

Abstract
Here, we investigated the antitumor effect of adenovirus-mediated gene transfer of LIGHT, the tumor-necrosis factor (TNF) superfamily member also known as TNFSF14, in the murine A20 B-cell lymphoma. LIGHT gene modification resulted in upregulated expression of Fas and the accessory molecule—intercellular adhesion molecule-1 (ICAM-1) on A20 cells and led to enhanced A20 cell apoptosis. LIGHT-modified A20 cells effectively stimulated the proliferation of T lymphocytes and interferon (IFN)-γ production in vitro. Immunization of BALB/c mice with a LIGHT-modified A20 cell vaccine efficiently elicited protective immunity against challenge with the parental tumor cell line. Adenovirus-mediated gene transfer of LIGHT by intratumoral injection exerted a very potent antitumor effect against pre-existing A20 cell lymphoma in BALB/c mice. This adenovirus-mediated LIGHT therapy induced substantial splenic natural killer (NK) and cytotoxic T lymphocyte (CTL) activity, enhanced tumor infiltration by inflammatory cells and increased chemokine expression of CC chemokine ligand 21 (CCL21), IFN-inducible protein-10 (IP-10) and monokine induced by IFN-γ (Mig) from tumor tissues. Thus, adenovirus-mediated LIGHT therapy might have potential utility for the prevention and treatment of B-cell lymphoma.
Keywords: adenovirus, B lymphoma, gene transfer, LIGHT/TNFSF14
INTRODUCTION
Malignant lymphoma may originate within lymphatic tissues following the malignant transformation of lymphocyte or tissue cells. Malignant lymphoma is divided into two categories, Hodgkin's disease and non-Hodgkin's lymphoma, based on the clinical and pathological characteristics. When the cell of origin is a B cell, the tumor is called a B-cell lymphoma.1 At present, B-cell lymphoma patients are mainly treated with chemotherapy and radiotherapy. Although patients with B cell-derived non-Hodgkin's lymphoma respond well to these treatments, they are rarely cured. Relapses may occur after a period of months or years and recurrent disease may no longer respond to treatment. High-dose chemoradiotherapy can induce longer remissions, but their substantial toxicity imparts a high risk of early patient mortality.2, 3 Fortunately, much progress has been made in recent years in the development of cancer vaccines and immunotherapy for lymphoma.4, 5, 6, 7
One of the major goals of tumor immunotherapy is the generation of tumor-specific T-cell responses that contribute to tumor eradication. The initiation of an antigen-specific T-cell immune response requires multiple signals, including the recognition of peptides by the T-cell receptor and the interaction of several T-cell surface molecules with their costimulatory ligands expressed on antigen presenting cells.8, 9 One of the mechanisms by which B-cell lymphomas evade immune surveillance is through the downregulation of important accessory molecules that are necessary for the efficient activation of antigen-specific T cells.10, 11, 12 Recent studies have focused on modifying the phenotype of tumor B cells, which may enhance their recognition by tumor-specific T cells. One of the most interesting approaches has been the genetic modification of tumor B cells to express costimulatory molecules.13, 14 Briones et al.13 found that the genetic modification of B-cell lymphoma cells with CD40L could be a useful strategy to promote systemic immunity against B-cell malignancies. Animals with pre-existing tumors also display inhibited tumor growth when transduced with CD40L, suggesting that this treatment confers a survival advantage.
LIGHT (also known as tumor-necrosis factor (TNF) superfamily member 14) is an acronym for ‘homologous to lymphotoxins, shows inducible expression, and competes with herpes simplex virus glycoprotein D for herpes virus entry mediator (HVEM), a receptor expressed by T lymphocytes'. LIGHT is a member of the TNF superfamily that is mainly expressed on activated T cells, natural killer (NK) cells, immature dendritic cells (DCs) and some tumor cell lines. LIGHT is a type-II transmembrane protein that assembles into a homotrimeric form and may bind to three receptors: HVEM, lymphotoxin-β receptor and decoy receptor 3/TNF receptor 6.15 Early studies have reported that LIGHT can selectively induce the apoptosis of some tumor cells, similar to other TNF superfamily members.16 Further, it was shown that the binding of LIGHT to T cells results in potent, CD28-independent costimulation, leading to enhanced T-cell proliferation and the secretion of interferon (IFN)-γ and granulocyte–macrophage colony-stimulating factor in vitro.17, 18 In vivo experiments have demonstrated that the upregulation of LIGHT can cause severe inflammation in non-lymphoid tissues while enforced LIGHT expression in tumors promoted lymphocyte infiltration and induced a potent antitumor immune response.19 Costello et al. recently found that LIGHT could induce the expression of some accessory molecules on human non-Hodgkin's lymphoma cells and that it renders B-cell lymphomas more immunogenic in vitro.14
In the present study, we examine whether adenovirus-mediated LIGHT gene transfer to murine A20 B-cell lymphoma would result in potent antitumor activity. We show that immunization with LIGHT-modified A20 vaccine efficiently elicits protective immunity against the parental tumor. Furthermore, intratumoral adenovirus-mediated LIGHT gene transfer into established A20 B-cell lymphoma resulted in a potent antitumor effect in BALB/c mice. Thus, genetic modification of tumor B cells with the costimulatory molecule LIGHT might represent a useful strategy for the prevention and treatment of B-cell lymphoma.
MATERIALS AND METHODS
Animals
Female wild-type BALB/c mice, 6–8 weeks old, were purchased from Joint Ventures Sipper BK Experimental Animal Company (Shanghai, China) and housed under specific pathogen-free conditions for all experiments. All animal usage was conducted according to protocols approved by the Zhejiang University Institutional Animal Care and Use Committee.
Cell lines
The human embryonic kidney 293 cell line, transformed with adenovirus type 5 (Ad5) E1A and E1B genes, was used to support the propagation of E1-deleted replication deficient adenoviruses. A20 is a BALB/c-derived B-cell lymphoma expressing major histocompatibility complex (MHC) class I and II H-2d molecules. YAC-1 is an NK-sensitive lymphoma cell line. All of the above cell lines were obtained from American Type Culture Collection (Rockville, MD, USA). Cells were cultured in RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal calf serum (HyClone, Logan, UT, USA), 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin.
Preparation of recombinant adenoviruses
Replication-deficient recombinant adenovirus vectors harboring the LacZ reporter gene (Ad LacZ) or the murine LIGHT gene (Ad LIGHT), which were constructed from human adenovirus serotype 5 using homologous recombination, were kindly provided by Professor Yangxin Fu (University of Chicago, Chicago, IL, USA). The recombinant adenovirus was released from 293 cells by three freeze-thaw cycles and subsequently propagated with 293 cells as previously described.20 The adenoviral titers were determined by plaque-forming assay on 293 cells. Briefly, serial 10-fold dilutions of adenovirus were added to 24-well plates (Corning, New York, USA) containing confluent 293 cell monolayers. After 48 h of incubation in a humidified atmosphere, the end point of 50% infectivity was determined based on the cytopathic effect observed. The recombinant adenovirus was diluted to a titer of 1×1010 plaque-forming units/ml in phosphate-buffered saline (PBS) and stored at −70 °C before subsequent use in experiments.
In vitro transduction of A20 with Ad LIGHT
A20 cells cultured in complete medium but containing only 2% fetal calf serum were infected with the Ad LIGHT or Ad LacZ at a multiplicity of infection (MOI) of 200 for 24 h. LIGHT expression was monitored by reverse transcription-polymerase chain reaction (RT-PCR) and western blot. Total RNA was extracted from adenovirus-infected A20 cells by TRIzol Reagent (Bio Basic Inc., Markham, ON, Canada) and reverse transcribed using a Reverse Transcription System Kit (MIB Fermentas, Vilnius, Lithuania) following the manufacturer's instructions. The cDNA, as a readout of the mRNA, was amplified by PCR using specific primers for LIGHT and hypoxanthine-guanine phosphoribosyltransferase (HPRT). Primers for LIGHT were 5′-GCA TCA ACG TCT TGG AGA CA-3′ (sense) and 5′-ATA CGT CAA GCC CCT CAA GA-3′ (antisense), with an expected product of 202 bp. Primers for HPRT were 5′-GTT GGA TAC AGG CCA GAC TTT GTT G-3′ (sense) and 5′-GAG GGT AGG CTG GCC TAT AGG CT-3′ (antisense), with an expected product of 252 bp. Thirty cycles of 30 s for denaturation at 94 °C, 30 s of annealing at 60 °C, and 1 min of extension at 72 °C were performed for both LIGHT and HPRT amplification. PCR products were visualized by electrophoresis in a 1.5% agarose gel containing 0.5 µg/ml ethidium bromide.
Following infection, cells were resuspended in complete medium and incubated for another 24 h. The expression of surface molecules and the frequency of Ad LIGHT-induced apoptosis of A20 cells were assessed by flow cytometry. Briefly, A20 cells were washed twice in PBS containing 1% bovine serum albumin and 0.05% sodium azide and stained for 30 min on ice with a panel of phycoerythrin-conjugated monoclonal antibodies (mAbs) specific for murine B7.1 (CD80) or Fas and a panel of FITC-conjugated mAbs specific for murine intercellular adhesion molecule-1 (ICAM-1, CD54), B7.2 (CD86), MHC class II or Annexin V and stained with propidium iodide for apoptosis detection (all from Pharmingen, San Diego, CA, USA). Appropriate isotype controls were used in each experiment. After incubation, the cells were washed and then fixed with 2% paraformaldehyde solution, and the cells were analyzed using a FACScan (Becton Dickson, Mountain View, CA, USA) with CellQuest software.
To investigate the in vivo growth of Ad LIGHT-modified A20 lymphoma, BALB/c mice were given A20 tumor cells that had been infected with adenoviruses at a MOI of 200. BALB/c mice were divided into three groups and subcutaneously inoculated with 2×105 cells/mouse of either A20, A20/LacZ or A20/LIGHT cells. The tumor size was measured every 2 days. Mean tumor diameter is expressed as (length+width)/2.
T-cell proliferation and IFN-γ production assays
The bioactivity of LIGHT produced by Ad LIGHT-infected A20 cells was assessed by costimulation of T-cell proliferation and by IFN-γ production. Briefly, spleens from BALB/c mice were harvested and minced, a single-cell suspension was prepared by filtration through sterile nylon wool, and red blood cells were lysed with 0.83% ammonium chloride. A20 cells to be used as stimulator cells in these assays were infected with Ad LIGHT or Ad LacZ at a MOI of 200, followed by treatment with mitomycin-C (100 µg/ml). Splenocytes were added to a 96-well plate coated with 0.3 µg/ml anti-CD3 (Pharmingen) and cultured for 48 h in the presence of A20 stimulator cells. Supernatants were collected and the amount of IFN-γ produced was measured with an ELISA Kit (R&D systems, Minneapolis, MN, USA). The proliferation of splenocytes was measured with an MTT Proliferation Assay according to the manufacturer's directions (Sigma, St. Louis, MO, USA).
Vaccination of LIGHT gene-modified B-lymphoma cells
A20 cells transfected with Ad LacZ (A20/LacZ) or Ad LIGHT (A20/LIGHT) and treated with mitomycin-C (100 µg/ml) for 1 h were used to prepare the tumor vaccine. BALB/c mice were divided into four groups and vaccinated subcutaneously (s.c.) with either PBS or 1×106 cells/mouse of A20, A20/LacZ or A20/LIGHT cells. The vaccination was repeated 2 weeks later. One week after the last vaccination, animals were challenged with live parental A20 cells (2×105 cells/mouse) in a volume of 0.2 ml s.c. on the opposite flank. After 10 days, three mice per group were killed and their splenic lymphocytes were isolated for use in cytotoxic T lymphocyte (CTL) assays. The remaining animals were followed over time to assess mean survival. Survival analysis was performed using Prism software (GraphPad, San Diego, CA, USA), and statistical differences were calculated using the log-rank test.
Intratumoral injection of Ad LIGHT in an established B lymphoma model
BALB/c mice were s.c. inoculated with 2×105 A20 cells. When the tumor nodules were palpable, the tumor-bearing BALB/c mice were divided into three groups and injected intratumorally with 0.1 ml PBS, Ad LacZ or Ad LIGHT (1×108 plaque-forming units virus/0.1 ml) at 1-week intervals for a total of three injections. Tumors were measured three times a week with a caliper, and tumor size was calculated according to the formula: mean tumor diameter=(width+length)/2. P values were determined using a two-tailed t-test. Survival of the mice was followed as described above.
In an independent experiment, tumor free mice that had survived more than 90 days after being cured by Ad LIGHT therapy were subcutaneously rechallenged on the contralateral site with 1×106 A20 parental tumor cells. Naive BALB/c mice receiving subcutaneous injections of 1×106 A20 cells were used as controls. The survival time of both groups was monitored.
Semiquantitative RT-PCR analysis for chemokine expression, including CC chemokine ligand 21 (CCL21), monokine induced by IFN-γ (Mig) and IFN-inducible protein-10 (IP-10), from tumor cells
The subcutaneous tumor mass was harvested from tumor-bearing mice killed 5 days after the last treatment. Total RNA was then extracted from tumor tissues using TRIzol Reagent (Bio Basic Inc.) according to the instructions of the manufacturer. One microgram of total RNA from each sample was reverse transcribed using a Reverse Transcription System Kit (MIB Fermentas) in a total volume of 20 µl. The cDNA, as readout of cellular mRNA, was amplified by PCR using specific primers for CCL21, Mig, IP-10 and HPRT. Primers for CCL21 were 5′-GGG AAT TCA TGG CTC AGA TGA TGA CTC-3′ (sense) and 5′-CCG AAT TCC TAT CCT CTT GAG GGC TG-3′ (antisense), with an expected product of 418 bp. Primers for Mig were 5′-ACT CAG CTC TGC CAT GAA GTC CGC-3′ (sense) 5′-AAA GGC TGC TCT GCC AGG GAA GGC-3′ (antisense), with an expected product of 479 bp. Primers for IP-10 were 5′-ACC ATG AAC CCA AGT GCT GCC GTC-3′ (sense) and 5′-GCT TCA CTC CAG TTA AGG AGC CCT-3′ (antisense), with an expected product of 312 bp. Primers for HPRT were 5′-GTT GGA TAC AGG CCA GAC TTT GTT G-3′ (sense) and 5′-GAG GGT AGG CTG GCC TAT AGG CT-3′ (antisense), with an expected product of 252 bp. Cycling conditions were as follows: 30 s for denaturation at 94 °C, 30 s of annealing at 56 °C, and 1 min of extension at 72 °C repeated for 30 cycles for CCL21 and HPRT or for 22 cycles for Mig and IP-10.
Histological examination
Subcutaneous tumor nodules were taken from tumor-bearing mice that were killed 5 days after the last treatment. The tumor samples were fixed in 10% formalin solution, dehydrated and embedded in paraffin. Thin-sliced sections were stained with hematoxylin and eosin.
Cytotoxic assay of CTL and NK cells
Splenic lymphocytes were isolated from tumor-bearing mice 5 days after the last injection of recombinant adenovirus. The erythrocytes were depleted with 0.83% ammonium chloride. Non-adherent lymphocytes were directly used as NK effector cells in cytolytic assays against YAC-1 cells. The lymphocytes were cocultured with inactivated A20 cells (previously treated with 100 µg/ml mitomycin-C for 1 h) for 6 days in the presence of recombinant murine IL-2 (20 U/ml) and then collected as CTL effector cells. A20 cells or P815 cells were used as specific or non-specific target cells, respectively. The NK or CTL activity was determined by a standard lactate dehydrogenase (LDH) release assay (Promega, Madison, MI, USA).21 The amount of LDH-released was detected by absorbance at 490 nm using an ELISA reader. Calculations were carried out according to the following formula: % of specific lysis=100×(experimental–effectorspontaneous–target spontaneous)/(target maximum–target spontaneous).
Statistical analysis
All experiments were run in triplicate and the results are provided as mean±SD of triplicate determinations or as representative data from two or three independent experiments. Statistical analysis was performed using analysis of variance and log-rank test (for survival analysis). Differences were considered statistically significant when the P value was <0.05.
RESULTS
Modification of A20 cells with Ad LIGHT in vitro
A20 cells were transfected with Ad LIGHT or Ad LacZ. LIGHT is effectively expressed in A20 cells based on mRNA and protein level by RT-PCR and western blot analysis, respectively (Figure 1a). To test whether ectopically expressed LIGHT was bioactive, we observed its ability to stimulate the proliferation of T lymphocytes. Compared with the control A20 cells, A20/LIGHT efficiently stimulated the proliferation of T lymphocytes (Figure 1b) and enhanced IFN-γ production in vitro (Figure 1c), indicating that LIGHT expression on A20 cells might promote the synergistic costimulation of T lymphocytes.
Figure 1.
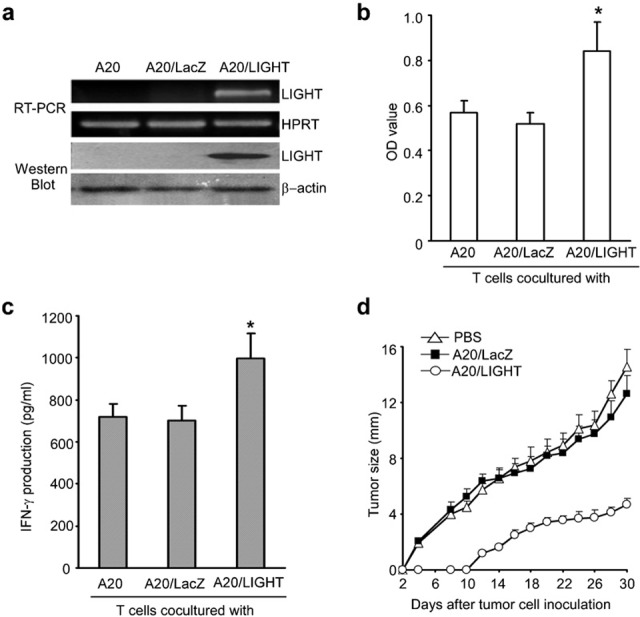
Ad LIGHT transfection of A20 cells. (a) LIGHT expression by A20 cells was detected by RT-PCR and western blot analysis 24 h after Ad LIGHT transfection. Twenty-four hours after adenovirus transfection, total RNA from gene-modified A20 cells was collected and reverse transcribed, and the cDNA was amplified by PCR using specific primers for LIGHT. A total of 1×106 A20 cells were collected 24 h after transfection with Ad LIGHT or Ad LacZ and LIGHT protein expression was measured by western blot analysis. One representative result of three independent experiments is shown. (b) A20 cells were infected with adenoviruses at a MOI of 200 followed by treatment with mitomycin-C (100 µg/ml). These cells were used as stimulator cells. Splenic T cells were cocultured with different A20 transfectants in the presence of plate-coated anti-CD3 mAb. The MTT assay was performed in triplicate to assess T-cell proliferation. (c) Culture supernatants were collected for the measurement of IFN-γ production. Data are shown as mean±SD. Similar results were obtained in three independent experiments. (d) Growth of Ad LIGHT-modified A20 lymphoma cells in vivo. Parental A20 cells were infected with gene-modified adenovirus at a MOI of 200. BALB/c mice were divided into three groups and subcutaneously inoculated with 2×105 A20, A20/LacZ or A20/LIGHT cells. The resultant tumor was measured every other day and mean tumor diameter was determined as follows: mean tumor diameter=(length+width)/2. *P<0.05. Ad, adenovirus; IFN, interferon; mAb, monoclonal antibody; MOI, multiplicity of infection; OD, optical density; PBS, phosphate-buffered saline; RT-PCR, reverse transcription-polymerase chain reaction.
To investigate the growth of Ad LIGHT-modified A20 lymphoma cells in vivo, 2×105 wild-type A20, A20/LacZ or A20/LIGHT cells were subcutaneously injected into BALB/c mice. Monitoring tumor growth indicated that the growth of LIGHT-modified A20 cells was significantly inhibited compared to the growth of LacZ-modified A20 or control wild-type A20 cells (Figure 1d; P<0.01).
We then used flow cytometry to test whether the modification of LIGHT expression affects the expression of other cell surface molecules on A20 tumor cells or the induction of cellular apoptosis. As shown in Figure 2a, LIGHT-expressing A20 cells exhibit upregulated ICAM-1 (CD54) expression, which may enhance the antigen-presenting capacity of B-cell lymphoma cells. Ectopic LIGHT expression also led to upregulated Fas expression on A20 cells (Figure 2a), but no change in the expression of CD80, CD86 or MHC class II molecules was observed. Furthermore, apoptosis of A20 cells transfected with Ad LIGHT was significantly induced compared with A20/LacZ or parental A20 (Figure 2b).
Figure 2.
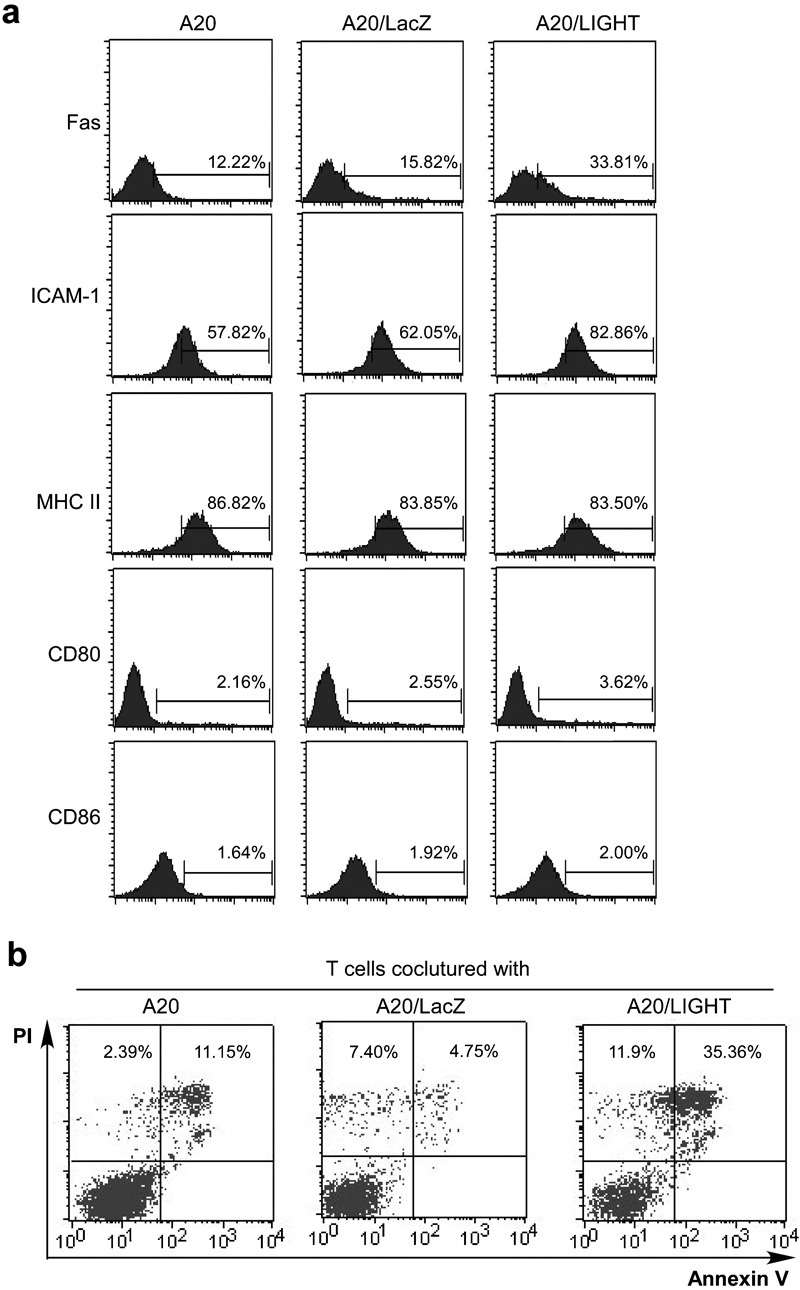
Phenotype of A20 cells after Ad LIGHT transfection. (a) Expression of surface molecules on A20 cells after Ad LIGHT transfection. A20 cells were infected with Ad LIGHT or Ad LacZ at a MOI of 200 for 24 h. Cells were then analyzed for the expression of Fas, MHC class II, CD80, CD86 and ICAM-1 by flow cytometry. (b) A20 cells were infected with Ad LacZ or Ad LIGHT, as in a. Cells were collected 24 h post-infection and stained with Annexin V and PI for the detection of apoptosis by flow cytometry. Ad, adenovirus; ICAM-1, intercellular adhesion molecule-1; MHC, major histocompatibility complex; MOI, multiplicity of infection; PI, propidium iodide.
Elicitation of protective immunity against tumor challenge by immunization with the manipulated vaccine
To prepare the tumor vaccine, A20 cells transfected with Ad LacZ (A20/LacZ) or Ad LIGHT (A20/LIGHT) were treated with mitomycin-C (100 µg/ml) for 1 h. BALB/c mice were then immunized twice s.c. with 1×106 of the manipulated A20 cell vaccines, with injections spaced 2 weeks apart. One week after the last inoculation, animals were challenged with live parental 2×105 A20 cells s.c. on the opposite flank. The results in Figure 3a illustrate that the growth of subcutaneous tumors in the A20/LIGHT immunized mice was significantly inhibited (42.86%) when compared with that in mice receiving A20/PBS (85.71%), A20/LacZ (85.71%) or in untreated control mice (100%) (P<0.01). The survival of mice in each group was also observed. The mice immunized with the A20/LIGHT vaccine and then challenged with parental A20 survived much longer than A20/PBS- or A20/LacZ-vaccinated mice or unvaccinated controls (Figure 3b; P<0.01).
Figure 3.
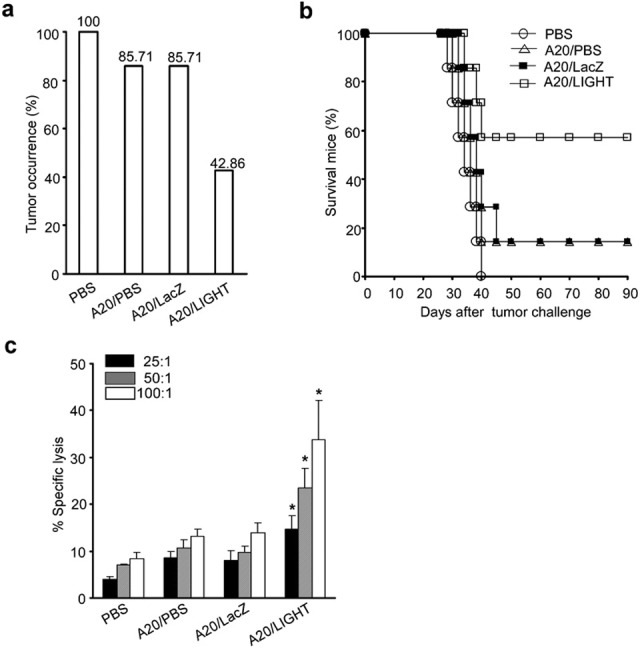
Immunization with the LIGHT gene-modified A20 vaccine elicits protective immunity against challenge with the parental tumor cell line. A20 cells were transfected with Ad LacZ or Ad LIGHT, followed by treatment with mitomycin-C (100 µg/ml) for 1 h, to prepare the tumor vaccine. BALB/c mice were divided into four groups and injected twice s.c. (2 weeks apart) with PBS or 1×106 A20 cells, A20/LacZ cells or A20/LIGHT cells. One week after the last vaccination, animals were challenged with live parental A20 cells (2×105 cells/mouse) in a volume of 0.2 ml s.c. on the opposite flank. (a) Tumor occurrence in mice vaccinated with PBS, A20 cells, A20/LacZ and A20/LIGHT. (b) Comparison of mean survival time among mice from different experimental groups following challenge with A20 cells. (c) Ten days after tumor challenge, three mice from each group were killed. Splenic lymphocytes from these mice were isolated and cocultured with inactivated A20 for 6 days in the presence of recombinant murine IL-2 (20 U/ml). Stimulated splenocytes were then collected for use as CTL effector cells in cultures at effector/target ratios of 25∶1, 50∶1 or 100∶1. The specific lysis against A20 cells was determined by a standard LDH release assay. Data are representative of three independent experiments. *P<0.05. Ad, adenovirus; CTL, cytotoxic T lymphocyte; LDH, lactate dehydrogenase; PBS, phosphate-buffered saline; s.c., subcutaneously.
To examine the generation of a CTL response against the parental A20 tumor cell line, mouse splenocytes were cocultured with inactivated parental A20 cells for 6 days. The CTL activity of splenocytes against the parental A20 cells during these cultures was determined by a standard LDH release assay. As shown in Figure 3c, the splenic CTL activity was markedly induced in mice that had received an A20/LIGHT vaccination compared to other control mice (P<0.05). The observed CTL response was tumor-specific because the same effector cells were not able to lyse cells of the syngeneic tumor cell line P815 (data not shown). This result suggested that tumor-specific immunity was induced significantly following vaccination with LIGHT gene-modified A20 cells.
The antitumor effects of adenovirus-mediated intratumoral gene transfer of LIGHT in an established murine B-cell lymphoma
We investigated the antitumor effects of adenovirus-mediated LIGHT gene transfer to pre-existing murine A20 B-cell lymphoma. Adult (6–8 weeks old), female BALB/c mice were subcutaneously inoculated with A20 cells. When the tumor nodules were palpable, 30 mice were randomly divided into three groups of 10 mice each and treated with PBS (blank control), Ad LacZ (blank virus control) or Ad LIGHT. The tumor size was measured every other day and mouse survival was observed. As shown in Figure 4a and b, tumor growth was effectively inhibited in mice receiving Ad LIGHT therapy. The tumor size and tumor weight in Ad LIGHT-treated mice were significantly suppressed compared to mice receiving Ad LacZ or PBS alone. In the PBS-treated group, tumors grew progressively and the tumor-bearing mice eventually died around 40 days after tumor inoculation. Tumor-bearing mice that had received Ad LIGHT treatment showed a significant reduction in tumor growth. In addition, 71.4% of the mice became tumor-free and survived for more than 90 days (Figure 4c).
Figure 4.
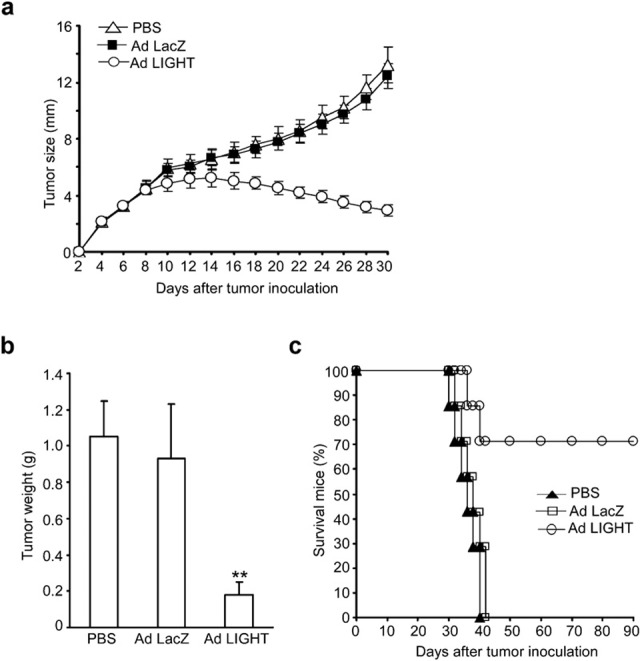
The antitumor effect of intratumoral Ad LIGHT injection on pre-existing A20 lymphoma. BALB/c mice were subcutaneously inoculated with A20 cells. When the tumor nodules were palpable, 30 mice were randomly divided into three groups (each contained 10 mice) and treated with PBS, Ad LacZ or Ad LIGHT. (a) The size of the tumor, measured every other day, is shown, where mean tumor diameter=(length+width)/2. (b) Five days after the last adenovirus injection, subcutaneous tumor nodules were taken from three killed tumor-bearing mice and weighed. **P<0.01. (c) The mean survival of remaining tumor-bearing mice was observed over time. Ad, adenovirus; PBS, phosphate-buffered saline.
Induction of splenic NK and CTL activity in tumor-bearing mice after Ad LIGHT therapy
Five days after the last treatment, mice were killed and splenocytes isolated for subsequent use in cytolytic assays against YAC-1 cells. Cytolytic assays were performed at effector/target ratios of 100∶1, 50∶1 and 25∶1, followed by analysis based on a standard LDH release assay. The NK activity of splenocytes from PBS- or Ad LacZ-treated mice was expectedly low. However, splenocyte NK activity was increased significantly in mice treated with Ad LIGHT (P<0.05; Figure 5a). Thus, an enhancement of non-specific immunity might be involved in the antitumor immune response following Ad LIGHT therapy.
Figure 5.
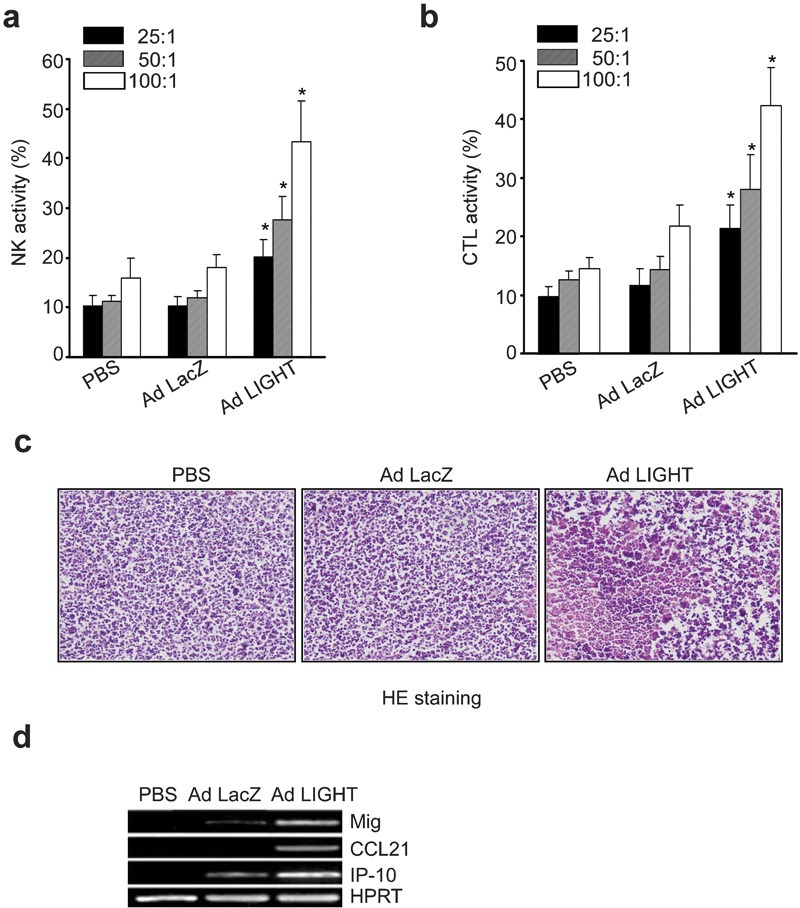
Intratumoral Ad LIGHT therapy elicits an effective immune response. (a) Five days after the last adenovirus injection, splenocytes were isolated from the tumor-bearing mice and used at different ratios as NK effector cells in cytolytic assays against YAC-1 target cells. NK activity was measured by a standard LDH assay. *P<0.05. (b) Spleen cells from the mice were cocultured with inactivated A20 cells (also treated with mitomycin-C, 100 µg/ml) for 6 days in the presence of recombinant mouse IL-2 (20 U/ml) and then collected for use as CTL effector cells. Splenocyte CTL activity was determined by a standard LDH release assay at effector/target ratios of 25∶1, 50∶1 and 100∶1 using A20 cells as target cells. *P<0.05. (c) Histological analysis of tumor tissues. Five days after the last injection of adenovirus, tumor nodules were removed for H&E staining. (d) Expression of chemokines (CCL21, IP-10 and Mig) in tumor tissues after adenovirus-mediated gene therapy. Total RNA was extracted from the subcutaneous tumor tissues of each treatment group. Semi-quantitative RT-PCR amplification was carried out using primers specific for CCL21, Mig or IP-10. PCR amplification of HPRT was also run as a control. One representative result of three independent experiments is shown. Ad, adenovirus; CCL21, CC chemokine ligand 21; CTL, cytotoxic T lymphocyte; H&E, hematoxylin and eosin; HPRT, hypoxanthine-guanine phosphoribosyltransferase; IP-10, interferon-inducible protein-10; LDH, lactate dehydrogenase; Mig, monokine induced by interferon-γ NK, natural killer; PBS, phosphate-buffered saline; RT-PCR, reverse transcription-polymerase chain reaction.
Splenocytes isolated from the killed mice were also restimulated in vitro with mitomycin-C-treated parental A20 cells for 6 days to induce CTL activity. These cells were then collected and used as effector cells. Their cytolytic activities against A20 cells were determined at effector/target ratios of 100∶1, 50∶1 and 25∶1 by a standard LDH release assay. As shown in Figure 5b, splenocytes from Ad LIGHT-treated mice showed increased CTL activity against A20 parental cells compared with mice that had received PBS or Ad LacZ treatment (P<0.05). Thereby, tumor-specific immunity was markedly augmented following intratumoral injection of Ad LIGHT.
Heightened infiltration of inflammatory cells and increased chemokine expression in the tumor mass after Ad LIGHT therapy
Subcutaneous tumor nodules were taken from tumor-bearing mice 5 days after the last treatment. Histological examination of the tumor mass demonstrated a high level of necrosis and a massive infiltration of inflammatory cells, including neutrophils, lymphocytes and monocytes, present inside tumors and in the surrounding tissue of mice that had received Ad LIGHT therapy. In contrast, relatively less infiltration was present in the tumor tissues of Ad LacZ- or PBS-treated groups (Figure 5c).
Enforced expression of LIGHT in tumor cells could trigger the upregulation of chemokines, including IP-10, Mig and CCL21.19 We used semiquantitative RT-PCR to examine the expression of each of these three chemokines in tumor tissues. We found IP-10 and Mig mRNA were strongly expressed in the tumor tissues of the Ad LIGHT-treated group. Very weak or undetectable IP-10 or Mig mRNA expression was detected from the tumor tissues of PBS- or Ad LacZ-treated mice. CCL21 mRNA was also detected in tumor tissues from Ad LIGHT-treated mice (Figure 5d). The high expression of IP-10 and Mig may contribute to the reduction of new blood vessels and further the retardation of tumor progression.
Protection against subsequent tumor rechallenge after Ad LIGHT therapy
To test whether intratumoral Ad LIGHT therapy could induce a systemic long-term protective immunity, mice from the Ad LIGHT-treated group that exhibited complete tumor regression were rechallenged on the contralateral site of the original tumor inoculation with 1×106 parental A20 cells. Tumor growth and mouse survival were observed. All of the mice that had been previously cured by Ad LIGHT therapy were protected from rechallenge with parental A20 cells and survived more than 90 days post-rechallenge. In contrast, naive mice challenged with the same number of A20 cells showed excessive tumor burden and all died around 40 days after the tumor challenge (Figure 6). Therefore, Ad LIGHT therapy induced the acquisition of a strong, long-term protective immunity against parental tumor cells in the host.
Figure 6.
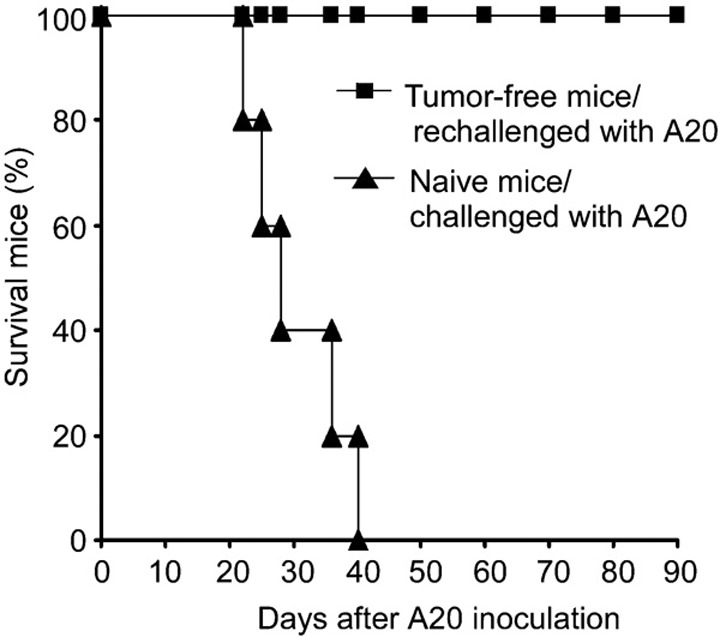
Ad LIGHT therapy induces long-term protective immunity. After 90 days, the tumor-free mice of the Ad LIGHT-treated group were subcutaneously rechallenged on the contralateral site with 1×106 A20 cells. Naive mice receiving subcutaneous injections of the same number of A20 cells were used as controls, with five mice per group. Survival curves of mice following A20 challenge are shown. Results are representative of three independent experiments. Ad, adenovirus.
DISCUSSION
Tumors evade immune surveillance by multiple mechanisms: the production of factors such as transforming growth factor-β and vascular endothelial growth factor, which inhibit DC activation and impair tumor-specific T-cell immunity; developing resistance to T cell-induced apoptosis; and the downregulation of MHC molecules.22, 23, 24 One reason why malignant B cells evade the immune system is that they lack expression of some important accessory molecules necessary for the efficient activation of antigen-specific T cells. Recent efforts have focused on changing the phenotype of B-cell tumors through the transduction of costimulatory molecules such as CD40L. Induced expression of costimulatory molecules may allow for enhanced recognition by tumor-specific T cells, leading to a more efficient immune response.13, 14
LIGHT possesses several functional attributes that make it an attractive candidate for cancer immunotherapy. The engagement of LIGHT with HVEM expressed on immature DCs induced the increased expression of activation markers and resulted in DC maturation. Mice deficient in LIGHT show defects in lymph node architecture and reduced cytotoxicity of CTLs.25, 26 In addition, LIGHT has been demonstrated to induce the expression of various chemokines and adhesion molecules that favor the infiltration of lymphocytes into tumor tissues.19 Interestingly, a recent study showed that LIGHT is constitutively expressed in some human melanoma cells and tumor-derived microvesicles and that tumors expressing LIGHT were associated with increased lymphocytic infiltration.27 It was recently found that tumor-localized LIGHT-treatment can generate tumor-specific CTLs that exit the primary tumor and may infiltrate distal tumors leading to the eradication of spontaneous metastases in a fibrosarcoma model and a metastatic breast cancer model.28
In this study, we used the BALB/c-derived A20 B-cell lymphoma model to investigate the antitumor effects of adenovirus-mediated LIGHT gene transfer on the established tumor burden and on the protective immunity elicited following inoculation with an Ad LIGHT-modified A20 vaccine. A20 cells transfected with Ad LIGHT were capable of stimulating T lymphocyte proliferation and IFN-γ production in vitro. LIGHT modification of A20 cells also induced the upregulated expression of ICAM-1 (CD54) and the death receptor Fas. ICAM-1 functions as an adhesion receptor for the β-integrins, LFA-1 and Mac1, expressed on leukocytes.29 ICAM-1 enhances leukocyte adhesion to endothelial cells and thus plays a pivotal role in the transendothelial migration of neutrophils to sites of inflammation. Engagement of ICAM-1 on antigen presenting cells or target cells by LFA-1 expressed on T cells provides an important costimulatory signal that promotes T-cell activation.30 B cells expressing high levels of surface ICAM-1 elicit significantly higher T-cell responses than those with low levels, suggesting that the expression level of ICAM-1 on B cells correlates with their costimulatory function.31 Stopeck et al.'s study on diffuse large B-cell lymphoma patients showed that the loss of CD86 and ICAM-1 expression is associated with a decrease in tumor-infiltrating T lymphocytes.12 The upregulation of ICAM-1 expression by LIGHT gene modification may therefore enhance the antigen-presenting capacity of A20 B-cell lymphoma cells, leading to increased T-cell activation. Upregulated Fas expression on A20 cells might enhance their sensitivity to Fas-induced apoptosis. In addition, LIGHT could directly induce apoptosis in A20 cells. The changes observed after LIGHT gene modification likely contribute to the in vivo antitumor effect.
In vivo immunization with Ad LIGHT-modified A20 vaccine efficiently elicited protective immunity against challenge by the parental A20 tumor. Indeed, subcutaneous tumor occurrence in A20/LIGHT-immunized mice was significantly inhibited and these mice survived much longer than control animals. In addition, tumor-specific splenic CTL activity was markedly augmented, indicating that vaccination with LIGHT gene-modified A20 cells significantly induced tumor-specific immunity. These results might provide a strategy for the prevention of tumor recurrence in B-cell lymphoma patients following radiotherapy and chemotherapy.
In our model of pre-existing A20 B-cell lymphoma, adenovirus-mediated LIGHT gene transfer also led to potent antitumor activity. Both tumor size and tumor weight were significantly suppressed in Ad LIGHT-treated mice compared to mice receiving Ad LacZ therapy or PBS alone. Tumor-bearing mice that had received an Ad LIGHT injection showed a significant reduction in tumor growth with 71.4% of the mice becoming tumor-free and surviving for greater than 90 days. Thus, Ad LIGHT therapy efficiently induced a potent local and systemic antitumor immune response in tumor-bearing mice, which was associated with strong CTL and NK activity of their splenocytes. Furthermore, when mice exhibiting complete tumor regression following Ad LIGHT treatment were given a secondary challenge with the parental A20 cell line, all mice were protected from rechallenge with a high dose, up to 1×106 cells, of parental A20 cells and they again exhibited prolonged survival (>90 days). Therefore, Ad LIGHT therapy led to a strong, long-term protective immunity against parental tumor cells. The induction of such a potent, long-lasting tumor-specific response is crucial for preventing tumor enlargement and maintaining a tumor-free state. These results indicate that adenovirus-mediated LIGHT gene transfer may be utilized as a potential therapy in the intervention of B-cell lymphoma.
The infiltration of inflammatory cells into tumor tissues is required for the elimination of pre-established tumors. Recently, Hisasa et al. reported that coexpression of LIGHT and CCL21 in a colon carcinoma resulted in profound infiltration by mature DCs and CD8+ T cells. They reasoned that the observed enhancement in CTL activity and IFN-γ production was mainly due to the efficient infiltration of the tumor by these immune cells.32 Previous findings have also demonstrated that in situ expression of LIGHT could promote increased infiltration by CD8+ T cells and NK cells.19, 33 In the present study, we noticed greater tumor necrosis and enhanced infiltration by inflammatory cells, within tumors and their surrounding tissue, in mice that had received Ad LIGHT treatment. We speculate that the intratumoral injection of Ad LIGHT triggered the infiltration of inflammatory cells, including NK cells and T cells. This notion is partly supported by the induced expression of CCL21 in tumor tissues following Ad LIGHT therapy. As CCL21 is chemotactic for lymphocytes,34 it is reasonable that induced expression of CCL21 by tumor cells promotes the rich infiltration by inflammatory cells that we observed. However, additional studies would be required to confirm the role of tumor-specific chemokine expression in lymphocyte infiltration of the tumor.
Here, we demonstrate that, in addition to the expression of CCL21, LIGHT also induces the expression of IP-10 and Mig in tumor tissues. IP-10 and Mig are two important chemokines that have been reported to possess antiangiogenic and antineoplastic activity. They may also modulate a variety of other functions, such as lymphocyte activation, differentiation and effector function.35, 36 Upregulation of IP-10 and Mig in the tumor microenvironment following Ad LIGHT injection may potentially reduce the formation of new tumor-induced blood vessels, as well as lead to the recruitment and retention of more activated T cells to the tumor tissues, thereby creating a favorable microenvironment for suppressing tumor growth. Nonetheless, the relative contribution of these two chemokines to the observed antitumor immunity requires further study.
In summary, we have shown that LIGHT modification of A20 cells induced the upregulation of ICAM-1 and Fas expression, as well as enhanced apoptosis of A20 cells. Immunization with LIGHT-modified A20 vaccine elicited efficient and protective immunity against challenge by the parental tumor. Adenovirus-mediated intratumoral expression of LIGHT exerted a very potent antitumor activity, inducing substantial specific systemic antitumor immune response by the host. We propose that such therapy might have potential utility for the prevention and treatment of human B-cell lymphoma.
Acknowledgments
We thank Professor Yangxin Fu of The University of Chicago for the gift of Ad LIGHT. This work was supported by grants from the National Natural Science Foundation of China (30328011 and 30872377), the National Basic Research Program of China (2004CB518802) and the Science and Technology Department of Zhejiang Province (2008C23044). We would like to acknowledge Jianping Pan and Dajing Xia for helpful suggestions and discussions.
References
- LaCasce AS, Kho ME, Friedberg JW, Niland JC, Abel GA, Rodriguez MA, et al. Comparison of referring and final pathology for patients with non-Hodgkin's lymphoma in the National Comprehensive Cancer Network. J Clin Oncol. 2008;26:5107–5112. doi: 10.1200/JCO.2008.16.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri T, Stakiw J, Pintilie M, Keating A, Crump M, Kuruvilla J. Utility of subsequent conventional dose chemotherapy in relapsed/refractory transplant-eligible patients with diffuse large B-cell lymphoma failing platinum-based salvage chemotherapy. Hematology. 2008;13:261–266. doi: 10.1179/102453308X343527. [DOI] [PubMed] [Google Scholar]
- Bernstein SH, Unger JM, Leblanc M, Friedberg J, Miller TP, Fisher RI. Natural history of CNS relapse in patients with aggressive non-Hodgkin's lymphoma: a 20-year follow-up analysis of SWOG 8516 – the Southwest Oncology Group. J Clin Oncol. 2009;27:114–119. doi: 10.1200/JCO.2008.16.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin's lymphoma. N Engl J Med. 2008;359:613–626. doi: 10.1056/NEJMra0708875. [DOI] [PubMed] [Google Scholar]
- Meziane el K, Bhattacharyya T, Armstrong AC, Qian C, Hawkins RE, Stern PL, et al. Use of adenoviruses encoding CD40L or IL-2 against B cell lymphoma. Int J Cancer. 2004;111:910–920. doi: 10.1002/ijc.20332. [DOI] [PubMed] [Google Scholar]
- Ulaner GA, Colletti PM, Conti PS. B-cell non-Hodgkin lymphoma: PET/CT evaluation after 90Y-ibritumomab tiuxetan radioimmunotherapy – initial experience. Radiology. 2008;246:895–902. doi: 10.1148/radiol.2463060588. [DOI] [PubMed] [Google Scholar]
- Sapra P, Allen TM. Improved outcome when B-cell lymphoma is treated with combinations of immunoliposomal anticancer drugs targeted to both the CD19 and CD20 epitopes. Clin Cancer Res. 2004;10:2530–2537. doi: 10.1158/1078-0432.ccr-03-0376. [DOI] [PubMed] [Google Scholar]
- Hurwitz AA, Kwon ED, van Elsas A. Costimulatory wars: the tumor menace. Curr Opin Immunol. 2000;12:589–596. doi: 10.1016/s0952-7915(00)00147-3. [DOI] [PubMed] [Google Scholar]
- Celli S, Garcia Z, Beuneu H, Bousso P. Decoding the dynamics of T cell-dendritic cell interactions in vivo. Immunol Rev. 2008;221:182–187. doi: 10.1111/j.1600-065X.2008.00588.x. [DOI] [PubMed] [Google Scholar]
- Schultze JL, Cardoso AA, Freeman GJ, Seamon MJ, Daley J, Pinkus GS, et al. Follicular lymphomas can be induced to present alloantigen efficiently: a conceptual model to improve their tumor immunogenicity. Proc Natl Acad Sci USA. 1995;92:8200–8204. doi: 10.1073/pnas.92.18.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyth-Dreese FA, Dellemijn TA, van Oostveen JW, Feltkamp CA, Hekman A. Functional expression of adhesion receptors and costimulatory molecules by fresh and immortalized B-cell non-Hodgkin's lymphoma cells. Blood. 1995;85:2802–2812. [PubMed] [Google Scholar]
- Stopeck AT, Gessner A, Miller TP, Hersh EM, Johnson CS, Cui H, et al. Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin Cancer Res. 2000;6:3904–3909. [PubMed] [Google Scholar]
- Briones J, Timmerman J, Levy R. In vivo antitumor effect of CD40L-transduced tumor cells as a vaccine for B-cell lymphoma. Cancer Res. 2002;62:3195–3199. [PubMed] [Google Scholar]
- Costello RT, Mallet F, Barbarat B, Schiano de Colella JM, Sainty D, Sweet RW, et al. Stimulation of non-Hodgkin's lymphoma via HVEM: an alternate and safe way to increase Fas-induced apoptosis and improve tumor immunogenicity. Leukemia. 2003;17:2500–2507. doi: 10.1038/sj.leu.2403175. [DOI] [PubMed] [Google Scholar]
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, et al. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest. 1998;102:1142–1151. doi: 10.1172/JCI3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, et al. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. doi: 10.4049/jimmunol.164.8.4105. [DOI] [PubMed] [Google Scholar]
- Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D, et al. Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat Med. 2000;6:283–289. doi: 10.1038/73136. [DOI] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141–149. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu H, Ju DW, He L, Pan JP, Xia DJ, et al. Intratumoral IL-18 gene transfer improves therapeutic efficacy of antibody-targeted superantigen in established murine melanoma. Gene Ther. 2001;8:542–550. doi: 10.1038/sj.gt.3301428. [DOI] [PubMed] [Google Scholar]
- Zoll B, Lefterova P, Csipai M, Finke S, Trojaneck B, Ebert O, et al. Generation of cytokine-induced killer cells using exogenous interleukin-2, -7 or -12. Cancer Immunol Immunother. 1998;47:221–226. doi: 10.1007/s002620050524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JS, Terabe M, Kang MJ, Chae H, Voong N, Yang YA, et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68:3915–3923. doi: 10.1158/0008-5472.CAN-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, et al. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol. 2008;180:4606–4614. doi: 10.4049/jimmunol.180.7.4606. [DOI] [PubMed] [Google Scholar]
- Lu B, Finn OJ. T-cell death and cancer immune tolerance. Cell Death Differ. 2008;15:70–79. doi: 10.1038/sj.cdd.4402274. [DOI] [PubMed] [Google Scholar]
- Ye Q, Fraser CC, Gao W, Wang L, Busfield SJ, Wang C, et al. Modulation of LIGHT-HVEM costimulation prolongs cardiac allograft survival. J Exp Med. 2002;195:795–800. doi: 10.1084/jem.20012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–1624. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortarini R, Scarito A, Nonaka D, Zanon M, Bersani I, Montaldi E, et al. Constitutive expression and costimulatory function of LIGHT/TNFSF14 on human melanoma cells and melanoma-derived microvesicles. Cancer Res. 2005;65:3428–3436. doi: 10.1158/0008-5472.CAN-04-3239. [DOI] [PubMed] [Google Scholar]
- Yu P, Lee Y, Wang Y, Liu X, Auh S, Gajewski TF, et al. Targeting the primary tumor to generate CTL for the effective eradication of spontaneous metastases. J Immunol. 2007;179:1960–1968. doi: 10.4049/jimmunol.179.3.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr Opin Immunol. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Dennig D, Lacerda J, Yan Y, Gasparetto C, O'Reilly RJ. ICAM-1 (CD54) expression on B lymphocytes is associated with their costimulatory function and can be increased by coactivation with IL-1 and IL-7. Cell Immunol. 1994;156:414–423. doi: 10.1006/cimm.1994.1186. [DOI] [PubMed] [Google Scholar]
- Hisada M, Yoshimoto T, Kamiya S, Magami Y, Miyaji H, Yoneto T, et al. Synergistic antitumor effect by coexpression of chemokine CCL21/SLC and costimulatory molecule LIGHT. Cancer Gene Ther. 2004;11:280–288. doi: 10.1038/sj.cgt.7700676. [DOI] [PubMed] [Google Scholar]
- Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 2006;107:1342–1351. doi: 10.1182/blood-2005-08-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- Zhang R, Tian L, Chen LJ, Xiao F, Hou JM, Zhao X, et al. Combination of MIG (CXCL9) chemokine gene therapy with low-dose cisplatin improves therapeutic efficacy against murine carcinoma. Gene Ther. 2006;13:1263–1271. doi: 10.1038/sj.gt.3302756. [DOI] [PubMed] [Google Scholar]