

Abstract
Immunoglobulin A (Iga)-isotype antibodies play an important role in immunity owing to their structure, glycosylation, localization and receptor interactions. Dysfunctions in this system can lead to multiple types of pathology. This review describes the characteristics of Iga and discusses the involvement of abnormalities in the Iga system on the development of celiac disease and Iga nephropathy.
Keywords: celiac disease, Iga nephropathy, immunoglobulin A, receptors
INTRODUCTION
Immunoglobulin A (Iga) is a relatively unique antibody isotype in humans due to its heterogeneity in molecular forms, subclasses and glycosylation, as well as its multiple roles, which vary from protection of mucosal surfaces to prevention of autoimmunity and development of inflammation.1, 2 Iga antibodies are by far the most abundant antibody isotype in humans and are differentially distributed between the systemic and mucosal immune system. Around 66 mg/kg of Iga are produced daily, particularly at epithelial sites, which considerably exceeds the production of all other immunoglobulin classes in the body.3 Serum Iga, the second most abundant isotype in circulation, consists mainly of monomers produced by bone marrow plasma cells,1 whereas secretory Iga (SIga) molecules are synthesized as dimers by local plasma cells before being transported to mucosal surfaces via epithelial cells, based on their interaction with the polymeric Ig receptor (pIgR).4 Signals delivered by monomeric serum Iga (mIga) are essential in controlling the immune system by preventing the development of autoimmunity and inflammation. Polymeric Iga (pIga) participates in immune responses by inducing the activation of eosinophils, neutrophils, monocytes and macrophages. However, aggregated IgA can also be deleterious to the host by inducing inflammatory diseases in various organs. This review highlights recent data that are important for understanding the role of IgA and its dysfunctions in the development of intestinal and renal diseases.
STRUCTURE OF IgA
IgA displays a T-shaped structure, which differs from the common Y shape of other Ig molecules.5 The monomeric structural unit of IgA comprises two identical heavy chains and two identical light chains corresponding to a total size of 160 kDa. In humans, the light chain contains one variable and one constant region, whereas the heavy chain is composed of three constant regions (CH1, CH2 and CH3). The latter has a molecular mass around 60 kDa, slightly larger than that of IgG because of its heavier glycosylation. The N-terminal variable region, the CH1 of one heavy chain and the entire light chain constitute the Fab fragment, which is responsible for antigen recognition. The remaining two or three constant domains of the heavy chains constitute the C-terminal Fc fragment, which mediates interactions with various receptors and effector molecules.6 The two Fab regions and the Fc region of a monomeric IgA molecule are separated by a flexible hinge region. With the exception of the upper domains of the Fc region (the CH1 domains), the domains are arranged in pairs, stabilized by numerous non-covalent trans interactions.7
In humans, IgAs are divided into closely related subclasses, IgA1 and IgA2, which differ by the absence of a 13-amino acid sequence in the hinge region of the IgA2 molecule.1 The lack of this region in IgA2 allows it to be resistant to the action of bacterial proteases (i.e., those from Streptococcus mutants, Neisseria meningitidis and Haemophilus influenzae) that cleave IgA1 in the hinge region and may underlie the predominance of IgA2 in mucosal secretions.7
IgA SYNTHESIS
The forms of IgA in serum and mucosal secretions, the two main compartments in which these antibodies are found, differ. In serum, IgA exists mainly in its monomeric form (about 85–90%). This form is produced in the bone marrow and in some peripheral lymphoid organs by plasma cells derived from B cells activated in lymph nodes.8 The ratio of IgA1/IgA2 in blood is about 10∶1. IgA constitutes one-fifth of the total Ig pool due to a rapid catabolism (half life: 3–6 days). In mucosal secretions (saliva, colostrum, gastrointestinal fluids and urine), local plasma cells produce IgA in its polymeric form (pIgA). pIgA exists almost exclusively as dimers linked by a polypeptide called the J chain, and the ratio of IgA1/IgA2 in mucosa is about 3∶2. The percentage about 12% of pIgA in the serum is low.9
Naive B-cell precursors of IgA-secreting plasma cells are activated in Peyer's patches and mesenteric lymph nodes. Class switching of naive B lymphocytes to produce IgA occurs following stimulation with transforming growth factor-β and other cytokines (interleukin-4 (IL-4), IL-6 and IL-10) in the organized lymphoid tissues of the gut-associated lymphoid tissue.10 After B-cell activation and differentiation, the resulting lymphoblasts upregulate the expression of the gut-homing receptors a4β7, CCR9 and/or CCR10, recirculate via the thoracic duct, and home to the intestinal lamina propria.11 Once in the lamina propria, plasma cells synthesize and secrete intact J chain-linked IgA dimers with increased avidity for antigen into the subepithelial space.12 To reach target antigens in the gut lumen, the IgA molecules must be transported across the epithelium. This process is performed by immature epithelial cells located at the base of the intestinal crypts, which express the pIgR on their basolateral surfaces. This receptor has a high affinity for J chain-linked polymeric immunoglobulins and transports the antibody by transcytosis to the laminal surface of the epithelium, where it is released by proteolytic cleavage of the extracellular domain of the pIgR. Part of the cleaved receptor remains associated with the IgA and is known as secretory component, while the resulting antibody is referred as SIgA.13
Interestingly, there are substantial differences in the IgA system between species, particularly in man compared to mouse and rat. While two IgA subclasses are recognized in humans, only one class exists in mice and rats and it contains a shorter hinge region.14 Serum IgA is mostly monomeric in humans and polymeric in mice. Clearance via the hepatobiliary route plays an important role in mice, but not in humans.1 Moreover, in humans intestinal SIgA originates only from the gut-associated lymphoid tissue, but is generated from two sources in mice: B2 lymphocytes in organized germinal centers of mucosal lymphoid tissues such as Peyer's patches (T lymphocyte-dependent IgA production) and B1 lymphocytes developed in the peritoneal cavity and distributed in the intestinal lamina propria (T lymphocyte-independent IgA production).7
IgA GLYCOSYLATION
IgA is the most glycosylated form of Ig. Both subclasses carry a number of N-linked carbohydrates, contributing 6–7% of molecular mass of IgA1 and 8–10% of IgA2.15 The heavy chains of IgA1 molecules contain a unique insertion in the hinge-region segment between the CH1 and CH2 region domains, unlike in IgA2. This hinge region has a high content of proline, serine and threonine residues, the last two being the sites of attachment of up to five O-linked glycan chains consisting of N-acetylgalactosamine (GalNAc).16 Galactose (Gal) may be linked to the GalNAc by a specific enzyme, β-1,3-galactosyltransferase, to form the disaccharide Gal-β-1,3-GalNAc. Gal chains may be extended with one or two sialic acid (NeuAc) units that are added by α-2,3-sialyltransferases. Sialic acid units may also be added to GalNAc by α-2,6-sialyltransferases. It is noteworthy, however, that the carbohydrate composition of the O-linked glycans in the hinge region of serum IgA1 is heterogeneous. The most common forms include GalNAc-Gal disaccharide and its mono- and di-sialylated forms. Gal-deficient variants with terminal GalNAc or sialylated GalNAc exist, but represent a minor percentage of the O-glycans in serum IgA1.16 SIgA contains a secretory component with seven N-glycosylation sites and a J chain with one N-glycan. Secretory component, through its carbohydrate residues, ensures the appropriate in vivo localization of SIgA by anchoring it to the mucosal lining of the epithelial surface and protects against bacterial infections.17
IgA FUCTION
SIgA molecules in the intestinal lumen serve a variety of functions at three anatomical levels in the mucosal epithelium. In the gut lumen, high-affinity IgA antibodies from T cell-dependent pathways bind to the layer of mucus, prevent the adherence and invasion of pathogenic microorganisms and neutralize pathogen toxins or enzymes.18 Conversely, low-affinity IgA emerging from T cell-independent pathways sequester commensal bacteria of the intestinal lumen through a process known as ‘immune exclusion'.10 SIgA can inhibit the entry of these bacteria by surrounding pathogens with a hydrophilic shell that is repelled by the mucin glycocalyx at mucosal surfaces.14 IgA in transit through the epithelium can bind to proinflammatory antigens that then are neutralized once in the luminal side. Moreover, SIgA can inhibit virus production via intracellular interception of viral antigens during transepithelial IgA transport.18 In the lamina propria, IgA bind and transport antigens into the lumen via the pIgR at the basolateral side of the epithelial cells by transcytosis or using FcαRI-bearing phagocytes.14 In addition to controlling pathogens and commensals, IgA antibodies inside epithelial cells neutralize microbial products that have proinflammatory activity, such as lipopolysaccharide.4 Hence, IgA can play a role both at normal conditions and in infection.
SIgA has little capacity to activate the classical pathway of complement or to act as an opsonin, so cannot induce inflammation. Its main function is to limit the access of pathogens to mucosal surfaces without risking inflammatory damage to these fragile tissues. It seems that our adaptive immune system drives the diversification of bacterial surface structures by exposing the bacteria in the gut to IgA.19 IgA can further participate in intestinal homeostasis by interacting with the local microbiota and contributing to restriction of these organisms to the gut lumen.10 In principle, deregulation of intestinal antibody responses to commensal bacteria might result in an excessive innate immune response, which in turn could precipitate or aggravate intestinal inflammation. For instance, in patients with Crohn's disease, IgG and IgA antibodies against Saccharomyces cerevisiae or microbial flagellin may play a role in shaping the microbiota20 with effects on both the host and the microorganism. Altogether, the available evidence suggests that IgA is important not only to sequester bacteria in the intestinal lumen, but also to shape the overall composition of the intestinal microbiota. Deregulation of these processes may trigger inflammatory disorders in the intestine like celiac disease (CD) or Crohn's disease or in other organs such as the kidney in IgA nephropathy (IgAN).
Whereas the role of SIgA in mucosal immunology is established, the function of serum IgA antibodies is largely unknown. In humans, IgA plays both pro- and anti-inflammatory roles and interfaces with the mucosal and systemic immune systems.21 In general, mIgA antibodies are particularly abundant in systemic compartments and the major role of serum mIgA in physiology is as a powerful anti-inflammatory effector of the immune system.22 Several groups have demonstrated that in the absence of antigen, serum IgA downregulates IgG-mediated phagocytosis, chemotaxis, bactericidal activity, oxidative burst activity and cytokine release.23, 24, 25, 26, 27, 28, 29 These IgA monomers may initiate FcαRI-mediated non-inflammatory responses against bacteria that breach the mucosal barrier.30 Another piece of data supporting the inhibitory role of serum mIgA is the observation that selective IgA-deficient patients frequently present allergies and autoimmunity.31 On the contrary, pIgA and IgA containing immune complexes (ICs) may trigger inflammatory responses by blood leukocytes through IgA Fc receptors in a non-mucosal context. Indeed, IgA polymers are frequently found in several autoimmune conditions and might be an aggravating factor in IgAN, as recently shown in a spontaneous model of this disease.32 At the same time, the interaction of serum IgA with FcαRI on tissue phagocytic cells can act as a second line of defense in the case of bacterial infections following penetration across the mucosal barrier.33 In any case, this dual nature of IgA molecules is mainly due to their heterogeneity in molecular forms and their interaction with IgA receptors.
IgA receptors
The anti-inflammatory action of serum mIgA was an enigma until the discovery of IgA Fc receptors. Fc receptors are defined by their specificity for the Fc fragment of immunoglobulin isotypes, and receptors for IgA are referred to as FcαR.14 Although they are not structurally related, there are five types of IgA receptors. Three are considered bona fide FcαRs. The first one is designated FcαRI (CD89) and is a receptor specific for IgA that is capable of binding both human IgA1 and IgA2 subclasses. The second type is the pIgR, which is expressed on the surface of epithelial cells in the mucosa and participates in the transcytosis of IgM and dimeric IgA across epithelial barriers. The third receptor type Fcα/μR binds IgA and IgM and is expressed by the majority of B cells and macrophages.2 The two alternative IgA receptors are the asialoglycoprotein receptor, expressed on hepatocytes, which recognizes terminal Gal residues on serum glycoproteins (including IgA) and conveys bound ligand for intracellular degradation, and the transferrin receptor (TfR), which selectively binds pIgA1 and is highly expressed on renal mesangial cells.34
Of these IgA receptors, the FcαRI plays an essential anti-inflammatory role in immunity allowing the transmission of inhibitory signals following binding of mIgA. FcαRI is expressed on cells of the myeloid lineage including: neutrophils; monocytes; tonsillar, splenic, and alveolar macrophages and Kupffer cells; eosinophils; and subpopulations of dendritic cells.35 FcαRI can bind both IgA1 and IgA2 with moderate affinity (Ka≈106 M−1) at the boundary between the CH2 and CH3 domains. This receptor has been shown to bind pIgA and IgA ICs with greater avidity than mIgA.21 Analysis of the FcαRI three-dimensional structure reveals that it binds IgA differently than other Ig isotypes. Its two Ig-like domains are oriented at right angles and two FcαRI molecules can bind one IgA molecule in the EC1 domain (Figure 1) at a site completely different from the other FcRs, yielding a 2∶1 stoichiometry compared to the 1∶1 stoichiometry of IgE and IgG to their FcRs.36 The FcαRI gene is not located in the FcR gene cluster but on chromosome 19, inside the leukocyte receptor cluster. Interestingly, as with IgA, FcαRI also differs substantially between species as mice fail to express an FcαRI homolog. This receptor is a transmembrane protein called α-chain in which the binding site is localized in the extra-cellular domain D1. This single α-chain does not transmit activation. However, the α-chain of FcαRI can be associated with a homodimer of γ-chains capable of activating the cell due to the presence of an immunoreceptor tyrosine-based activation motif (ITAM)-signaling motif.37
Figure 1.
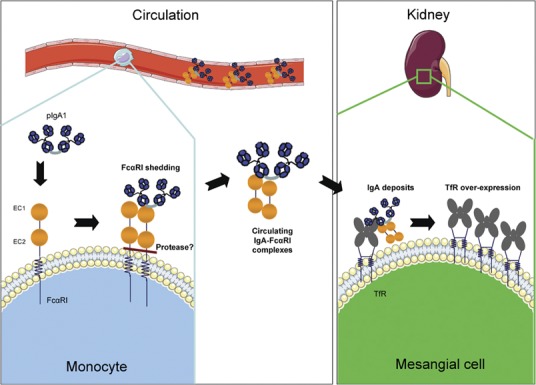
IgA–FcαRI complexes and IgA nephropathy. Polymeric and hypoglycosylated IgA1 can bind FcαRI expressed by myeloid cells and induce its shedding. IgA–FcαRI complexes are formed in circulation and deposited in the mesangium. Binding of these complexes to TfR induces TfR overexpression on mesangial cells and proliferation and activation of these cells. IgA, immunoglobulin A; TfR, transferrin receptor.
We have demonstrated for the first time that monovalent targeting of FcαRI by mIgA inhibits IgG-mediated phagocytosis in human monocytes,30 which explains the previously reported inhibitory functions of serum mIgA.23, 24, 25, 27, 28, 29 More importantly, we have been able to show that FcαRI-mediated inhibition is not mediated by conventional inhibitory motifs such as the immunoreceptor tyrosine-based inhibition motif. While γ-less FcαRI recycles mIgA, thus playing an essential role in mIgA homeostasis, FcRγ-associated FcαRI mediates either activating or inhibitory responses.30 This receptor acts as a dual receptor, depending on whether the ligand is present as a multimer or a monomer. Hence, binding of mIgA to FcαRI leads to reduced phosphorylation of an ITAM embedded within the FcRγ chain and elicits recruitment of src homology domain 2-containing protein-tyrosine phosphatase-1, a signal inhibitor that prevents inflammation by interfering with the activation of multiple signaling pathways. Conversely, crosslinking of the FcαRI by pIgA causes inflammation by triggering full phosphorylation of FcγR and subsequent recruitment of Syk, a protein tyrosine kinase linked to multiple proinflammatory signaling pathways.30, 38
Thus, IgA-containing ICs that are retrotranscytosed across epithelial cells may initiate non-inflammatory or inflammatory immune responses, depending on the monomeric or polymeric nature of the IgA. Monovalent targeting of FcαRI anti-FcαRI Fab fragments prevented asthma or nephritis development in FcαRI transgenic mice.30, 38 Enhanced FcαRI surface expression has been observed on eosinophils from allergic patients39 and on monocytes from patients with gram-negative bacteremia.40 Increased levels of IgA antibodies against allergens and bacterial antigens have been documented in the sputum of atopic asthmatic individuals. Whether increased FcαRI expression exerts a protective or harmful role in these diseases remains to be established.
Another receptor that leads to the implication of IgA in mucosal immunity is the TfR, also known as CD71. TfR is a homodimeric transmembrane glycoprotein that is ubiquitously expressed except on erythrocytes. TfR is also expressed on the surface of mesangial cells and binds to IgA1, with a higher affinity to polymeric hypoglycosylated forms, compared with FcαRI. We, together with Nadine Cerf-Bensoussan's group, have recently shown that TfR can also mediate the apical-to-basolateral transcytosis of antigens bound to IgA across duodenal epithelial cells.41
IgA DYSFUNCTIONS IN INTESTINAL AND RENALl DISEASESZ
Dysfunction in the intestine: the example of Celiac Disease
An important aspect of IgA is its utility as a diagnostic biomarker in several diseases, such as celiac and Crohn's diseases. CD is an autoimmune disease triggered by a dysregulated immune response to wheat gluten proteins, particularly gliadins, in genetically predisposed individuals. CD is characterized by villous atrophy, crypt hyperplasia and increased infiltration of intraepithelial lymphocytes.42
The pathogenic mechanism of this disease is now becoming clear. Intact gliadin 33-mers or p31-49 peptides complexed with IgA are retrotranscytosed from the apical to the basolateral side of epithelial cells via TfR, an IgA receptor expressed on these cells (Figure 2).41 These peptides would be fully degraded if transported via fluid-phase transcytosis, indicating that epithelial cells must tightly control retrotranscytosis to avoid tissue damage. Such control is lost in CD, given that intestinal epithelial cells from celiac patients display an increased expression of TfR.41
Figure 2.
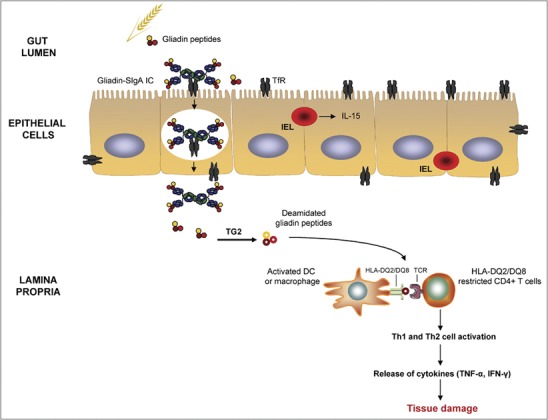
Role of IgA in the pathogenesis of celiac disease. Intact SIgA–gliadin peptide complexes are retrotranscytosed across the lamina propria via TfR, which is abnormally expressed on the apical pole of epithelial cells. Tissue TG2 selectively deamidates gliadin peptides, which bind with high affinity to HLA-DQ2 or HLA-DQ8 molecules of antigen-presenting cells. These cells present deamidated gliadin peptides to HLA-DQ2- or HLA-DQ8-restricted populations of CD4+ T cells that become activated and release inflammatory mediators that lead to tissue damage. IL-15, produced by IELs, also participates in the activation of cytotoxicity against enterocytes. IC, immune complex; IEL, intraepithelial lymphocyte; IFN, interferon; IgA, immunoglobulin A; IL, interleukin; SIgA, secretory IgA; TfR, transferrin receptor; TG2, transglutaminase 2; TNF, tumor necrosis factor.
This TfR-mediated retrotranscytosis triggers intestinal inflammation as a result of a progressive accumulation of toxic gliadin peptides in subepithelial areas, initiating CD enteropathy. In the lamina propria, gliadin peptides encounter tissue transglutaminase 2 (TG2), the celiac autoantigen,43 which selectively deamidates gluten protein, converting glutamine into glutamic acid (Figure 2).44 This post-translational process may link the immune response against wheat with autoimmunity in the gut. The antigen-presenting cells present deamidated gliadin peptides to HLA-DQ2- or HLA-DQ8-restricted populations of CD4+ T cells that then become activated and differentiate into Th1 and Th2 cells.42 These cells release mediators, such as interferon-γ and tumor necrosis factor-α,45 that ultimately lead to tissue damage and the production of autoantibodies.46 During the innate immune response in CD, IL-15 is upregulated on the surface of enterocytes. IL-15, in turn, induces the secretion of interferon-γ, expression of natural killer receptors on intraepithelial cells and the activation of cytotoxicity against enterocytes.47
Untreated celiac patients typically have increased IgA and IgG antibodies against gliadin, endomysium (the substrate of TG2) and TG2 in their serum. IgA antibodies against both gliadin and TG2 are more specific for CD than are the corresponding IgG antibodies.48 It has been reported that the production of anti-TG2 IgA autoantibodies may occur in CD patients with HLA types typical of CD even in the absence of clear morphological changes.49 Celiac autoantibodies are produced in the small intestinal mucosa, and it has been recognized that the small intestinal epithelial membrane of CD patients contains deposited IgA.50 Korponay-Szabo et al. have shown that deposited extracellular IgA in the small intestinal mucosa targets TG2 and connective tissue in untreated CD patients51 and have proposed that anti-TG2 IgA antibodies may be predictors of CD.49 However, it should be pointed out that IgA deficiency is more frequent among celiac patients (1 in 40) than the general population (1 in 400), suggesting a secondary rather than a primary involvement of IgA in the pathogenesis of this intestinal disease.52
Dysfunction in the kidney: the example of IgA Nephropathy
IgAN is the most common form of glomerulonephritis in the developed world (ranging from 20 to 40% of primary glomerulonephritis) and it is an important cause of end-stage kidney failure. The presence of mesangial IgA became a major criterion for the diagnosis of IgAN, while features of the disease include mesangial cell proliferation, matrix expansion and clinical symptoms of renal injury, such as hematuria and proteinuria.53, 54
The pathogenesis of IgAN seems to be linked to abnormalities of the IgA system. Indeed, recurrence of IgA deposits in IgAN patients after transplantation of a normal kidney indicates that circulating rather than local kidney abnormalities are crucial for the development of IgAN.55 The level of serum IgA is two- to threefold higher in approximately half of IgAN patients.54, 56 Increased circulating macromolecular IgA molecules were identified as ICs containing pIgA56, 57 of the IgA1 subclass, but not of the IgA2 subclass.57 Idiotypic determinants are shared between the circulating complexes and the mesangial deposits;58 however, disease-specific idiotypes have not been identified.59 Furthermore, mesangial IgA deposits have been shown to be primarily of the IgA1 subclass and to be composed largely of pIgA.60 IgA size seems to play a significant role in the pathogenicity of the IgA IC. Indeed, polymeric but not monomeric IgA1 induces TfR hyperexpression on human mesangial cells,61 leading to increased production of cytokines (IL-6, transforming growth factor-β and tumor necrosis factor-α), mesangial expansion and pIgA1 mesangial deposition.62, 63
Studies employing lectins, which bind to glycosidic residues, revealed that circulating and mesangial IgA1 molecules from IgAN patients possess truncated O-glycans.64 These aberrantly glycosylated molecules are deposited in the mesangium in IgAN patients.65 Moreover, hypoglycosylation of IgA may lead to self-aggregation and formation of IgA1–IgA1 and IgA1–IgG ICs.66, 67, 68 Interestingly, it has been shown that dimers and polymers of IgA aggregate more readily than monomers,69 which might explain the propensity of this form to aggregate and deposit in tissues. Our group has also shown that aberrant glycosylation of IgA1 and IC formation constitute essential factors favoring mesangial IgA1–TfR interactions as initial steps in IgAN pathogenesis.61
The formation of the circulating IC may involve at least three distinct mechanisms: the self-aggregation of aberrantly glycosylated IgA1; the formation of complexes with soluble FcαRI; and the induced aggregation by interaction of IgA1 with other circulating proteins. As discussed above, the first mechanism is directly linked to the abnormal structure of IgA1. The second mechanism relies on an interaction with myeloid cell-expressed FcαRI, which binds mIgA of both IgA subclasses with low affinity and pIgA with high avidity.14 IgAN patients exhibit increased IgA1–FcαRI complexes in their serum compared to normal individuals.70 The mechanism proposed to explain this phenomenon involves the shedding of the FcαRI extracellular domain.71 Cleavage is promoted by aggregation of FcRγ-less FcαRI.32 To demonstrate the role of FcαRI in IgAN, we generated human FcαRI transgenic mice, which have been shown to spontaneously develop IgAN.70 Moreover, injection of IgA from IgAN patients into mice that were both immunodeficient and transgenic for FcαRI induced a massive hematuria and IgA deposits.70 These results indicate that alteration of the IgA–FcαRI interaction may play a key role in the development of IgAN. The third mechanism for the formation of IgA complexes in IgAN may involve IgA binding to other circulating proteins such as fibronectin, collagen or laminin,72, 73, 74 suggesting an additional mechanism by which IgA complexes bind to mesangium through adhesion to extracellular matrix proteins. In favor of this hypothesis, knockout mice for uteroglobin, a serum protein that controls fibronectin levels, develop IgAN.75 However, no alterations of uteroglobin have been found in patients.76
The physiopathologic mechanisms of IgAN remain partially unknown. Our hypothesis, based on our experimental data and on data from the literature, is that a defect in the glycosylation of IgA1 from patients with IgAN induces an alteration in the interaction between IgA1 and FcαRI expressed by myeloid cells (Figure 1). As a consequence, cleavage of the extracellular domain of γ-less FcαRI and the formation of nephrotoxic circulating IgA1–FcαRI complexes may occur. Specific antigens may also contribute to the formation of macromolecular IgA1. A feedback loop that is largely responsible for the progression of the disease and its chronicity could take place, leading to a decrease in FcαRI membrane expression and to the decreased clearance of pIgA1. TfR was identified as the mesangial receptor for these IgA1 complexes. A second loop could then take place including IgA1 deposition, inducing an increase in TfR expression, which would, in turn, increase the deposition of the complexes. These deposits would be responsible for the mesangial cell proliferation and for the increased local inflammation after kidney infiltration by leukocytes expressing γ-associated FcαRI, which could be aggravated by defective negative regulation through the FcRγ ITAM and by genetic factors with the final consequence of renal failure.
IgA: a common link between the two diseases?
As described above, IgA plays an important role in various inflammatory diseases, such as CD and IgAN. Two principal questions are raised: (i) Is there a correlation between these two diseases? (ii) How is IgA involved in this possible common pathogenic mechanism? Various studies have shown that IgAN occurs as a primary disease but can be associated with Henoch Shonlein purpura, CD or inflammatory bowel disease.2 Indeed, the prevalence of CD in the general population is 0.5–1% depending on the geographical region and this percentage increases to 4% in patients with IgAN.77 Moreover, several cases have been recently reported of patients with nephrotic syndrome and CD.78, 79, 80 Alternatively, CD may be a risk factor for renal disease, the majority of which would be IgAN. Indeed, glomerular mesangial deposits of IgA occur frequently in untreated CD and they are associated with circulating IC containing IgA.81 However, in this situation IgA seems to be deposited without being able to induce clinically overt glomerulonephritis.
Coppo et al. have observed the presence of IgA antigliadin antibodies, associated with elevated IgA levels, in the serum of IgAN patients.82 In agreement with this finding, another study in patients with IgAN found increased production of serum antigliadin IgA antibodies, suggesting hyperactivity of IgA producing B cells associated with mucosal atrophy.83 Moreover, anti-endomysium IgA and IgG antibodies were detected in the serum of IgAN patients.84 These data suggest that dietary components (e.g. gliadin) may play a role in IgAN by promoting IgA IC formation and perhaps favoring mesangial localization via lectin interactions and these authors propose a possible association between IgAN and CD.77, 81 This suggestion is supported by the finding that treatment of IgAN patients and model animals with a gluten-free diet decreased IgA IC and antigliadin IgA antibody levels in the serum and ameliorated clinical symptoms of the disease, such as proteinuria and hematuria.82, 83, 85, 86 However, progression to renal failure was still observed.
In regard to mucosal immunity in IgAN, Kovács et al. have observed that IgAN patients compared to controls presented significantly higher intestinal permeability, related to increased proteinuria, microhematuria and serum IgA levels.87 Thus, elevated intestinal permeability in IgAN patients may play a role in the pathogenesis of the disease and adversely influence its progression. No increase in celiac-type HLA-DQ was observed in patients with IgAN, indicating that the increased permeability found in IgAN patients may have a role in predisposing patients to CD.77 This elevated intestinal permeability may be associated with the increased number of intestinal intraepithelial T lymphocytes observed in patients with IgAN, suggesting a possible role for breakdown of oral tolerance in the pathophysiology of IgAN.88 Another study demonstrated the presence of rectal mucosal sensitivity to gluten in one-third of IgAN patients without any signs of CD, suggesting that subclinical inflammation to gluten might be involved in the pathogenesis of IgAN in a subgroup of patients.89
The pathogenic mechanism that links IgAN and CD is not yet clear. Lectins, particularly gliadins, can bind polymeric IgA1 containing Gal and GalNac residues (Figure 3).82 This reaction leads to the formation of macromolecular IgA1 ICs. Gliadin can also bind mesangial cells via lectin bonds favoring the bridging of pIgA1 and IgA1 ICs to these cells and thus enhancing both IgA1 mesangial trapping and in situ IgA1 deposit formation. Moreover, gliadin binding to mesangial cells modulates the production of immunological mediators and hemodynamic factors (an increase in tumor necrosis factor-α and inhibition of prostaglandin E2 production).90 These changes might stimulate mesangial cell growth and mesangial matrix production, contributing to IgAN pathogenesis.
Figure 3.
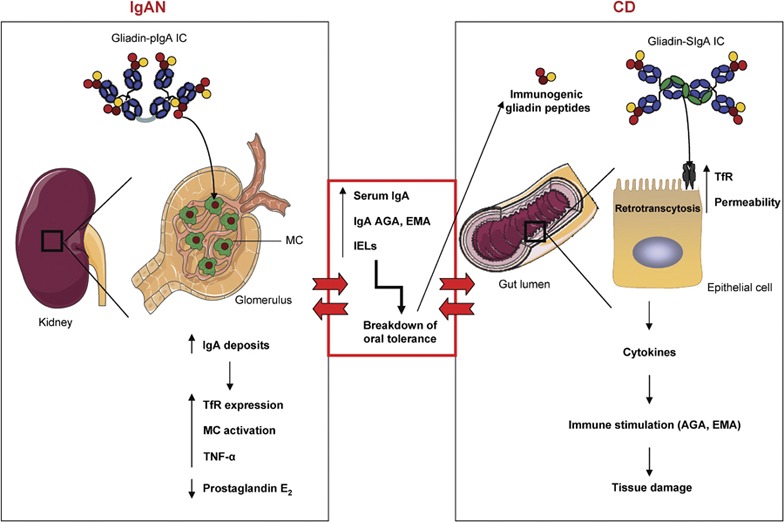
The IgA system constitutes a common link between CD and IgAN. In IgAN patients, gliadins bind pIgA-containing Gal and GalNAc residues, leading to the formation of macromolecular gliadin–IgA ICs. These complexes are deposited on the glomelular mesangium via lectin bonds between gliadin and MCs. This binding induces the overexpression of the mesangial IgA receptor TfR, activates mesangial cells and enhances the in situ IgA deposits. Moreover, binding modulates the production of immunological mediators by mesangial cells (increases in TNF-α and inhibition of prostaglandin E2 production), contributing to IgAN pathogenesis. These patients present with elevated serum IgA, increased production of celiac-specific antibodies (AGA and EMA) and an increase in the number of IELs in the intestine. These dysfunctions are responsible for the breakdown of oral tolerance, which in turn results in perturbations of epithelial cell function and abnormal processing of dietary antigens (immunogenic gliadin peptides). These peptides are retrotranscytosed into the lamina propria via TfR, which is overexpressed on epithelial cells, and induce the release of proinflammatory cytokines. These modifications result in increased intestinal permeability, stimulation of the mucosal immune system and tissue damage, which leads to CD development. Alternatively, the increased intestinal permeability and the presence of circulting IgA-gliadin ICs observed in CD may be a risk factor for the development of IgAN. AGA, antigliadin antibody; CD, celiac disease; EMA, anti-endomysium antibody; Gal, galactose; IgA, immunoglobulin A; IgAN, IgA nephropathy; IC, immune complex; IEL, intraepithelial lymphocyte; MC, mesangial cell; pIgA, polymeric IgA; TfR, transferrin receptor; TNF, tumor necrosis factor. GalNAc, N-acetylgalactosamine.
The possible role of a breakdown in oral tolerance in the pathophysiology of IgAN has been already proposed; however, there is no clear evidence of the triggering factor in this process.88 It seems that the breakdown of oral tolerance may be favored by perturbations of epithelial cell function, resulting in abnormal processing of dietary antigens, such as gliadin, which renders them immunogenic rather than tolerogenic (Figure 3). In this case, the cytokines produced by immune system activation can influence the epithelial cell secretory component and class II antigen presentation. These modifications lead to intestinal permeability changes, allowing increased antigen uptake and presentation along with stimulation of the mucosal immune system, resulting in mucosal inflammatory diseases like CD.82 Furthermore, we have recently shown that the gliadin peptide 31–49 crosses the intestinal barrier in CD patients without being degraded by the lysosomal pathway, using a mechanism similar to that observed in IgAN (overexpression of CD71 at the apical surface of enterocytes).41 To conclude, the efficacy of a gluten-free diet together with the observed increase in intestinal permeability and in serum IgA reactivity to gliadin and the increased levels of salivary and serum SIgA have raised the possibility that the mucosal immune system plays a role in the physiopathology of IgAN.88
CONCLUDING REMARKS
The IgA and IgA receptors play a significant role in vivo in maintaining the integrity of immune responses in the systemic and mucosal compartments. Dysfunctions in IgA, such as abnormal glycosylation, self-aggregation, receptor shedding or production of ICs with dietary antigens, result in inflammatory diseases that can affect both the intestine and the kidney. CD and IgAN exhibit common IgA abnormalities, an essential molecular mechanism that may open new avenues for understanding pathogenesis and developing new therapeutic approaches for these diseases.
References
- Kerr MA. The structure and function of human IgA. Biochem J. 1990;271:285–296. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro RC. Role of IgA and IgA Fc receptors in inflammation. J Clin Immunol. 2011;30:1–9. doi: 10.1007/s10875-009-9338-0. [DOI] [PubMed] [Google Scholar]
- Solomon A. Monoclonal immunoglobulins as biomarkers of cancer. Cancer Markers. 1980;1:57–87. [Google Scholar]
- Fernandez MI, Pedron T, Tournebize R, Olivo-Marin JC, Sansonetti PJ, Phalipon A. Anti-inflammatory role for intracellular dimeric immunoglobulin a by neutralization of lipopolysaccharide in epithelial cells. Immunity. 2003;18:739–749. doi: 10.1016/s1074-7613(03)00122-5. [DOI] [PubMed] [Google Scholar]
- Boehm MK, Woof JM, Kerr MA, Perkins SJ. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling. J Mol Biol. 1999;286:1421–1447. doi: 10.1006/jmbi.1998.2556. [DOI] [PubMed] [Google Scholar]
- Mix E, Goertsches R, Zett UK. Immunoglobulins–basic considerations. J Neurol. 2006;53 Suppl 5:V9–V17. doi: 10.1007/s00415-006-5002-2. [DOI] [PubMed] [Google Scholar]
- Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol. 2006;208:270–282. doi: 10.1002/path.1877. [DOI] [PubMed] [Google Scholar]
- Kutteh WH, Prince SJ, Mestecky J. Tissue origins of human polymeric and monomeric IgA. J Immunol. 1982;128:990–995. [PubMed] [Google Scholar]
- Mestecky J. Immunobiology of IgA. Am J Kidney Dis. 1988;12:378–383. doi: 10.1016/s0272-6386(88)80029-5. [DOI] [PubMed] [Google Scholar]
- Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740–750. doi: 10.1016/j.immuni.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- Mestecky J, Zikan J, Butler WT. Immunoglobulin M and secretory immunoglobulin A: presence of a common polypeptide chain different from light chains. Science. 1971;171:1163–1165. doi: 10.1126/science.171.3976.1163. [DOI] [PubMed] [Google Scholar]
- Brandtzaeg P, Prydz H. Direct evidence for an integrated function of J-chain and secretory component in epithelial transport of immunoglobulins. Nature. 1984;311:71–73. doi: 10.1038/311071a0. [DOI] [PubMed] [Google Scholar]
- Monteiro RC, van de Winkel JG. IgA Fc receptors. Annu Rev Immunol. 2003;21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011. [DOI] [PubMed] [Google Scholar]
- Tomana M, Niedermeier W, Mestecky J, Skvaril F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry. 1976;13:325–328. doi: 10.1016/0019-2791(76)90342-6. [DOI] [PubMed] [Google Scholar]
- Mestecky J, Tomana M, Moldoveanu Z, Julian BA, Suzuki H, Matousovic K, et al. Role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press Res. 2008;31:29–37. doi: 10.1159/000112922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo EM, Morrison SL. IgA: an immune glycoprotein. Clin Immunol. 2005;116:3–10. doi: 10.1016/j.clim.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Corthesy B. Roundtrip ticket for secretory IgA: role in mucosal homeostasis. J Immunol. 2007;178:27–32. doi: 10.4049/jimmunol.178.1.27. [DOI] [PubMed] [Google Scholar]
- Fagarasan S. Evolution, development, mechanism and function of IgA in the gut. Curr Opin Immunol. 2008;20:170–177. doi: 10.1016/j.coi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wines BD, Hogarth PM. IgA receptors in health and disease. Tissue Antigens. 2006;68:103–114. doi: 10.1111/j.1399-0039.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol. 2008;1:11–22. doi: 10.1038/mi.2007.6. [DOI] [PubMed] [Google Scholar]
- van Epps DE, Williams RC., Jr Suppression of leukocyte chemotaxis by human IgA myeloma components. J Exp Med. 1976;144:1227–1242. doi: 10.1084/jem.144.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Epps DE, Reed K, Williams RC., Jr Suppression of human PMN bactericidal activity by human IgA paraproteins. Cell Immunol. 1978;36:363–376. doi: 10.1016/0008-8749(78)90280-0. [DOI] [PubMed] [Google Scholar]
- Wilton JM. Suppression by IgA of IgG-mediated phagocytosis by human polymorphonuclear leucocytes. Clin Exp Immunol. 1978;34:423–428. [PMC free article] [PubMed] [Google Scholar]
- Wolf HM, Fischer MB, Puhringer H, Samstag A, Vogel E, Eibl MM. Human serum IgA downregulates the release of inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) in human monocytes. Blood. 1994;83:1278–1288. [PubMed] [Google Scholar]
- Nikolova EB, Russell MW. Dual function of human IgA antibodies: inhibition of phagocytosis in circulating neutrophils and enhancement of responses in IL-8-stimulated cells. J Leukoc Biol. 1995;57:875–882. doi: 10.1002/jlb.57.6.875. [DOI] [PubMed] [Google Scholar]
- Wolf HM, Hauber I, Gulle H, Samstag A, Fischer MB, Ahmad RU, et al. Anti-inflammatory properties of human serum IgA: induction of IL-1 receptor antagonist and Fc alpha R (CD89)-mediated down-regulation of tumour necrosis factor-alpha (TNF-alpha) and IL-6 in human monocytes. Clin Exp Immunol. 1996;105:537–543. doi: 10.1046/j.1365-2249.1996.d01-793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olas K, Butterweck H, Teschner W, Schwarz HP, Reipert B. Immunomodulatory properties of human serum immunoglobulin A: anti-inflammatory and pro-inflammatory activities in human monocytes and peripheral blood mononuclear cells. Clin Exp Immunol. 2005;140:478–490. doi: 10.1111/j.1365-2249.2005.02779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Schaffer FM, Monteiro RC, Volanakis JE, Cooper MD. IgA deficiency. Immunodefic Rev. 1991;3:15–44. [PubMed] [Google Scholar]
- Kanamaru Y, Arcos-Fajardo M, Moura IC, Tsuge T, Cohen H, Essig M, et al. Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur J Immunol. 2007;37:1116–1128. doi: 10.1002/eji.200636826. [DOI] [PubMed] [Google Scholar]
- van Egmond M, Damen CA, van Spriel AB, Vidarsson G, van Garderen E, van de Winkel JG. IgA and the IgA Fc receptor. Trends Immunol. 2001;22:205–211. doi: 10.1016/s1471-4906(01)01873-7. [DOI] [PubMed] [Google Scholar]
- Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med. 2001;194:417–425. doi: 10.1084/jem.194.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten MA, van Egmond M. The Fc receptor for IgA (FcalphaRI, CD89) Immunol Lett. 2004;92:23–31. doi: 10.1016/j.imlet.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. . Nature. 2003;423:614–620. doi: 10.1038/nature01685. [DOI] [PubMed] [Google Scholar]
- Launay P, Patry C, Lehuen A, Pasquier B, Blank U, Monteiro RC. Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcRgamma association and protects against degradation of bound ligand. J Biol Chem. 1999;274:7216–7225. doi: 10.1074/jbc.274.11.7216. [DOI] [PubMed] [Google Scholar]
- Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol. 2008;180:2669–2678. doi: 10.4049/jimmunol.180.4.2669. [DOI] [PubMed] [Google Scholar]
- Monteiro RC, Hostoffer RW, Cooper MD, Bonner JR, Gartland GL, Kubagawa H. Definition of immunoglobulin A receptors on eosinophils and their enhanced expression in allergic individuals. J Clin Invest. 1993;92:1681–1685. doi: 10.1172/JCI116754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamolera M, Launay P, Montenegro V, Rivero MC, Velasco IT, Monteiro RC. Enhanced expression of Fc alpha receptor I on blood phagocytes of patients with gram-negative bacteremia is associated with tyrosine phosphorylation of the FcR-gamma subunit. Shock. 2001;16:344–348. doi: 10.1097/00024382-200116050-00004. [DOI] [PubMed] [Google Scholar]
- Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Lebreton C, Menard S, Candalh C, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205:143–154. doi: 10.1084/jem.20071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagnoff MF. Overview and pathogenesis of celiac disease. Gastroenterology. 2005;128:S10–S18. doi: 10.1053/j.gastro.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995;37:766–776. doi: 10.1136/gut.37.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periolo N, Chernavsky AC. Coeliac disease. Autoimmun Rev. 2006;5:202–208. doi: 10.1016/j.autrev.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology. 2000;119:996–1006. doi: 10.1053/gast.2000.18149. [DOI] [PubMed] [Google Scholar]
- Sollid LM, Lundin KE. Diagnosis and treatment of celiac disease. Mucosal Immunol. 2009;2:3–7. doi: 10.1038/mi.2008.74. [DOI] [PubMed] [Google Scholar]
- Salmi TT, Collin P, Jarvinen O, Haimila K, Partanen J, Laurila K, et al. Immunoglobulin A autoantibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming coeliac disease. Aliment Pharmacol Ther. 2006;24:541–552. doi: 10.1111/j.1365-2036.2006.02997.x. [DOI] [PubMed] [Google Scholar]
- Picarelli A, Maiuri L, Frate A, Greco M, Auricchio S, Londei M. Production of antiendomysial antibodies after in-vitro gliadin challenge of small intestine biopsy samples from patients with coeliac disease. Lancet. 1996;348:1065–1067. doi: 10.1016/S0140-6736(96)03060-7. [DOI] [PubMed] [Google Scholar]
- Korponay-Szabo IR, Halttunen T, Szalai Z, Laurila K, Kiraly R, Kovacs JB, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53:641–648. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- Berger J, Hinglais N. Les dépôts intercapillaires d'IgA-IgG. J Urol Néphol. 1968;74:694–695. [PubMed] [Google Scholar]
- Galla JH. IgA nephropathy. Kidney Int. 1995;47:377–387. doi: 10.1038/ki.1995.50. [DOI] [PubMed] [Google Scholar]
- Berger J, Yaneva H, Nabarra B, Barbanel C. Reccurence of mesangial deposition of IgA after renal transplantation. Kidney Int. 1975;7:232–241. doi: 10.1038/ki.1975.35. [DOI] [PubMed] [Google Scholar]
- Valentijn RM, Radl J, Haaijman JJ, Vermeer BJ, Weening JJ, Kauffmann RH, et al. Circulating and mesangial secretory component-binding IgA-1 in primary IgA nephropathy. Kidney Int. 1984;26:760–766. doi: 10.1038/ki.1984.213. [DOI] [PubMed] [Google Scholar]
- Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28:78–87. doi: 10.1016/j.semnephrol.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cabrero J, Egido J, Mampaso F, Rivas MC, Hernando L. Characterization of circulating idiotypes containing immune complexes and their presence in the glomerular mesangium in patients with IgA nephropathy. Clin Exp Immunol. 1989;76:204–209. [PMC free article] [PubMed] [Google Scholar]
- van den Wall Bake AW, Bruijn JA, Accavitti MA, Crowley-Nowick PA, Schrohenloher RE, Julian BA, et al. Shared idiotypes in mesangial deposits in IgA nephropathy are not disease-specific. Kidney Int. 1993;44:65–74. doi: 10.1038/ki.1993.214. [DOI] [PubMed] [Google Scholar]
- Monteiro RC, Halbwachs-Mecarelli L, Roque-Barreira MC, Noel LH, Berger J, Lesavre P. Charge and size of mesangial IgA in IgA nephropathy. Kidney Int. 1985;28:666–671. doi: 10.1038/ki.1985.181. [DOI] [PubMed] [Google Scholar]
- Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, et al. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol. 2004;15:622–634. doi: 10.1097/01.asn.0000115401.07980.0c. [DOI] [PubMed] [Google Scholar]
- Moura IC, Arcos-Fajardo M, Gdoura A, Leroy V, Sadaka C, Mahlaoui N, et al. Engagement of transferrin receptor by polymeric IgA1: evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol. 2005;16:2667–2676. doi: 10.1681/ASN.2004111006. [DOI] [PubMed] [Google Scholar]
- Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–13. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118:629–639. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- Kokubo T, Hiki Y, Iwase H, Horii A, Tanaka A, Nishikido J, et al. Evidence for involvement of IgA1 hinge glycopeptide in the IgA1–IgA1 interaction in IgA nephropathy. J Am Soc Nephrol. 1997;8:915–919. doi: 10.1681/ASN.V86915. [DOI] [PubMed] [Google Scholar]
- Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119:1668–1677. doi: 10.1172/JCI38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almogren A, Kerr MA. Irreversible aggregation of the Fc fragment derived from polymeric but not monomeric serum IgA1—implications in IgA-mediated disease. Mol Immunol. 2008;45:87–94. doi: 10.1016/j.molimm.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Launay P, Grossetete B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger's disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med. 2000;191:1999–2009. doi: 10.1084/jem.191.11.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro RC, Moura IC, Launay P, Tsuge T, Haddad E, Benhamou M, et al. Pathogenic significance of IgA receptor interactions in IgA nephropathy. Trends Mol Med. 2002;8:464. doi: 10.1016/s1471-4914(02)02405-x. [DOI] [PubMed] [Google Scholar]
- van den Wall Bake AW, Kirk KA, Gay RE, Switalski LM, Julian BA, Jackson S, et al. Binding of serum immunoglobulins to collagens in IgA nephropathy and HIV infection. Kidney Int. 1992;42:374–382. doi: 10.1038/ki.1992.298. [DOI] [PubMed] [Google Scholar]
- Cederholm B, Wieslander J, Bygren P, Heinegard D. Circulating complexes containing IgA and fibronectin in patients with primary IgA nephropathy. Proc Natl Acad Sci USA. 1988;85:4865–4868. doi: 10.1073/pnas.85.13.4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Karai M, Osawa G, Sato M, Koshikawa S. Antimouse laminin antibodies in IgA nephropathy and various glomerular diseases. Nephron. 1990;56:285–296. doi: 10.1159/000186156. [DOI] [PubMed] [Google Scholar]
- Zheng F, Kundu GC, Zhang Z, Ward J, DeMayo F, Mukherjee AB. Uteroglobin is essential in preventing immunoglobulin A nephropathy in mice. Nature Med. 1999;5:1018–1025. doi: 10.1038/12458. [DOI] [PubMed] [Google Scholar]
- Coppo R, Chiesa M, Cirina P, Peruzzi L, Amore A. In human IgA nephropathy uteroglobin does not play the role inferred from transgenic mice. Am J Kidney Dis. 2002;40:495–503. doi: 10.1053/ajkd.2002.34890. [DOI] [PubMed] [Google Scholar]
- Collin P, Syrjanen J, Partanen J, Pasternack A, Kaukinen K, Mustonen J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am J Gastroenterol. 2002;97:2572–2576. doi: 10.1111/j.1572-0241.2002.06025.x. [DOI] [PubMed] [Google Scholar]
- Jhaveri KD, D'Agati VD, Pursell R, Serur D. Coeliac sprue-associated membranoproliferative glomerulonephritis (MPGN) Nephrol Dial Transplant. 2009;24:3545–3548. doi: 10.1093/ndt/gfp353. [DOI] [PubMed] [Google Scholar]
- Biyikli NK, Gokce I, Cakalagoglu F, Arbak S, Alpay H. The co-existence of membranoproliferative glomerulonephritis type 1 and coeliac disease: a case report. Pediatr Nephrol. 2009;24:1247–1250. doi: 10.1007/s00467-009-1118-9. [DOI] [PubMed] [Google Scholar]
- Prasad D, Khara HS, Gupta M, Sterman P. Celiac disease associated membranous nephropathy—a rare cause or coincidence? A case report. Cases J. 2009;2:7018. doi: 10.4076/1757-1626-2-7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack A, Collin P, Mustonen J, Reunala T, Rantala I, Laurila K, et al. Glomerular IgA deposits in patients with celiac disease. Clin Nephrol. 1990;34:56–60. [PubMed] [Google Scholar]
- Coppo R, Amore A, Roccatello D. Dietary antigens and primary immunoglobulin A nephropathy. J Am Soc Nephrol. 1992;2:S173–S180. doi: 10.1681/ASN.V210s173. [DOI] [PubMed] [Google Scholar]
- Fornasieri A, Sinico RA, Maldifassi P, Bernasconi P, Vegni M, D'Amico G. IgA-antigliadin antibodies in IgA mesangial nephropathy (Berger's disease) Br Med J. 1987;295:78–80. doi: 10.1136/bmj.295.6590.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci A, Fofi C, Bartoli B, Simonetti BM, Pecci G, Sabbatella L, et al. Antiendomysial antibodies in Berger's disease. Am J Kidney Dis. 2002;39:1176–1182. doi: 10.1053/ajkd.2002.33387. [DOI] [PubMed] [Google Scholar]
- La Villa G, Pantaleo P, Tarquini R, Cirami L, Perfetto F, Mancuso F, et al. Multiple immune disorders in unrecognized celiac disease: a case report. World J Gastroenterol. 2003;9:1377–1380. doi: 10.3748/wjg.v9.i6.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppo R, Roccatello D, Amore A, Quattrocchio G, Molino A, Gianoglio B, et al. Effects of a gluten-free diet in primary IgA nephropathy. Clin Nephrol. 1990;33:72–86. [PubMed] [Google Scholar]
- Kovacs T, Kun L, Schmelczer M, Wagner L, Davin JC, Nagy J. Do intestinal hyperpermeability and the related food antigens play a role in the progression of IgA nephropathy? I. Study of intestinal permeability. Am J Nephrol. 1996;16:500–505. doi: 10.1159/000169050. [DOI] [PubMed] [Google Scholar]
- Rostoker G, Delchier JC, Chaumette MT. Increased intestinal intra-epithelial T lymphocytes in primary glomerulonephritis: a role of oral tolerance breakdown in the pathophysiology of human primary glomerulonephritides. Nephrol Dial Transplant. 2001;16:513–517. doi: 10.1093/ndt/16.3.513. [DOI] [PubMed] [Google Scholar]
- Smerud HK, Fellstrom B, Hallgren R, Osagie S, Venge P, Kristjansson G. Gluten sensitivity in patients with IgA nephropathy. Nephrol Dial Transplant. 2009;24:2476–2481. doi: 10.1093/ndt/gfp133. [DOI] [PubMed] [Google Scholar]
- Amore A, Emancipator SN, Roccatello D, Gianoglio B, Peruzzi L, Porcellini MG, et al. Functional consequences of the binding of gliadin to cultured rat mesangial cells: bridging immunoglobulin A to cells and modulation of eicosanoid synthesis and altered cytokine production. Am J Kidney Dis. 1994;23:290–301. doi: 10.1016/s0272-6386(12)80987-5. [DOI] [PubMed] [Google Scholar]