer; HLA DQ2 or DQ8 genotypes; celiac enteropathy at duodenal biopsy and response to a GFD. In case a HLA genotyping is not performed the rule can be modified to ‘3 out of 4'.
Outlook
Due to the increase in prevalence and the implications of an undiagnosed and untreated CD which may lead to such harsh complications as refractory CD or lymphoma, it is important to stress that all available information (genetics, histology and serology) is combined to obtain a correct diagnostic result. With the advancing knowledge in the pathomechanism of CD, the newly developed diagnostic antigens (neo-epitopes) take this progress into account mimicking the physiological antibody generation process. The neo-epitopes thus offer an improved prognostic value especially in cases where the classical antigens and histology are lacking in sensitivity as shown in prospective studies where they have been preceding these antigens by 6 months and more. Further studies have to evaluate the value of these novel antigens which shall help to melt down the celiac iceberg.
References
- Losowsky MS. A history of coeliac disease. Dig Dis. 2008;26:112–120. doi: 10.1159/000116768. [DOI] [PubMed] [Google Scholar]
- van Berge-Henegouwen GP, Mulder CJ. Pioneer in the gluten free diet: Willem-Karel Dicke 1905–1962, over 50 years of gluten free diet. Gut. 1993;34:1473–1475. doi: 10.1136/gut.34.11.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulley JW. Observation on the aetiology of idiopathic steatorrhoea: jejunal and lymph-node biopsies. Br Med J. 1954;2:1318–1321. doi: 10.1136/bmj.2.4900.1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue') Gastroenterology. 1992;102:330–354. [PubMed] [Google Scholar]
- Oberhuber G, Granditsch G, Vogelsang H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999;11:1185–1194. doi: 10.1097/00042737-199910000-00019. [DOI] [PubMed] [Google Scholar]
- Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- Dube C, Rostom A, Sy R, Cranney A, Saloojee N, Garritty C, et al. The prevalence of celiac disease in average-risk and at-risk Western European populations: a systematic review. Gastroenterology. 2005;128:S57–S67. doi: 10.1053/j.gastro.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Vilppula A, Kaukinen K, Luostarinen L, Krekela I, Patrikainen H, Valve R, et al. Increasing prevalence and high incidence of celiac disease in elderly people: a population-based study. BMC Gastroenterol. 2009;9:49–. doi: 10.1186/1471-230X-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffenberg EJ, MacKenzie T, Barriga KJ, Eisenbarth GS, Bao F, Haas JE, et al. A prospective study of the incidence of childhood celiac disease. J Pediatr. 2003;143:308–314. doi: 10.1067/s0022-3476(03)00282-8. [DOI] [PubMed] [Google Scholar]
- Lohi S, Mustalahti K, Kaukinen K, Laurila K, Collin P, Rissanen H, et al. Increasing prevalence of coeliac disease over time. Aliment Pharmacol Ther. 2007;26:1217–1225. doi: 10.1111/j.1365-2036.2007.03502.x. [DOI] [PubMed] [Google Scholar]
- Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- Green PHR, Stavropoulos SN, Panagi SG, Goldstein SL, Mcmahon DJ, Absan H, et al. Characteristics of adult celiac disease in the USA: results of a national survey. Am J Gastroenterol. 2001;96:126–131. doi: 10.1111/j.1572-0241.2001.03462.x. [DOI] [PubMed] [Google Scholar]
- Logan I, Bowlus CL. The geoepidemiology of autoimmune intestinal diseases. Autoimmun Rev. 2010;9:A372–A378. doi: 10.1016/j.autrev.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Silano M, Agostoni C, Guandalini S. Effect of the timing of gluten introduction on the development of celiac disease. World J Gastroenterol. 2010;16:1939–1942. doi: 10.3748/wjg.v16.i16.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akobeng AK, Ramanan AV, Buchan I, Heller RF. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child. 2006;91:39–43. doi: 10.1136/adc.2005.082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostoni C, Decsi T, Fewtrell M, Goulet O, Kolacek S, Koletzko B, et al. Complementary feeding: a commentary by the ESPGHAN Committee on Nutrition. J Pediatr Gastroenterol Nutr. 2008;46:99–110. doi: 10.1097/01.mpg.0000304464.60788.bd. [DOI] [PubMed] [Google Scholar]
- Plot L, Amital H. Infectious associations of Celiac disease. Autoimmun Rev. 2009;8:316–319. doi: 10.1016/j.autrev.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. 2006;101:2333–2340. doi: 10.1111/j.1572-0241.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- Greco L, Romino R, Coto I, Di CN, Percopo S, Maglio M, et al. The first large population based twin study of coeliac disease. Gut. 2002;50:624–628. doi: 10.1136/gut.50.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Mowat AM. Coeliac disease–a meeting point for genetics, immunology, and protein chemistry. Lancet. 2003;361:1290–1292. doi: 10.1016/s0140-6736(03)12989-3. [DOI] [PubMed] [Google Scholar]
- Festen EA, Szperl AM, Weersma RK, Wijmenga C, Wapenaar MC. Inflammatory bowel disease and celiac disease: overlaps in the pathology and genetics, and their potential drug targets. Endocr Metab Immune Disord Drug Targets. 2009;9:199–218. doi: 10.2174/187153009788452426. [DOI] [PubMed] [Google Scholar]
- Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol. 2002;283:G996–G1003. doi: 10.1152/ajpgi.00136.2002. [DOI] [PubMed] [Google Scholar]
- Fleckenstein B, Molberg O, Qiao SW, Schmid DG, von der MF, Elgstoen K, et al. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. 2002;277:34109–34116. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998. [DOI] [PubMed]
- Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de WY, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- Quarsten H, Molberg O, Fugger L, McAdam SN, Sollid LM. HLA binding and T cell recognition of a tissue transglutaminase- modified gliadin epitope. Eur J Immunol. 1999;29:2506–2514. doi: 10.1002/(SICI)1521-4141(199908)29:08<2506::AID-IMMU2506>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- Szabolcs M, Sipka S, Csorba S. In vitro cross-linking of gluten into high-molecular-weight polymers with transglutaminase. Acta Paediatr Hung. 1987;28:215–227. [PubMed] [Google Scholar]
- Tack GJ, Verbeek WH, Schreurs MW, Mulder CJ. The spectrum of celiac disease: epidemiology, clinical aspects and treatment. Nat Rev Gastroenterol Hepatol. 2010;7:204–213. doi: 10.1038/nrgastro.2010.23. [DOI] [PubMed] [Google Scholar]
- Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346:180–188. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- Holtmeier W, Henker J, Riecken EO, Zimmer KP.Definitions of celiac disease–statement of an expert group from the German Society for Celiac Disease Z Gastroenterol 200543751–754.German. [DOI] [PubMed] [Google Scholar]
- Collin P, Reunala T, Pukkala E, Laippala P, Keyrilainen O, Pasternack A. Coeliac disease-associated disorders and survival. Gut. 1994;35:1215–1218. doi: 10.1136/gut.35.9.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Green PH. Extraintestinal manifestations of celiac disease. Curr Gastroenterol Rep. 2006;8:383–389. doi: 10.1007/s11894-006-0023-7. [DOI] [PubMed] [Google Scholar]
- Hansen D, Brock-Jacobsen B, Lund E, Bjorn C, Hansen LP, Nielsen C, et al. Clinical benefit of a gluten-free diet in type 1 diabetic children with screening-detected celiac disease: a population-based screening study with 2 years' follow-up. Diabetes Care. 2006;29:2452–2456. doi: 10.2337/dc06-0990. [DOI] [PubMed] [Google Scholar]
- Stenson WF, Newberry R, Lorenz R, Baldus C, Civitelli R. Increased prevalence of celiac disease and need for routine screening among patients with osteoporosis. Arch Intern Med. 2005;165:393–399. doi: 10.1001/archinte.165.4.393. [DOI] [PubMed] [Google Scholar]
- Cerqueira RM, Rocha CM, Fernandes CD, Correia MR. Celiac disease in Portuguese children and adults with Down syndrome. Eur J Gastroenterol Hepatol. 2010;22:868–871. doi: 10.1097/meg.0b013e3283328341. [DOI] [PubMed] [Google Scholar]
- Whitaker JK, West J, Holmes GK, Logan RF. Patient perceptions of the burden of coeliac disease and its treatment in the UK. Aliment Pharmacol Ther. 2009;29:1131–1136. doi: 10.1111/j.1365-2036.2009.03983.x. [DOI] [PubMed] [Google Scholar]
- Mitea C, Havenaar R, Drijfhout JW, Edens L, Dekking L, Koning F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: implications for coeliac disease. Gut. 2008;57:25–32. doi: 10.1136/gut.2006.111609. [DOI] [PubMed] [Google Scholar]
- Catassi C, Bearzi I, Holmes GK. Association of celiac disease and intestinal lymphomas and other cancers. Gastroenterology. 2005;128:S79–S86. doi: 10.1053/j.gastro.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Sugai E, Moreno ML, Hwang HJ, Cabanne A, Crivelli A, Nachman F, et al. Celiac disease serology in patients with different pretest probabilities: is biopsy avoidable. World J Gastroenterol. 2010;16:3144–3152. doi: 10.3748/wjg.v16.i25.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo L. Celiac disease. World J Gastroenterol. 2006;12:6585–6593. doi: 10.3748/wjg.v12.i41.6585. [DOI] [PubMed] [Google Scholar]
- Sweis R, Pee L, Smith-Laing G. Discrepancies between histology and serology for the diagnosis of coeliac disease in a district general hospital: is this an unrecognised problem in other hospitals. Clin Med. 2009;9:346–348. doi: 10.7861/clinmedicine.9-4-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostom A, Dube C, Cranney A, Saloojee N, Sy R, Garritty C, et al. The diagnostic accuracy of serologic tests for celiac disease: a systematic review. Gastroenterology. 2005;128:S38–S46. doi: 10.1053/j.gastro.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Matthias T, Pfeiffer S, Selmi C, Eric GM. Diagnostic challenges in celiac disease and the role of the tissue transglutaminase-neo-epitope. Clin Rev Allergy Immunol. 2010;38:298–301. doi: 10.1007/s12016-009-8160-z. [DOI] [PubMed] [Google Scholar]
- Lewis NR, Scott BB. Meta-analysis: deamidated gliadin peptide antibody and tissue transglutaminase antibody compared as screening tests for coeliac disease. Aliment Pharmacol Ther. 2010;31:73–81. doi: 10.1111/j.1365-2036.2009.04110.x. [DOI] [PubMed] [Google Scholar]
- Cataldo F, Marino V, Ventura A, Bottaro G, Corazza GR. Prevalence and clinical features of selective immunoglobulin A deficiency in coeliac disease: an Italian multicentre study. Italian Society of Paediatric Gastroenterology and Hepatology (SIGEP) and ‘Club del Tenue' Working Groups on Coeliac Disease. Gut. 1998;42:362–365. doi: 10.1136/gut.42.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo F, Lio D, Marino V, Picarelli A, Ventura A, Corazza GR. IgG(1) antiendomysium and IgG antitissue transglutaminase (anti-tTG) antibodies in coeliac patients with selective IgA deficiency. Working Groups on Celiac Disease of SIGEP and Club del Tenue. Gut. 2000;47:366–369. doi: 10.1136/gut.47.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich W, Esslinger B, Schuppan D. Pathomechanisms in celiac disease. Int Arch Allergy Immunol. 2003;132:98–108. doi: 10.1159/000073710. [DOI] [PubMed] [Google Scholar]
- Lewis NR, Scott BB. Systematic review: the use of serology to exclude or diagnose coeliac disease (a comparison of the endomysial and tissue transglutaminase antibody tests) Aliment Pharmacol Ther. 2006;24:47–54. doi: 10.1111/j.1365-2036.2006.02967.x. [DOI] [PubMed] [Google Scholar]
- Skovbjerg H, Koch C, Anthonsen D, Sjostrom H. Deamidation and cross-linking of gliadin peptides by transglutaminases and the relation to celiac disease. Biochim Biophys Acta. 2004;1690:220–230. doi: 10.1016/j.bbadis.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Skovbjerg H, Noren O, Anthonsen D, Moller J, Sjostrom H. Gliadin is a good substrate of several transglutaminases: possible implication in the pathogenesis of coeliac disease. Scand J Gastroenterol. 2002;37:812–817. [PubMed] [Google Scholar]
- Fleckenstein B, Qiao SW, Larsen MR, Jung G, Roepstorff P, Sollid LM. Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J Biol Chem. 2004;279:17607–17616. doi: 10.1074/jbc.M310198200. [DOI] [PubMed] [Google Scholar]
- Vermeersch P, Geboes K, Marien G, Hoffman I, Hiele M, Bossuyt X. Diagnostic performance of IgG anti-deamidated gliadin peptide antibody assays is comparable to IgA anti-tTG in celiac disease. Clin Chim Acta. 2010;411:931–935. doi: 10.1016/j.cca.2010.02.060. [DOI] [PubMed] [Google Scholar]
- Volta U, Granito A, Parisi C, Fabbri A, Fiorini E, Piscaglia M, et al. Deamidated gliadin peptide antibodies as a routine test for celiac disease: a prospective analysis. J Clin Gastroenterol. 2010;44:186–190. doi: 10.1097/MCG.0b013e3181c378f6. [DOI] [PubMed] [Google Scholar]
- Dewar D, Pereira SP, Ciclitira PJ. The pathogenesis of coeliac disease. Int J Biochem Cell Biol. 2004;36:17–24. doi: 10.1016/s1357-2725(03)00239-5. [DOI] [PubMed] [Google Scholar]
- Dahle C, Hagman A, Ignatova S, Strom M. Antibodies against deamidated gliadin peptides identify adult coeliac disease patients negative for antibodies against endomysium and tissue transglutaminase. Aliment Pharmacol Ther. 2010;32:254–260. doi: 10.1111/j.1365-2036.2010.04337.x. [DOI] [PubMed] [Google Scholar]
- Reeves GE, Squance ML, Duggan AE, Murugasu RR, Wilson RJ, Wong RC, et al. Diagnostic accuracy of coeliac serological tests: a prospective study. Eur J Gastroenterol Hepatol. 2006;18:493–501. doi: 10.1097/00042737-200605000-00006. [DOI] [PubMed] [Google Scholar]
- Remes-Troche JM, Rios-Vaca A, Ramirez-Iglesias MT, Rubio-Tapia A, Andrade-Zarate V, Rodriguez-Vallejo F, et al. High prevalence of celiac disease in Mexican Mestizo adults with type 1 diabetes mellitus. J Clin Gastroenterol. 2008;42:460–465. doi: 10.1097/MCG.0b013e318046ea86. [DOI] [PubMed] [Google Scholar]
- Lutteri L, Sagot C, Chapelle JP.Anti-deamidated gliadin peptides antibodies and coeliac disease: state of art and analysis of false-positive results from five assays Ann Biol Clin (Paris) 201068149–156.French. [DOI] [PubMed] [Google Scholar]
- Tonutti E, Visentini D, Fabris D, Blasone N, Molinaro P, Pavan E, et al. Antibodies to the Transglutaminase-deamidated gliadin peptides complex: a new serological approach to the diagnosis of celiac disease.Presented at 7th International Congress on Autoimmunity; 5–9 May 2010; Ljubliana, Slovenia. Kenes International: Geneva, Switzerland, 2010.
- Tjon JM, van BJ, Koning F. Celiac disease: how complicated can it get. Immunogenetics. 2010;62:641–651. doi: 10.1007/s00251-010-0465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catassi C, Fasano A. Celiac disease diagnosis: simple rules are better than complicated algorithms. Am J Med. 2010;123:691–693. doi: 10.1016/j.amjmed.2010.02.019. [DOI] [PubMed] [Google Scholar]