

Abstract
Receptor-interacting protein (RIP) kinases are a group of threonine/serine protein kinases with a relatively conserved kinase domain but distinct non-kinase regions. A number of different domain structures, such as death and caspase activation and recruitment domain (CARD) domains, were found in different RIP family members, and these domains should be keys in determining the specific function of each RIP kinase. It is known that RIP kinases participate in different biological processes, including those in innate immunity, but their downstream substrates are largely unknown. This review will give an overview of the structures and functions of RIP family members, and an update of recent progress in RIP kinase research.
Keywords: apoptosis, kinase, necrosis, RIP, TNF
Introduction
Cytokine stimulation, pathogen infection, DNA damage or inflammation can initiate cellular signaling pathways that lead to diverse responses, including immune cell activation and death. The receptor-interacting protein (RIP) kinase family members have emerged as essential sensors of intracellular and extracellular stresses. They have been demonstrated to play an important role in not only inflammation and other immune responses, but also in death-inducing processes.1, 2
The RIP kinases now contain seven members, all of which share a homologous kinase domain but have different functional domains (Figure 1). RIP1 contains a C-terminal death domain, through which RIP1 can be recruited to signaling complexes that initiate different pathways. RIP2 bears a caspase activation and recruitment domain (CARD). RIP3 has a C-terminus that is unique among all known protein domains. However, a RIP homotypic interaction motif (RHIM) found in the intermediate domain of RIP1 was also identified in the C-terminus of RIP3. The RHIM domain is likely to mediate protein–protein interactions, since it is required for the interaction between RIP3 and RIP1.3 RIP4 and RIP5 are characterized by the ankyrin repeats in their C-terminus. RIP6 and RIP7 have a leucine-rich repeat motif that may have a role in the recognition of damage-, pathogen- or stress-associated molecular patterns.1 Additionally, they both harbor Ros of complex proteins/C-terminal of Roc domains (Figure 1). Upon binding of GTP to the Roc domain, the RIP6 protein kinase activity is stimulated. Evidence was provided that the C-terminal of Roc domain might play a role in transmitting stimulating signals to the kinase domain.4 It is reasonable to deduce that the C-terminal domains of RIP family members play key roles in determining the diverse functions of different RIP kinases.
Figure 1.
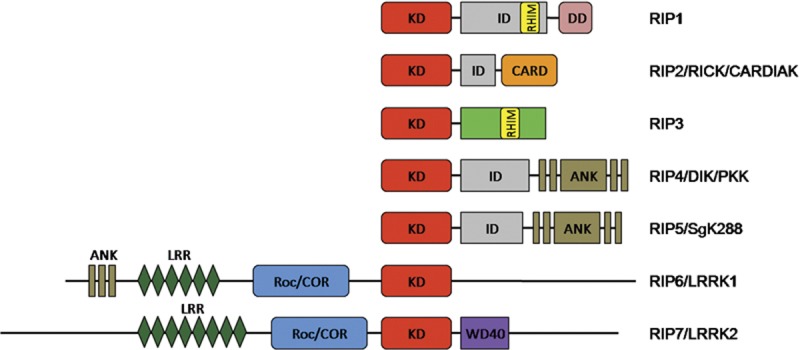
Domain organization of the RIP kinase family. The members of the RIP kinase family all share a homologous kinase domain (KD). RIP1 and RIP2 have a C-terminal death domain (DD) and caspase activation and recruitment domain (CARD), respectively. RIP3 has a unique C-terminus that is diverse from any other known proteins. RIP homotypic interaction motif (RHIM) is found both in RIP1 and RIP3. RIP4 and RIP5 contain C-terminal ankyrin repeats (ANK) domains, which are also observed in RIP6. RIP6 and RIP7 are characterized by the presence of leucine-rich repeat (LRR) motifs and Ros of complex proteins/C-terminal of Roc (Roc/COR) domains. RIP7 further harbors a WD40 motif in its C terminus. CARDIAK, CARD-containing IL-1β converting enzyme-associated kinase; DIK, protein kinase C-δ-interacting protein kinase; ID, intermediate domain; LRRK, leucine-rich repeat kinase; PKK, protein kinase C-associated kinase; RICK, RIP-like-interacting caspase-like apoptosis-regulatory protein kinase; RIP, receptor-interacting protein; SgK288, Sugen kinase 288.
RIP1
RIP1, the first member of the RIP kinase family, was initially identified in 1995 as a novel protein kinase with a C-terminal death domain (Figure 1), through which it interacts with the death domain of receptor Fas (CD95). Therefore, it was designated as ‘receptor-interacting protein'.5 As more and more RIP kinases were characterized, it became called ‘RIP1'. The death domain of RIP1 was shown to bind to several death receptors, such as tumor-necrosis factor (TNF) receptor 1 (TNF-R1), TNF-related apoptosis-inducing ligand receptor 1 and 2, and TNF-receptor-related apoptosis-mediated protein.5, 6, 7 RIP1 also interacts with adaptor proteins, such as TNF-receptor-associated death domain (TRADD), Fas-associated death domain (FADD), RIP-associated ICH-1/CED-3-homologous protein with death domain, Toll/IL-1 receptor domain-containing adaptor inducing interferon-β(TRIF), TNF-receptor-associated factor 1 (TRAF1), TRAF2, TRAF3 and A20.8, 9, 10, 11, 12, 13, 14, 15 In addition, the intermediate domain and death domain of RIP1 enable it to associate with a variety of other kinases, such as RIP3, focal adhesion kinase, mitogen-activated protein/extracellular signal-regulated kinase kinase 1 (MEKK1) and MEKK3.16, 17, 18, 19, 20
RIP1 is constitutively expressed in many tissues. However, TNF-α treatment or T-cell activation also make the expression of RIP1 inducible.5, 21 RIP1 expression is important for T-cell survival, because mice lacking RIP1 display extensive apoptosis in lymphoid tissue. Cultured RIP1 deletion cells are also more sensitive to TNF-α-induced cell death. RIP1-deficient mice die within 3 days of birth.22 All of these indicate that RIP1 has a crucial role in regulating cell death and controlling the homeostasis of a tissue or organism.
The death receptors TNF-R1, Fas and TNF-related apoptosis-inducing ligand receptor mediate apoptosis in many cell systems. When these receptors bind to their ligands, they polymerize and initiate signaling pathways that either lead to an inflammatory response or cell death. Binding of TNF-α to its receptor, TNF-R1, induces the formation of two sequential signaling complexes, with RIP1 being a component of both complexes.23 The initial plasma membrane-bound complex (complex I) is composed of TNF-R1, TRADD, RIP1, TRAF2 and cellular inhibitor of apoptosis 1 (cIAP1). This induces rapid activation of nuclear factor-kappa B (NF-κB). As a consequence, NF-κB promotes the expression of several anti-apoptotic proteins, including TRAF1, TRAF2, cIAP1, cIAP2 and notably cellular FADD-like IL-1β-converting enzyme inhibitory protein, a potent inhibitor of death receptor-induced apoptosis.24, 25 And here, TRAF2 and RIP1 also mediate the activation of mitogen-activated protein kinases (MAPKs), such as p38, c-Jun N-terminal kinase (JNK) and ERK (Figure 2). ERK activation requires the kinase activity of RIP1, which appear not to be essential for the activation of p38, JNK, and NF-κB signaling.22, 26 Subsequently, a second complex (so-called complex II) is formed, which contains TRADD, RIP1, FADD, procaspase-8 and caspase-10. The activation of FADD and caspase-8 then initiate apoptosis. However, complex II only triggers apoptosis when complex I-mediated NF-κB activation is too low to induce a sufficient level of anti-apoptotic signaling. If the caspases are blocked, necrotic cell death ensues.23, 27 Recently, the critical role of RIP1 in mediating programmed necrosis has been addressed (Figure 2).28, 29, 30 It has been reported that the homologous kinase RIP3 can interact with and phosphorylate RIP1 upon TNF stimulation.28, 31 RIP3 and RIP1 form a phosphorylation-driven necrosome that finally leads to necrotic cell death.28
Figure 2.
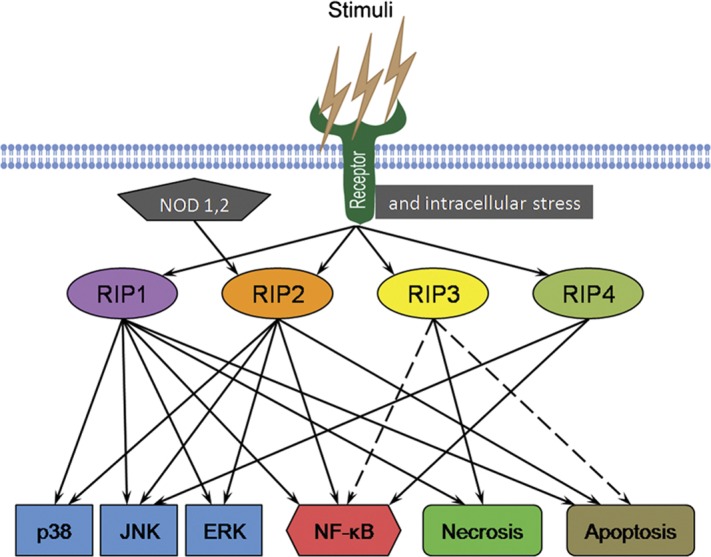
Network of the RIP kinases in the multiple cellular signaling pathways. RIP1 is a key mediator of several signaling pathways that lead to the activation of MAPKs and NF-κB, as well as cell death. RIP2 is critical for signaling from NOD-like receptors and can trigger MAPKs and NF-κB activation. The role of RIP3 is major in the induction of necrosis: it may participate in the process of apoptosis and regulate NF-κB signaling. RIP4 mainly functions in the activation of JNK and NF-κB pathways. ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-kappa B; NOD, nucleotide-binding oligomerization domain; RIP, receptor-interacting protein.
The role of FADD in TNF-α-induced caspase-independent signaling is controversial: FADD seems necessary for TNF-α-induced death in mouse embryonic fibroblasts,32 but it is not responsible for the TNF-α-induced cell death of Jurkat.33 The activated caspase-8 can cleave the kinase RIP1 that is essential for anti-apoptotic NF-κB signaling, showing that RIP1 functions at the cross-roads of cell survival and death, as well as the cross-roads between complex I and complex II signaling.
The modification of RIP1, such as phosphorylation and ubiquitination, determines RIP1's function in the prosurvival and death-inducing signaling pathways. Upon TNF stimulation, RIP1 is autophosphorylated and ubiquitinated. The ubiquitination of RIP1 can occur through linking to both Lys48 and Lys63 of ubiquitin.15 The Lys63-linked polyubiquitination of RIP1 by cIAP1 and cIAP2 initiates the TNF-induced activation of MAPKs and NF-κB, which promote the expression of prosurvival proteins. However, the Lys63-linked RIP1 ubiquitination is negatively regulated by deubiquitinases (A20) and cylindromatosis.34 A20 recruits to the TNF-R1 complex, removes the Lys63-linked ubiquitin chains from RIP1, and promotes ubiquitination of RIP1 in Lys48 linkage, which causes RIP1 degradation by the 26S proteasome complex,15 resulting in the termination of NF-κB signaling and potentiation of cell death.
RIP2
RIP2 is also called RIP-like-interacting caspase-like apoptosis-regulatory protein kinase or CARD-containing IL-1β converting enzyme-associated kinase. It was first described by three independent groups as a novel RIP-like kinase that has a role in NF-κB activation and apoptosis (Figure 2).35, 36, 37
RIP2 is characterized by an N-terminal kinase domain and a C-terminal CARD domain (Figure 1). Just like RIP1, the kinase activity of RIP2 is not required for the NF-κB response.37 RIP2 can interact with several TRAF members, such as TRAF1, TRAF2, TRAF5 and TRAF6.36, 37 RIP2 also associates with the anti-apoptotic proteins cIAP1 and Cflip.35, 36, 37 In addition to NF-κB, overexpression of RIP2 activates the MAPKs ERK2 and JNK (Figure 2). The activation of ERK2 but not JNK requires RIP2 kinase activity.37, 38 Experimental data have also demonstrated that RIP2 has a role in p38 MAPK activation in the stressed heart (Figure 2).39 Cytokine production induced by IL-1, IL-18 and Toll receptors is reduced in RIP2-deficient cells.40, 41 Furthermore, T-cell receptor-triggered proliferation is severely compromised in the RIP2-knockout animals.41
Nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and NOD2 (also called CARD-4 and CARD-15, respectively) are two components of the innate immune system from the NOD-like receptor protein family and are involved in sensing the presence of pathogens.42 Upon activation by specific bacterial peptides derived from peptidoglycans, NOD1 and NOD2 interact with the kinase RIP2 via CARD–CARD interaction to induce NF-κB activation and to regulate innate immune responses.42, 43 RIP2's kinase activity is critical for protein stability, and thus plays a central role in the preservation of NOD1- and NOD2-mediated innate immune responses.44 Many molecules that interact with RIP2 are demonstrated to be involved in the regulation of the NF-κB signaling pathway. Evidence was provided that the X-linked inhibitor of apoptosis protein (XIAP) interacts with RIP2 via its BIR2 domain and is involved in the NOD signaling. XIAP-deficient cells exhibit a marked reduction in NF-κB activation induced by microbial NOD ligands and by overexpression of NOD1 or NOD2. Both NOD1 and NOD2 associate with XIAP in a RIP2-dependent manner, suggesting a role for XIAP in regulating innate immune responses by interacting with NOD1 and NOD2 through interaction with RIP2.45 The LIM-domain-containing protein TRIP6 can functionally associate with RIP2. Overexpression of TRIP6 potentiates RIP2-mediated NF-κB activation triggered by TNF, IL-1, TLR2 and NOD1.46 Additionally, an MAPK, MEKK4, binds to RIP2 to sequester RIP2 from the NOD2 signaling pathway, inhibiting the NOD2-driven NF-κB activation.47 Moreover, ITCH, an E3 ubiquitin ligase, is found to K63-ubiquitinate RIP2 to inhibit NOD2-induced NF-κB activation. However, ITCH E3 ligase activity is responsible for optimal NOD2-induced p38 and JNK activation, suggesting an important role in inflammatory signaling.48 RIP2 has also been shown to interact with the CARD of caspase-1 and to induce IL-1β maturation, as well as induce Fas-mediated programmed cell death by enhancing caspase-8 activity.49
Recently, Krieg and colleagues have identified a novel alternative mRNA splice variant of RIP2, encoding a protein designated RIP2-β.50 It is comprised of only a portion of the N-terminal kinase domain, lacking the intermediate region and C-terminal CARD domain. These structural changes in RIP2-β are correlated with a loss of activation with respect to NF-κB and MAPK activation, IL-1β secretion and caspase-8-mediated apoptotic cell death.50
Furthermore, evidence has been provided showing that RIP2's protein level is inducible upon certain stimulation. For instance, in response to the inflammatory cytokines TNF-α, IL-1β and interferon-γ, the increase in RIP2 transcription and translation can be detected in endothelial cells.51
RIP3
RIP3 is the third RIP kinase family member to be described. RIP3 is comprised of an N-terminal kinase domain similar to that found in other RIP kinases, an RHIM domain and a unique C-terminal domain that differs from all known protein domains (Figure 1). Overexpression of RIP3 could induce apoptosis and NF-κB activation in some cell lines (Figure 2).18, 20, 52, 53 However, evidence also indicates that RIP3 interacts with and then phosphorylates RIP1 to diminish TNF-induced, RIP1-dependent NF-κB activation.31 So, the activation of NF-κB by RIP3 is controversial. Indeed, RIP3-deficient mice do not exhibit any alteration in the level of NF-κB activation induced by TNF-α.54
TNF-α induces apoptosis in many types of cells, which can be blocked by the pan caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (zVAD), but TNF-α can still trigger some cell lines to undergo necrosis when the activities of caspases are inhibited by zVAD treatment.1 Similar to RIP1, the kinase activity of RIP3 is required for this caspase-independent cell death.55
Recently, we find that, similar to RIP1, RIP3 is required for caspase-independent necrotic cell death triggered by TNF treatment (Figure 2).30 Through microarray analysis, we have revealed that RIP3 is not expressed in A cells (one NIH-3T3 cell line) which undergo apoptosis upon TNF stimulation, whereas it is highly expressed in N cells (another NIH-3T3 cell line) which undergo necrosis when treated with TNF. The apoptotic cell death of A cells is protected by the pan caspase inhibitor zVAD, while the necrotic cell death of N cells is not blocked but rather enhanced by zVAD. Small hairpin RNA knockdown of RIP3 in N cells prevents necrosis in the presence of zVAD. We further uncovered that RIP3 is also required for TNF-induced necrosis in L929 fibroblasts and zVAD-induced necrosis in TNF-/lipopolysaccharide-stimulated primary peritoneal macrophages. Deletion and point mutations analysis reveals that the kinase activity and RIP1-interacting RHIM domain in RIP3 are both required for necrotic signaling.30
The essential role of RIP3 in caspase-independent necrosis has also been reported in Cell by two other groups.28, 29 Using an RNA interference screen, Cho and colleagues identified that RIP1 and RIP3 are among 10 kinases required for cellular programmed necrosis in response to TNF treatment, and during virus infection. The kinase RIP3 regulates necrosis-specific RIP1 phosphorylation. The phosphorylation of RIP1 and RIP3 can stabilize their interaction within the pronecrotic complex, activate the pronecrotic kinase activity and trigger downstream reactive oxygen species (ROS) production which finally leads to necrotic cell death.28 Through a genome-wild RNA interference screen, another group also demonstrated that both RIP1 and RIP3 are the key mediators of TNF-induced necrosis in mouse L929 fibroblasts and human HT-29 cells (treated with a combination of TNF-α, Smac mimetic and zVAD). They report that, upon induction of necrosis, RIP3 is recruited to RIP1 to form a necrosis-inducing complex, which is in complex II downstream of the TNF receptor.29
Although there are many potential sources of ROS, ROS derived from mitochondria has been proved to play a crucial role in the execution of caspase-independent cell death.1 Our experimental data have revealed that RIP3 is required for ROS production in N cells, L929 fibroblasts and primary peritoneal macrophages during the process of necrosis triggered by different death stimuli.
By means of liquid chromatography-tandem mass spectrometry analysis, we identify several cellular metabolic enzymes in the RIP3 immunocomplex that are immunoprecipitated from N cells treated with TNF+zVAD for 2 h. Further experiments show that RIP3 can directly interact with and enhance the enzymatic activity of liver glycogen phosphorylase (PYGL), glutamate ammonia ligase (GLUL) and glutamate dehydrogenase 1 (GLUD1) (Figure 3). PYGL catalyzes the rate-limiting degradation of glycogen by releasing glucose-1-phosphate, thereby having a key role in utilizing reserved glycogen as an energy source. GLUL is a cytosolic enzyme that catalyzes the condensation of glutamate and ammonia to form permeable glutamine. Glutamine can transfer into the mitochondria to function as an energy substrate. GLUD1 is found in the mitochondrial matrix and converts glutamate to α-ketoglutarate. So, both GLUL and GLUD1 are essential for the use of glutamate or glutamine as substrates for ATP production by means of oxidative phosphorylation. Knockdown of PYGL, GLUL or GLUD1 partially inhibits TNF+zVAD-induced ROS production and necrosis in N cells. Because increasing glucose by breaking down glycogen and promoting the use of glutamate and glutamine as energy substrates all increase energy metabolism, the role of RIP3 in rendering cells more permissive to TNF-induced necrosis should at least partly occur through increasing energy metabolism-associated ROS production (Figure 3).
Figure 3.

Schematic for the mechanism of RIP3/RIP1-mediated necrosis. TNF induces apoptosis in the absence of RIP3, while in cells that have sufficient RIP3 expression, the gateways to using glycogen and glutamate or glutamine are readily opened after TNF stimulation. zVAD may enhance RIP3 function, as RIP3 and RIP1 are reported to be cleaved by caspases. The enhanced metabolism should be accompanied by increased ROS production from the mitochondrial respiration chain, which is likely responsible for the function of RIP3/RIP1 in mediating necrosis. FADD, Fas-associated death domain; GLUD, glutamate dehydrogenase; GLUL, glutamate ammonia ligase; PYGL, phosphorylase, glycogen, liver; RIP, receptor-interacting protein; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; TNF, tumor-necrosis factor; TNF-R1, tumor-necrosis factor receptor 1; TRADD, TNF-receptor-associated death domain; TRAF, TNF-receptor-associated factor; zVAD, benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone.
RIP4
RIP4 was first identified as protein kinase C (PKC)-δ-interacting protein kinase (DIK), as it is a kinase that can interact with PKC-δ, a member of the PKC family, in a yeast two-hybrid system.56 The ortholog of DIK in mice also interacts with PKC-β, another PKC isoform, and thus is referred to as protein kinase C-associated kinase (PKK).57 DIK/PKK contains an N-terminal RIP-like kinase domain and C-terminal ankyrin repeats domains linked to an intermediate region (Figure 1).57, 58, 59, 60 Therefore, DIK and PKK were renamed human and mouse RIP4, respectively.58, 59
Overexpression of RIP4 leads to the activation of NF-κB and JNK signaling (Figure 2).59, 60 RIP4-mediated NF-κB activation requires IKK-α and IKK-β, but not IKK-γ, a regulatory subunit of the IκB kinase (IKK) complex. Interestingly, the kinase-dead RIP4 mutant blocks NF-κB activation triggered by phorbol ester and Ca2+-ionophore, but not TNF-α, IL-1β or NOD1. Inhibition of NF-κB activation by dominant negative RIP4 can be reconstituted by coexpression of PKC-β, suggesting a functional link between RIP4 and PKC-β. RIP4-induced NF-κB activation is independent of Bcl10 and is not suppressed by dominant negative Bimp1, two components of the PKC signaling pathway.60 Moreover, RIP4 binds to several members of the TRAF protein family, and dominant negative TRAF1, TRAF3 and TRAF6 inhibit RIP4-induced NF-κB activation.59 Similar to RIP1, RIP4 is also cleaved by caspases at Asp340 and Asp378 in the intermediate region during the process of apoptosis, resulting in the inhibition of its activity of promoting NF-κB signaling.59 RIP4 is phosphorylated by two specific MAPKs, MEKK2 and MEKK3, and this interaction may partly be mediated through a critical activation loop residue, Thr184.61
The transgenic mice overexpressing a catalytically inactive form of RIP4 in lymphoid cells results in a decreased number of peripheral B cells.62 In RIP4-deficient mice, the differentiation of keratinocytes is severely impaired, and no cornified layer can be observed.58 Mice lacking RIP4 are perinatally lethal due to abnormal epidermal differentiation. Moreover, RIP4-knockout mice have shorter hindlimbs and tails when compared with their wild-type counterparts, indicating a defective morphogenesis. Interestingly, the phenotype of keratinocyte defects in RIP4-deficient mice is similar to that of mice lacking IKK-α, another kinase in NF-κB signaling.63, 64, 65 Taken together, these data suggest that RIP4 and IKK-α may function in a common pathway to regulate epidermal development and homeostasis.
RIP5
The Sugen kinase 288 exhibits high sequence homology with RIP4. They both share an N-terminal kinase domain and C-terminal ankyrin repeats (Figure 1), with an overall homology of 35%. Therefore, the kinase, Sugen kinase 288, has been proposed to be a RIP kinase family member, and designated as RIP5. The similar structures may suggest similar functions between RIP5 and RIP4. Overexpression of RIP5 induces cell death with characteristic apoptotic morphology, including DNA fragmentation.66 However, the biological function of RIP5 is still largely unknown.
RIP6 and RIP7
Another two kinases, leucine-rich repeat kinases LRRK1 and LRRK2, are closely related to the RIP kinases based on structural studies, and are thus referred to as RIP6 and RIP7, respectively.3 Similar to RIP4 and RIP5, RIP6 is characterized by the ankyrin repeats domains, which are not observed in RIP7. Unlike the other RIP kinases, RIP6 and RIP7 harbor additional domains, such as leucine-rich repeat and Ros of complex proteins/C-terminal of Roc domains. RIP7 further has WD40 repeats in its C terminus (Figure 1). The high resemblance in structural organization between RIP6 and RIP7 suggests that they might have conserved biological functions. Indeed, experimental studies show that both of these two proteins correlate with a susceptibility to Parkinson's disease.67, 68, 69, 70
Mutations in RIP7 have been identified as the most genetic causes of autosomal dominant inherited Parkinson's disease.68 The mutation types and the frequencies of RIP7 distribute unevenly in different populations.71 These mutations result in amino-acid changes throughout the protein and alterations in both its enzymatic properties and interactions. The best-characterized mutations in RIP7 to date are G2019S and R1441C/G. G2019S mutation results in the increased kinase activity, whereas R1441C/G mutations cause defective GTPase activity and also influence kinase activity.72 Moreover, all the variants of RIP7 and their contribution to Parkinson's disease have been recently examined and summarized by Paisan-Ruiz.73 The kinase activity, GTP-binding (GTPase) and WD40 domain of RIP7 are all reported to be implicated in RIP7's neurotoxicity.74, 75, 76, 77, 78 Although much effort has been made in searching for the molecular mechanism underlying RIP7's function,79, 80, 81, 82, 83, 84, 85 the pathogenesis of RIP7 mutations in Parkinson's disease remains unclear.
Perspectives
Much progress has been made in understanding the biological functions of the RIP kinase family members during the past several years. These RIP kinases are involved in many cellular signaling pathways, regulating inflammatory responses and cell death or survival. The commonality of RIP family members is their role in modulating the NF-κB pathway and cell death program. Despite the homology in structural features, the different RIP kinases participate in diverse biological processes due to their specific domains. RIP1 is the most intensively studied RIP kinase and is known to play a crucial role in death receptor-initiated intracellular signaling. The biological function of RIP2 in the immune responses was revealed in recent years and the key role of RIP3 in necrotic cell death was discovered last year. The information regarding the function of RIP6 and RIP7 is not yet clear, but their participation in the development of Parkinson's disease has been demonstrated. Although the study of other RIP kinase family members is currently limited, we anticipate that their functional importance will be revealed in the coming years. Current knowledge of RIP kinases has suggested therapeutic potential in targeting RIP family kinases in the treatment of diseases of inflammation, ischemia and neurodegeneration.
References
- Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J Biol Chem. 2003;278:39251–39258. doi: 10.1074/jbc.M305833200. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Boldin MP, Goncharov TM, Wallach D. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: proteins binding to the death domains of the two receptors also bind to each other. J Exp Med. 1996;183:1271–1275. doi: 10.1084/jem.183.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, et al. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 1997;57:615–619. [PubMed] [Google Scholar]
- Duan H, Dixit VM. RAIDD is a new ‘death' adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24. doi: 10.1006/excr.1999.4733. [DOI] [PubMed] [Google Scholar]
- Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs) Oncogene. 2001;20:6482–6491. doi: 10.1038/sj.onc.1204788. [DOI] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Kim JW, Joe CO, Choi EJ. Role of receptor-interacting protein in tumor necrosis factor-alpha -dependent MEKK1 activation. J Biol Chem. 2001;276:27064–27070. doi: 10.1074/jbc.M009364200. [DOI] [PubMed] [Google Scholar]
- Kurenova E, Xu LH, Yang X, Baldwin AS, Jr, Craven RJ, Hanks SK, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–4371. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. 1999;274:16871–16875. doi: 10.1074/jbc.274.24.16871. [DOI] [PubMed] [Google Scholar]
- Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- Yu PW, Huang BC, Shen M, Quast J, Chan E, Xu X, et al. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr Biol. 1999;9:539–542. doi: 10.1016/s0960-9822(99)80239-5. [DOI] [PubMed] [Google Scholar]
- Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lin Y, Li J, Pober JS, Min W. RIP1-mediated AIP1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ASK1-JNK/p38 activation. J Biol Chem. 2007;282:14788–14796. doi: 10.1074/jbc.M701148200. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- Kuang AA, Diehl GE, Zhang J, Winoto A. FADD is required for DR4- and DR5-mediated apoptosis: lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J Biol Chem. 2000;275:25065–25068. doi: 10.1074/jbc.C000284200. [DOI] [PubMed] [Google Scholar]
- Lawrence CP, Chow SC. FADD deficiency sensitises Jurkat T cells to TNF-alpha-dependent necrosis during activation-induced cell death. FEBS Lett. 2005;579:6465–6472. doi: 10.1016/j.febslet.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–12300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–16975. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, et al. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- Navas TA, Baldwin DT, Stewart TA. RIP2 is a Raf1-activated mitogen-activated protein kinase kinase. J Biol Chem. 1999;274:33684–33690. doi: 10.1074/jbc.274.47.33684. [DOI] [PubMed] [Google Scholar]
- Jacquet S, Nishino Y, Kumphune S, Sicard P, Clark JE, Kobayashi KS, et al. The role of RIP2 in p38 MAPK activation in the stressed heart. J Biol Chem. 2008;283:11964–11971. doi: 10.1074/jbc.M707750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- Viala J, Sansonetti P, Philpott DJ. Nods and ‘intracellular' innate immunity. C R Biol. 2004;327:551–555. doi: 10.1016/j.crvi.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Nembrini C, Kisielow J, Shamshiev AT, Tortola L, Coyle AJ, Kopf M, et al. The kinase activity of Rip2 determines its stability and consequently Nod1- and Nod2-mediated immune responses. J Biol Chem. 2009;284:19183–19188. doi: 10.1074/jbc.M109.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106:14524–14529. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bin LH, Li F, Liu Y, Chen D, Zhai Z, et al. TRIP6 is a RIP2-associated common signaling component of multiple NF-kappaB activation pathways. J Cell Sci. 2005;118:555–563. doi: 10.1242/jcs.01641. [DOI] [PubMed] [Google Scholar]
- Clark NM, Marinis JM, Cobb BA, Abbott DW. MEKK4 sequesters RIP2 to dictate NOD2 signal specificity. Curr Biol. 2008;18:1402–1408. doi: 10.1016/j.cub.2008.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol. 2009;19:1255–1263. doi: 10.1016/j.cub.2009.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Krieg A, Le Negrate G, Reed JC. RIP2-beta: a novel alternative mRNA splice variant of the receptor interacting protein kinase RIP2. Mol Immunol. 2009;46:1163–1170. doi: 10.1016/j.molimm.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Yin X, Krikorian P, Logan T, Csizmadia V. Induction of RIP-2 kinase by proinflammatory cytokines is mediated via NF-kappaB signaling pathways and involves a novel feed-forward regulatory mechanism. Mol Cell Biochem; 333:251–259. doi: 10.1007/s11010-009-0226-y. [DOI] [PubMed] [Google Scholar]
- Pazdernik NJ, Donner DB, Goebl MG, Harrington MA. Mouse receptor interacting protein 3 does not contain a caspase-recruiting or a death domain but induces apoptosis and activates NF-kappaB. Mol Cell Biol. 1999;19:6500–6508. doi: 10.1128/mcb.19.10.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Ma L, Yang Y, Wu M. Truncated RIP3 (tRIP3) acts upstream of FADD to induce apoptosis in the human hepatocellular carcinoma cell line QGY-7703. Biochem Biophys Res Commun. 2006;347:558–565. doi: 10.1016/j.bbrc.2006.06.118. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Bhr C, Rohwer A, Stempka L, Rincke G, Marks F, Gschwendt M. DIK, a novel protein kinase that interacts with protein kinase Cdelta. Cloning, characterization, and gene analysis. J Biol Chem. 2000;275:36350–36357. doi: 10.1074/jbc.M004771200. [DOI] [PubMed] [Google Scholar]
- Chen L, Haider K, Ponda M, Cariappa A, Rowitch D, Pillai S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J Biol Chem. 2001;276:21737–21744. doi: 10.1074/jbc.M008069200. [DOI] [PubMed] [Google Scholar]
- Holland P, Willis C, Kanaly S, Glaccum M, Warren A, Charrier K, et al. RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr Biol. 2002;12:1424–1428. doi: 10.1016/s0960-9822(02)01075-8. [DOI] [PubMed] [Google Scholar]
- Meylan E, Martinon F, Thome M, Gschwendt M, Tschopp J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-kappa B and is processed during apoptosis. EMBO Rep. 2002;3:1201–1208. doi: 10.1093/embo-reports/kvf236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Ruland J, McAllister-Lucas LM, Lucas PC, Yamaoka S, Chen FF, et al. Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-kappa B activation induced by phorbol ester. J Biol Chem. 2002;277:31871–31876. doi: 10.1074/jbc.M202222200. [DOI] [PubMed] [Google Scholar]
- Moran ST, Haider K, Ow Y, Milton P, Chen L, Pillai S. Protein kinase C-associated kinase can activate NFkappaB in both a kinase-dependent and a kinase-independent manner. J Biol Chem. 2003;278:21526–21533. doi: 10.1074/jbc.M301575200. [DOI] [PubMed] [Google Scholar]
- Cariappa A, Chen L, Haider K, Tang M, Nebelitskiy E, Moran ST, et al. A catalytically inactive form of protein kinase C-associated kinase/receptor interacting protein 4, a protein kinase C beta-associated kinase that mediates NF-kappa B activation, interferes with early B cell development. J Immunol. 2003;171:1875–1880. doi: 10.4049/jimmunol.171.4.1875. [DOI] [PubMed] [Google Scholar]
- Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, et al. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, et al. Limb and skin abnormalities in mice lacking IKKalpha. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- Zha J, Zhou Q, Xu LG, Chen D, Li L, Zhai Z, et al. RIP5 is a RIP-homologous inducer of cell death. Biochem Biophys Res Commun. 2004;319:298–303. doi: 10.1016/j.bbrc.2004.04.194. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, et al. Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem. 2007;102:93–102. doi: 10.1111/j.1471-4159.2007.04523.x. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Toft M, Ross OA, White LR, Aasly JO, Farrer MJ. Variants in the LRRK1 gene and susceptibility to Parkinson's disease in Norway. Neurosci Lett. 2007;416:299–301. doi: 10.1016/j.neulet.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Elbaz A. LRRK2: bridging the gap between sporadic and hereditary Parkinson's disease. Lancet Neurol. 2008;7:562–564. doi: 10.1016/S1474-4422(08)70118-2. [DOI] [PubMed] [Google Scholar]
- Anand VS, Braithwaite SP. LRRK2 in Parkinson's disease: biochemical functions. FEBS J. 2009;276:6428–6435. doi: 10.1111/j.1742-4658.2009.07341.x. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C. LRRK2 gene variation and its contribution to Parkinson disease. Hum Mutat. 2009;30:1153–1160. doi: 10.1002/humu.21038. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen ND, Peng Y, Ho CC, Rideout HJ, Petrey D, Liu P, et al. The WD40 Domain Is Required for LRRK2 Neurotoxicity. PLoS One. 2009;4:e8463. doi: 10.1371/journal.pone.0008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benamer HT.The ancestry of LRRK2 Gly2019Ser parkinsonism Lancet Neurol 20087769–770.author reply 770–761. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP. LRRK2 in Parkinson's disease: building an understanding of disease etiology. FEBS J. 2009;276:6427. doi: 10.1111/j.1742-4658.2009.07340.x. [DOI] [PubMed] [Google Scholar]
- Webber PJ, West AB. LRRK2 in Parkinson's disease: function in cells and neurodegeneration. FEBS J. 2009;276:6436–6444. doi: 10.1111/j.1742-4658.2009.07342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen KK, Wang L, Aasly JO, White LR, Matson WR, Henchcliffe C, et al. Metabolomic profiling in LRRK2-related Parkinson's disease. PLoS One. 2009;4:e7551. doi: 10.1371/journal.pone.0007551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Goldberg MS. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS One. 2009;4:e5949. doi: 10.1371/journal.pone.0005949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner CJ, Schumacher A, Boldt K, Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]