

Fibro-adipogenic progenitors (FAPs) are components of the skeletal muscle regenerative environment that strongly influence the pathogenesis of muscular dystrophies. Saccone et al. show that HDAC, muscle-specific microRNAs (myomiRs), and SWI/SNF chromatin remodeling complex component BAF60 variants function together to regulate the fate of FAPs in dystrophic muscles of mdx mice. The results shed light on differences between the muscle-regenerative capacities of early and late stage mdx mice and provide a molecular rationale to promote the therapeutic regeneration of dystrophic muscles.
Keywords: SWI/SNF chromatin remodeling, BAF60, microRNA, HDAC, FAPs, muscular dystrophy
Abstract
Fibro-adipogenic progenitors (FAPs) are important components of the skeletal muscle regenerative environment. Whether FAPs support muscle regeneration or promote fibro-adipogenic degeneration is emerging as a key determinant in the pathogenesis of muscular diseases, including Duchenne muscular dystrophy (DMD). However, the molecular mechanism that controls FAP lineage commitment and activity is currently unknown. We show here that an HDAC–myomiR–BAF60 variant network regulates the fate of FAPs in dystrophic muscles of mdx mice. Combinatorial analysis of gene expression microarray, genome-wide chromatin remodeling by nuclease accessibility (NA) combined with next-generation sequencing (NA-seq), small RNA sequencing (RNA-seq), and microRNA (miR) high-throughput screening (HTS) against SWI/SNF BAF60 variants revealed that HDAC inhibitors (HDACis) derepress a “latent” myogenic program in FAPs from dystrophic muscles at early stages of disease. Specifically, HDAC inhibition induces two core components of the myogenic transcriptional machinery, MYOD and BAF60C, and up-regulates the myogenic miRs (myomiRs) (miR-1.2, miR-133, and miR-206), which target the alternative BAF60 variants BAF60A and BAF60B, ultimately directing promyogenic differentiation while suppressing the fibro-adipogenic phenotype. In contrast, FAPs from late stage dystrophic muscles are resistant to HDACi-induced chromatin remodeling at myogenic loci and fail to activate the promyogenic phenotype. These results reveal a previously unappreciated disease stage-specific bipotency of mesenchimal cells within the regenerative environment of dystrophic muscles. Resolution of such bipotency by epigenetic intervention with HDACis provides a molecular rationale for the in situ reprogramming of target cells to promote therapeutic regeneration of dystrophic muscles.
Duchenne muscular dystrophy (DMD) is an inherited muscular disease that remains fatal despite the increasing knowledge of the biochemical and functional consequences of dystrophin absence in skeletal muscles (Dalkilic and Kunkel 2003). DMD pathogenesis is characterized by an early stage regenerative response that counterbalances muscle loss but eventually declines as the disease progresses such that initial myofiber repair becomes biased toward the formation of fibrotic scars and fat infiltration (Dalkilic and Kunkel 2003; Mozzetta et al. 2009; Serrano et al. 2011). Whether the decline occurs because the principal effectors of muscle regeneration—the muscle stem (satellite) cells (MuSCs)—exhibit an intrinsic failure to respond to regeneration cues or because their microenvironment (the satellite cell niche) deteriorates remains controversial (Brack and Rando 2012; Yin et al. 2013a).
It is becoming apparent that reciprocal interactions between distinct cellular components of the regeneration machinery contribute to establish either a productive or a hostile environment for compensatory regeneration of dystrophic muscles (Judson et al. 2013). In particular, the recent identification of multipotent mesenchymal cells residing in skeletal muscle interstitium, termed fibro-adipogenic progenitors (FAPs) (Joe et al. 2010; Uezumi et al. 2010; Pannérec et al. 2013), that influence the activity of MuSCs has revealed an interesting novel cellular contributor to DMD pathogenesis (Uezumi et al. 2011; Mozzetta et al. 2013). FAPs appear to convert environmental cues into signals that modulate muscle regeneration through functional interactions with MuSCs. For instance, muscle injury-derived signals, such as those from type 2 innate immunity (Heredia et al. 2013), stimulate FAPs to produce paracrine factors that promote MuSC-mediated regeneration (Joe et al. 2010). In contrast, in degenerating muscles such as dystrophic muscles at advanced stages of disease, FAPs turn into fibro-adipocytes, which mediate fat deposition and fibrosis (Uezumi et al. 2011), thereby disrupting the environment conducive for muscle regeneration. We reported recently that changes in FAP activity during disease progression contribute to the exhaustion of muscle regeneration and the progressive fibro-adipogenic degeneration of dystrophic muscles and determine the loss of response to HDAC inhibitors (HDACis) in old mdx mice (Mozzetta et al. 2013).
Previous studies showed that pharmacological interventions such as HDACi treatment can counter the progressive decline in muscle function by promoting regeneration, specifically by targeting key events downstream from the genetic defect (Minetti et al. 2006; Colussi et al. 2008; Consalvi et al. 2013; for review, see Mozzetta et al. 2009; Consalvi et al. 2011). In the permissive environment of young mdx mice, FAPs support MuSC-mediated regeneration, and this correlates with the ability of HDACis to promote muscle regeneration and inhibit fibro-adipogenic degeneration. At late stages of disease progression (old mdx mice), FAPs constitutively inhibit the myogenic potential of MuSCs and become resistant to HDACis (Mozzetta et al. 2013). This evidence indicates that FAP function is influenced by environmental changes that occur in dystrophic muscle during disease progression. Importantly, FAPs from regenerating muscles, such as those of young mdx mice, restored the ability of HDACis to promote satellite cell-mediated formation of myotubes and endogenous muscle regeneration in old mdx mice (Mozzetta et al. 2013). These data suggest that the intrinsic myogenic potential of MuSCs is not altered during disease progression in mdx mice and that extrinsic changes in muscle environment compromise productive interactions between FAPs and MuSCs. Collectively, the data reported above point to FAPs as a novel cell population within the MuSC niche that can be exploited to improve regeneration and limit pathological events, such as fibro-adipose infiltration. As such, the alternative ability of FAPs to support muscle regeneration or promote fibro-adipogenic degeneration might be an important determinant in the pathogenesis of DMD.
Manipulating FAP function might be a powerful means to alter disease progression; however, the molecular basis for the alternative FAP phenotypes remains unknown. Prior work had suggested that the phenotypical plasticity of FAPs in relation to the environmental changes might be under epigenetic control (for review, see Giordani and Puri 2013). Here, we describe a novel molecular network that mediates HDACis’ ability to reprogram the FAP phenotype and supports compensatory regeneration at early stages of DMD progression in mdx mice, thereby providing the rationale for the pharmacological control of the FAP phenotype to counter DMD progression.
Results
HDACis promote a myogenic phenotype in FAPs of young mdx mice
We began to decipher the molecular events underlying the ability of FAPs to mediate the beneficial effects of HDACis in young mdx mice (Mozzetta et al. 2013) by performing a gene expression microarray analysis. RNA was collected from FAPs immediately after their isolation from muscles of young (1.5-mo-old) mdx mice treated with TSA or untreated. Among the up-regulated transcripts, those implicated in muscle determination and differentiation as well as in muscle contraction and metabolism scored as the top category in FAPs isolated from TSA-treated mdx mice (Fig. 1A,B; Supplemental Fig. S1A,B). The complete list of up-regulated and down-regulated genes is accessible through Gene Expression Omnibus (GEO) series accession number GSE34843 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE34843). Interestingly, in TSA-treated FAPs, we noted the induction of the muscle-specific determination factor MyoD and the SWI/SNF chromatin remodeling complex subunit BAF60C (encoded by Smarcd3) that is required for skeletal myogenesis and MYOD-mediated activation of muscle gene transcription (Simone et al. 2004; Lickert et al. 2004; Forcales et al. 2012; Albini et al. 2013). Although the muscle gene transcripts induced by TSA in FAPs from young mdx mice were expressed at lower levels than in MuSCs (Fig. 1B), MyoD-expressing FAPs eventually differentiated into MYHC-positive myotubes rather than Oil red O-positive adipocytes when cultured in adipogenic conditions (adipogenic differentiation medium) for 6 d (Fig. 1C–F). Gene ontology and network analysis revealed the up-regulation of components of pathways (e.g., Wnt and Notch) implicated in muscle development and adult myogenesis (Brack and Rando 2012; von Maltzahn et al. 2012) as well as signals recently involved in FAP activation by eosinophils during muscle regeneration (Supplemental Fig. S1A,B; Heredia et al. 2013). Quantitative RT–PCR (qRT–PCR) analysis confirmed the induction of muscle genes in FAPs from TSA-treated mdx mice over the very low levels detectable in the same cells from control mdx mice (Supplemental Fig. S1C) but not from wild-type mice (data not shown), as previously reported by Joe et al. (2010). Genes involved in adipogenic differentiation (e.g., Pparγ) and fibrogenesis (e.g., Fibronectin [FINC]) that are typically induced in FAPs by culture in adipogenic medium (Joe et al. 2010; Uezumi et al. 2010) were down-regulated by exposure to TSA (Supplemental Fig. S1C). Western blot analysis confirmed the expression of MYOD and the late contractile protein MYHC in FAPs from young mdx mice exposed to TSA, while the adipogenic protein Perilipin was down-regulated under the same conditions (Fig. 1C).
Figure 1.

HDACis induce the myogenic phenotype in FAPs isolated from young mdx mice. (A) Heat map representation of 6132 genes with a fold change of at least ±2 and a P-value <0.05 that were selected and hierarchically clustered for conditions and genes in a microarray analysis of gene expression of FAPs from 1.5-mo-old mdx mice treated with either 0.5 mg/kg TSA or vehicle (control) for 45 d. The replicate sample names are indicated at the bottom (CTR1 and CTR2, control samples; TSA1 and TSA2, TSA samples), and the color scale for the log2 expression levels are at the top. (B) Manually assembled list of muscle genes up-regulated by TSA in FAPs isolated from mdx mice. Fold changes in gene expression in the first column are relative to FAPs isolated from control mdx mice; fold changes in the second column are relative to MuSCs isolated from control mdx mice compared with FAPs isolated from TSA-treated mdx mice. Pax7* levels are 8.8-fold higher in TSA-treated cells despite levels that were not detectable in one sample of control-treated cells. (C) Myogenic differentiation was assessed by Western blot analysis for MYOD and MYHC, while adipogenic differentiation was assessed by Western blot for Perilipin. (D) Immunofluorescence staining for MYOD in FAPs isolated from TSA-treated or control 1.5-mo-old mdx mice cultured in growth medium (GM) for 4 d. (Left panel) Representative images of MYOD (green) staining are shown. Nuclei were counterstained with DAPI (blue). (Right panel) Graph showing the percentage of nuclei that were MYOD-positive. (E) Immunofluorescence for MYHC expression in FAPs isolated from TSA-treated or control 1.5-mo-old mdx mice and cultured in adipogenic differentiation medium (DM). (Left panel) Representative images of MYHC (green) staining. Nuclei were counterstained with DAPI (blue). (Right panel) Graph showing the quantification of the myogenic differentiation index as a percentage of nuclei that were MYHC-positive. (F) Oil red O staining in FAPs isolated from TSA-treated or control 1.5-mo-old mdx mice and cultured in adipogenic differentiation medium (DM). (Left panel) Representative images of Oil red O staining (red). (Right panel) Graph showing the quantification of the Oil red O area (measured as pixels squared) per field. Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01.
The up-regulation of muscle genes observed in FAPs from TSA-treated mdx mice could, in theory, be due to contamination with sporadic MuSCs during FACS sorting and their subsequent clonigenic expansion. However, a large proportion of MYOD-expressing FAPs was detected already within 4 d after their isolation from TSA-treated mdx mice (Fig. 1D), timing incompatible with a potential clonigenic expansion of contaminating MuSCs. We further investigated this issue by evaluating the ability of TSA to promote MYOD expression in FAPs cultured after isolation from mdx mice. FAPs sorted from 1.5-mo-old mdx mice did not show expression of α7INTEGRIN, a surface marker that is used for FACS-mediated isolation of MuSCs and was uniformly detected in MuSCs sorted from mdx mice (Supplemental Fig. S2A). Conversely, FAPs sorted from mdx mice showed expression of SCA1, the surface marker that is used for FACS-mediated isolation of FAPs and was not detected in MuSCs (Supplemental Fig. S2B). When this “pure” population of α7INTEGRINneg/SCA1pos FAPs was exposed to TSA in vitro, we could observe MYOD expression in SCA1-expressing cells within the first 48 h (Supplemental Fig. S2C), demonstrating that FAPs of mdx muscles have an HDACi-inducible intrinsic myogenic potential.
Collectively, these data support the conclusion that, in addition to the reported ability to support the myogenic potential of MuSCs (Joe et al. 2010; Mozzetta et al. 2013), FAPs from young mdx mice show a “latent” myogenic phenotype that is derepressed by HDACis at the expense of their fibro-adipogenic potential. The induction of the myogenic program by TSA appears to be a distinctive feature of FAPs from young mdx mice, as the same phenotype was not observed in FAPs isolated from uninjured muscles of wild-type or old mdx mice exposed to TSA (see Supplemental Fig. S3D; data not shown).
HDACis promote genome-wide changes of chromatin structure and focal remodeling at muscle loci in FAPs from young mdx mice
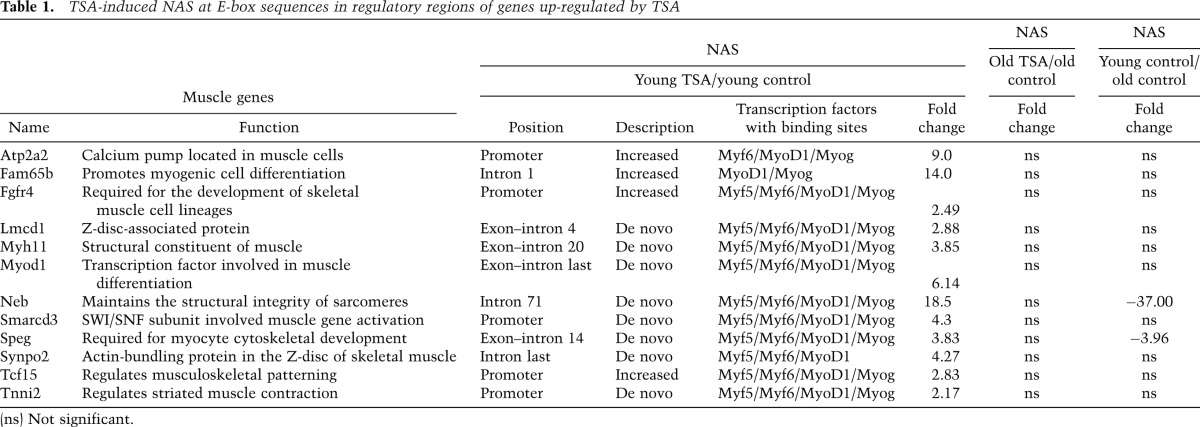
The induction of BAF60C and MyoD in FAPs from TSA-treated mdx young mice suggests that these two factors could assemble to form the pioneer complex that recruits the SWI/SNF complex, which promotes chromatin remodeling at muscle loci, as previously shown in skeletal myoblasts (Forcales et al. 2012) and during conversion of human embryonic stem cells into skeletal muscle cells (Albini et al. 2013). We used a nuclease accessibility (NA) assay coupled to next-generation sequencing (NA-seq) to monitor chromatin remodeling at the genome-wide level in FAPs from TSA-treated mdx mice, as compared with untreated controls. An increased frequency of NA sites (NASs) was detected in FAPs from young mdx mice exposed to TSA (Fig. 2A), a treatment leading to the functional and histological recovery of dystrophic muscles (Minetti et al. 2006; Mozzetta et al. 2013) and to the up-regulation of muscle genes (see Fig. 1). In contrast, FAPs from old mdx mice, which are resistant to the beneficial activity of HDACis (Mozzetta et al. 2013), did not show any statistically significant increase in NASs in response to TSA (Fig. 2A). The complete list of NASs in FAPs from young and old mdx mice with or without TSA treatment is accessible through GEO series accession number GSE52062 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52062). Among the total NASs detected in FAPs from TSA-treated young mdx mice, ∼70% were induced de novo, while 30% were previously existing NASs that were further increased by TSA (Fig. 2B). Interestingly, the E-box sequences typically bound by myogenic basic helix–loop–helix factors and detected in previous chromatin immunoprecipitation (ChIP) combined with deep sequencing (ChIP-seq) analysis of MYOD–chromatin binding in C2C12 myoblasts (Cao et al. 2010) were found to be associated with high frequency with both de novo and “increased” NASs detected in FAPs from TSA-treated young mdx mice (Fig. 2C,E). When we matched the NASs activated by TSA in FAPs from young mdx mice with the changes in gene expression revealed by microarray analysis (Fig. 1B), we identified 3.04% of the total NASs that were found on putative regulatory elements (within 20 kb of the transcription start site [TSS]) of up-regulated and down-regulated genes (Fig. 2D,E). The remaining 96.5% of the TSA-induced NASs apparently did not coincide with up-regulated or down-regulated genes (Fig. 2D,E). Global gene ontology analysis of the overlap between NASs and genes up-regulated in FAPs from young mdx mice treated with TSA revealed a strong enrichment in muscle-related genes and components of signaling pathways, such as the WNT signaling, that promote skeletal myogenesis (Table 1; Supplemental Fig. S3A–C). Among these genes, we again noted MyoD and BAF60C (Table 1). We focused on these two key activators of skeletal myogenesis as “proxies” to validate the ability of TSA to activate the myogenic program in FAPs via histone hyperacetylation and chromatin remodeling at muscle-specific loci. Indeed, qRT–PCR analysis confirmed the ability of TSA to induce the expression of Myod1 and Baf60c in FAPs from young but not old mdx mice (Supplemental Fig. S3D). We used ChIP with anti-acetylated H3 (AcH3) antibodies and formaldehyde-assisted isolation of regulatory element (FAIRE) to monitor the histone acetylation and chromatin structure at the NASs detected in Myod1 and Baf60c genes. This analysis showed an increased histone acetylation and chromatin accessibility in coincidence with the NASs detected at Myod1 and Baf60c loci (Fig. 2F). In contrast, TSA could not induce the expression of Hoxa7 (Supplemental Fig. S3D), which was not annotated among the genes up-regulated in FAPs from TSA-treated young mdx mice. The lack of Hoxa7 induction correlated with the absence of NASs and lack of histone hyperacetylation and chromatin remodeling on the regulatory sequence of Hoxa7 in FAPs from TSA-treated mdx mice (Fig. 2F). Collectively, the results of the combined gene expression profile and NA-seq analysis revealed a latent myogenic feed-forward circuitry in FAPs from young mdx mice that is derepressed by HDACis and relies on the activation of the core components of the transcriptional myogenic machinery, MYOD and BAF60C. Conversely, the constitutive repression of MYOD and BAF60C appears to compromise the activation of this network in FAPs from old mdx mice.
Figure 2.

HDACi remodeling of FAPs from young mdx mice at muscle loci. (A) Box plots showing the distributions of P-values of TSA-induced (TSA vs. control [CTRL]) and TSA-repressed (control vs. TSA) accessible regions in FAPs from young (1.5-mo-old) mdx mice (top panel) and old (5-mo-old) mdx mice (bottom panel) treated with vehicle (CTRL) or 0.5 mg/kg TSA for 15 d. (B) Pie chart showing the distribution of the statistically significant TSA-induced NASs detected in FAPs from young mdx mice, of which 70.4% were de novo (meaning regions that were not accessible prior to treatment), while 29.6% were previously existing NASs that are further increased after TSA treatment. (C) Table showing the percentage of myogenic E-boxes in the global NAS analysis. Along with the myogenic transcription factor (TF) gene symbol, logos representing published binding preferences are reported. The percentages showing a putative binding site for each transcription factor in de novo and increased NASs after HDACi treatment are indicated. NASs with a myogenic E-box were compared with MYOD-binding sites (ChiP-seq data) in proliferating and differentiating myoblasts (C2C12 growth medium [GM] and differentiation medium [DM]) and are shown in the bottom panel of E. The overlap is reported as percentage. (D) Pie chart showing the percentage of NASs found near genes up-regulated (3%) or down-regulated (0.5%) by TSA or not related to changes in gene expression (96.5%). (E) Heat maps showing quantitative information for 305 NASs residing either in gene bodies or within 20 kb upstream of TSSs of TSA-regulated genes in young mdx mice and for the corresponding genes. The top panel shows the FPKM (fragments per kilobase of accessible region per million fragments sequenced) in FAPs from control (CTR) and TSA-treated mdx mice (red, more accessible). The second panel reports the position of the NASs relative to genes. The third panel shows the log2 ratio among mRNAs detected by microarrays in FAPs from control and TSA-treated mdx mice. The bottom panel shows the NAS correlation with MYOD chromatin-binding regions detected by previous ChIP-seq data (Cao et al. 2010) in proliferating and differentiating myoblasts (C2C12 growth medium [GM] and differentiation medium [DM]). (F) NA-seq validation. (Top panel) Schematic representation of mbaf60c, mMyod1, and mHoxa7 loci with the NASs and primers used to amplify these regions. (Bottom panel) On the left are graphs showing ChIP analysis for acetylated histone 3 (AcH3), and on the right are graphs showing FAIRE analysis at the NA regions of Baf60c and Myod1 in FAPs isolated from control (CTR) or TSA-treated mdx mice. Hoxa7 was used as a negative control. Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01.
Table 1.
TSA-induced NAS at E-box sequences in regulatory regions of genes up-regulated by TSA
Identification of HDACi-induced myogenic microRNAs (myomiRs) that target the BAF60 variants A and B
We next analyzed the genomic distribution of the largest portion of TSA-induced NASs (>95%) that did not associate with putative regulatory elements within 20 kb of the TSSs of up-regulated and down-regulated genes. Supplemental Figure 4 shows that most of these NASs are associated with noncoding genomic sequences that included miRs, long noncoding RNAs (lincRNAs), and long and short interspersed noncoding elements (LINEs and SINEs, respectively). Among these noncoding transcripts, we focused initially on miRs, although they represented only a small percentage (1%), since a previous report showed a HDAC-dependent control of miRs in myoblasts isolated from dystrophic muscles (Cacchiarelli et al. 2010). To establish a functional relationship between HDACis, activation of miR, and BAF60-mediated chromatin modifications that activate the myogenic program in FAPs from young mdx mice, we used high-throughput screening (HTS) of a library of miRs against the untranslated region (UTR) sequences of the Baf60 (Smarcd) variants a, b, and c linked to the luciferase (Fig. 3A). We initially performed this screening in HEK293 cells. Among the miRs identified by this screening, we noted two miRs that have been previously implicated in the activation of the myogenic program—miR-1.2 and miR-206 (Sokol and Ambros 2005; Chen et al. 2006; Kim et al. 2006)—as BAF60B targeting miRs (Fig. 3B; Supplemental Fig. S5A). In a complementary approach, among the miRs induced by TSA in FAPs from young mdx mice, small RNA sequencing (RNA-seq) analysis again identified miR-206 as well as miR-133a, another miR that has been implicated in the regulation of skeletal myogenesis (Chen et al. 2006) (Fig. 3C). The complete list of miRs expressed in FAPs from young mdx mice with or without TSA treatment is accessible through GEO series accession number GSE51933 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51933). miR-1.2, miR-133, and miR-206, collectively referred to as myomiRs, are components of a miR cluster that regulates a wide array of target transcripts in skeletal muscle cells during development and adult life and have been implicated in the pathogenesis of MDs and other muscular disorders (Eisenberg et al. 2009; Greco et al. 2009; Williams et al. 2009). Thus, the combination of two independent approaches (HTS and small RNA-seq) indicated a potential relationship between TSA-induced myomiRs and regulation of BAF60 variants in FAPs (Fig. 3D).
Figure 3.

Identification of miRs targeting the alternative BAF60A and BAF60B variants. (A) Schematic representation of the pGK plasmid transfected into HEK293 cells for miR sensor screening. The 3′ UTRs of target genes were cloned downstream from the luciferase coding sequence. (B) Volcano plots representing primary data for each of the three miR screens. The significance versus fold change for each of the 875 miRs tested is plotted. miR activities plotted as percent inhibition relative to siRNA against Smarcd1 (Baf60a), Smarcd2 (Baf60b), or Smarcd3 (Baf60c) (100% inhibition) and scrambled sequence control (0% inhibition) (X-axis) and P-value from t-test (Y-axis). miRs that block Luc-Smarcd1, Luc-Smarcd2, or Luc-Smarcd3 production (>20% cutoff for Smarcd1 and Smarcd2; >40% cutoff for Smarcd3; P < 0.11) are shown in green. From this group of hits, miR-1 and miR-206 are highlighted in yellow and purple, respectively. The primary screen was performed using 10 nM miRs in triplicate. (C) Manually assembled list representing small RNA-seq results of miRs induced in FAPs isolated from TSA-treated versus control (CTR) 1.5-mo-old mdx mice. (D) Representation of overlapping results from miR HTS described in B and small RNA-seq described in C. (E) BAF60A, BAF60B, and BAF60C protein levels in TSA-treated versus control (CTR) FAPS were measured by Western blot analysis. (F) MyomiR (miR1.2as, miR133, and miR206) endogenous expression in MuSCs during differentiation (growth medium [GM] > differentiation medium [DM]) (left panel) and in FAPs isolated from young 1.5-mo-old mdx mice treated with vehicle (CTR) or TSA for 15 d (right panel). (G) BAF60 subunits’ relative expression in MuSCs during differentiation (left panel) and in FAPs isolated as described in E after control (CTR) or TSA treatment (right panel). Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
By using a bioinformatic search for predictive UTR targets of these myomiRs, we consistently noted the presence of Baf60a and Baf60b (but not Baf60c) UTR sequences (Supplemental Fig. S5B). Indeed, Munsterberg and colleagues (Goljanek-Whysall et al. 2012) have recently identified BAF60B among the targets of miR-1 and miR-206 in skeletal myoblasts. We directly tested the effect of myomiRs on the Baf60a, Baf60b, or Baf60c 3′ UTR linked to the luciferase gene in HEK293 cells. All myomiRs (miR-1.2, miR-133, and miR-206), when independently expressed by transfection, down-regulated the luciferase activity of Baf60a-UTR-luc and Baf60b-UTR-luc constructs coexpressed in these cells (Supplemental Fig. 5C). This effect was specific, as mutations that disrupted the recognition sequence of myomiRs in Baf60a and Baf60b mRNAs were sufficient to abrogate miR-mediated reduction of luciferase activity (Supplemental Fig. S5C). In contrast, Baf60c-UTR-luc was not reduced by myomiR expression in HEK293 cells (Supplemental Fig. S5C). We further investigated whether exposure to a HDACi (TSA) could replicate the effect of myomiRs on Baf60-UTR-luc activity in C2C12 myoblasts, which provide a more relevant biological context. Supplemental Figure S5D shows that TSA down-regulated the activity of Baf60a-UTR-luc and Baf60b-UTR-luc but not Baf60c-UTR-luc.
We next established a direct correlation between myomiR induction and the expression levels of BAF60 variants in FAPs isolated from young mdx mice exposed or not to TSA. BAF60A and BAF60B protein levels were down-regulated, while BAF60C was up-regulated, in FAPs isolated from muscles of mdx mice exposed to TSA (Fig. 3E). The up-regulation of myomiRs in FAPs of mdx mice exposed to TSA (Fig. 3F, right panel) coincided with the down-regulation of Baf60a and Baf60b transcripts (Fig. 3G, right panel). In contrast, Baf60c transcripts were induced in FAPs of mdx mice exposed to TSA (Fig. 3G, right panel), consistent with the lack of a myomiR recognition sequences in Baf60c UTR and with the ability of TSA to promote chromatin remodeling and transcription at the Baf60c locus (see Fig. 2). Of note, a pattern of myomiR and BAF60 variant modulation similar to that detected in FAPs exposed to TSA was also observed in differentiating MuSCs isolated from young mdx mice (Fig. 3F,G, left panels), once again suggesting interesting analogies between MuSCs and the FAPs from regenerating muscles of mdx mice exposed to TSA.
We then used antagomirs to evaluate the specificity of myomiR-mediated down-regulation of BAF60A and BAF60B in TSA-treated FAPs. Transfection of specific antagomirs blunted the TSA-mediated down-regulation of Baf60a and Baf60b, but not Baf60c, and prevented the expression of muscle differentiation markers (such as Myh3) in FAPs isolated from young mdx mice (Fig. 4A). Consistently, blockade of each myomiR by ectopic expression of specific antagomiRs completely reverted TSA-mediated induction of MYHC-positive myotubes (Fig. 4B,C) and the inhibition of adipogenic differentiation in FAPs from young mdx mice (Fig. 4B,D). These results indicate the essential function of myomiRs in mediating the HDACi-directed conversion of FAPs into the myogenic lineage.
Figure 4.

AntagomiRs against myomiRs counteract the HDACi activity in FAPs. (A) qRT–PCR detecting relative amounts of Baf60a, Baf60b, Baf60c, and Myh3 transcripts in FAPs from 1.5-mo-old mdx mice transfected in vitro with antagomir-1 (a-miR1), antagomir-133 (a-miR133), antagomir-206 (a-miR206), or scrambled antagomir (mock) in either the absence (black bars, CTR) or presence of TSA added after transfection (gray bars, TSA). (B) FAPs’ myogenic and adipogenic potential after TSA treatment and/or antagomiR transfection was assessed by immunofluorescence for MYHC (green) and Oil red O (red). Nuclei were counterstained with DAPI (blue). (C) Graph showing the quantification of the myogenic differentiation of FAPs in the experiment represented in B. We measured the percentage of nuclei in MYHC-positive cells per field. (D) Graph showing the quantification of the adipogenic differentiation of FAPs in the experiment represented in B. We measured the percentage of the Oil red O-positive cells per field.
Reprogramming of FAPs into a promyogenic phenotype by modulation of the myomiR–BAF60 network
We next tested whether ectopic expression of individual myomiRs could affect the function and phenotype of FAPs isolated from young mdx mice. Previous studies have used coculture experiments to show that the promyogenic activity of FAPs relies on their ability to implement MuSC-mediated formation of multinucleated myotubes during muscle regeneration (Joe et al. 2010). Moreover, a transwell coculture assay demonstrated that FAPs from young mdx mice promote the ability of MuSCs to differentiate into myotubes, possibly via soluble factors, with this activity being further augmented by HDACis (Mozzetta et al. 2013). We therefore used transwell cocultures to test whether transfection of myomiRs would directly influence the ability of FAPs to functionally interact with MuSCs. The myomiRs were individually expressed in FAPs isolated from young mdx mice (Fig. 5A), prior to transwell coculture with MuSCs (Fig. 5B). Notably, each myomiR increased the ability to promote the formation of larger, MYHC-expressing multinucleated myotubes from cocultured MuSCs (Fig. 5C,D). The effect of each myomiR was comparable with that of TSA and resulted in an increased ability of FAPs to stimulate MuSC fusion into myotubes, as compared with mock transfected, control FAPs (Fig. 5C,D). In a parallel assay, we monitored the effect of myomiR expression on the intrinsic ability of mdx-derived FAPs to adopt a fibro-adipogenic or the alternative myogenic lineage that was observed in response to TSA (Fig. 1). Lineage and differentiation potential were scored by the formation of MYHC-positive myotubes and/or perilipin-expressing adipocytes under adipogenic culture conditions. A significant proportion (∼10%) of FAPs from young mdx mice typically differentiated into perilipin-expressing adipocytes (Fig. 5E–G). Interestingly, a smaller proportion of cells (∼3%) expressed MYHC, indicating a latent myogenic potential in FAPs from young mdx mice (Fig. 5E,F) that was not detected in FAPs isolated from uninjured muscles of wild-type or old mdx mice (data not shown). Treatment with TSA increased the formation of MYHC-positive myotubes (Fig. 5E,F) and the concomitant inhibition of perilipin-expressing adipocytes (Fig. 5E–G). Ectopic expression of each myomiR replicated the effect of TSA, with increased formation of MYHC-positive myotubes and the reciprocal reduction of perilipin-expressing adipocytes (Fig. 5E–G). These data support the notion that TSA-mediated up-regulation of myomiRs switches FAP fate toward the myogenic lineage.
Figure 5.

MyomiR expression induced a myogenic phenotype in FAPs and enhanced their ability to stimulate MuSC fusion into myotubes. (A) qRT–PCR to check miR1.2, miR133, and miR206 expression in FAPs isolated from young mdx mice transfected with psp65-miR1.2, psp65-miR133a, and psp65-miR206 constructs as compared with FAPs transfected with the psp65-null vector (CTR). (B) Transwell coculture scheme. MuSCs FACS-isolated from young mdx mice (MuSCs CTR) were seeded at the bottom of the well. FAPs isolated from young mdx mice (control [CTR] or TSA-treated for 15 d) were seeded on a transwell insert. Control FAPs were transfected with psp65-null vector (CTR), psp65-miR1, psp65-miR133, and psp65-miR206. Six hours after FAP transfection, all transwells were put in coculture with MuSCs. (C) The myogenic differentiation of MuSCs—alone or after coculture with FAPs—was assessed by immunostaining for MYHC (green). Nuclei were counterstained with DAPI (blue). (D) Graph showing the fusion index of the experiment represented in C. We measured the percentage of nuclei that were in MYHC− or MYHC+ mononucleated myotubes (n < 2), the percentage of nuclei that were inside myotubes containing between two and five nuclei (2 < n < 5), and the percentage of nuclei inside myotubes containing more than five nuclei (n > 5). (E) FAPs’ myogenic and adipogenic potential after TSA treatment or myomiR transfection was assessed by immunofluorescence for MYHC (green) and PERILIPIN (red). Nuclei were counterstained with DAPI (blue). (F) Graph showing the quantification of the myogenic differentiation of FAPs in the experiment represented in E. We measured the percentage of nuclei in MYHC-positive cells. (G) Graph showing the quantification of the adipogenic differentiation of FAPs in the experiment represented in E. We measured the percentage of the Perilipin-positive cells per field. Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01.
Lineage determination is programmed at the transcription level by changes in chromatin structure that are typically promoted by the SWI/SNF chromatin remodeling complex. The heterogeneous composition of SWI/SNF has been recently regarded as a key determinant of localized chromatin remodeling to support lineage-specific transcription and ultimately the decision to adopt alternative phenotypes within mesodermal-derived populations (for review, see Puri and Mercola 2012). In both skeletal and cardiac muscle, the formation of the BAF60C-based SWI/SNF complex is essential for the transcriptional activation of cardiac or skeletal myogenic genes (Lickert et al. 2004; Takeuchi and Bruneau 2009; Forcales et al. 2012; Albini et al. 2013). Moreover, reconfiguring the SWI/SNF composition by miR-mediated targeting of alternative variants of BAF subunits has previously been shown to regulate neurogenesis (Lessard et al. 2007; Yoo et al. 2011). Thus, we hypothesized that myomiR-mediated down-regulation of BAF60A and BAF60B would favor the formation of a BAF60C-based SWI/SNF complex and subsequently evoke a myogenic program in FAPs. We therefore tested the hypothesis that reducing the levels of Baf60a and Baf60b could exert an effect equivalent to myomiR expression in FAPs from young mdx mice. Indeed, shRNAi-mediated down-regulation of Baf60a and Baf60b was sufficient to replicate the effect of ectopic expression of myomiRs in FAPs shown in Figure 5. Reducing the levels of Baf60a or Baf60b by shRNAi (Fig. 6A) enhanced FAPs’ ability to promote MuSC-mediated formation of myotubes in transwell coculture experiments (Fig. 6B–D) and promoted FAP differentiation into MYHC-positive myotubes with an efficiency comparable with that of TSA (Fig. 6E,F) or ectopic myomiR expression (cf. Figs. 6E,F and 5E,F). Importantly, shRNAi-mediated down-regulation of Baf60a and Baf60b reduced the extent of adipogenic differentiation of FAPs (Fig. 6E–G), as also observed with overexpression of myomiRs (cf. Figs. 6E–G and 5E–G). In contrast, shRNAi-mediated knockdown of Baf60c did not alter the phenotype of FAPs from young mdx mice (Fig. 6E) but abrogated the promyogenic effects of TSA (Fig. 6C,D), indicating an essential role of BAF60C in the activation of the myogenic program upon HDAC blockade.
Figure 6.

Down-regulation of Baf60a or Baf60b exerts an effect equivalent to myomiR expression in FAPs. (A) qRT–PCR to check Baf60a, Baf60b, and Baf60c down-regulation in FAPs isolated from young mdx mice and transfected with pSuper short hairpin Baf60a (shBaf60a), pSuper short hairpin Baf60b (shBaf60b), and pSuper short hairpin Baf60c (shBaf60c), as compared with FAPs transfected with the pSuper-null vector (CTR). (B) Transwell coculture scheme: MuSCs FACS-isolated from young MDX mice (MuSCs CTR) were seeded at the bottom of the well. FAPs isolated from young mdx mice (control [CTR] or TSA-treated for 15 d) were seeded on a transwell insert. FAPs were transfected with control (CTR), shBaf60a, shBaf60b, and shBaf60c pSuper vectors. Six hours after transfection, FAPs were put in coculture with MuSCs. (C) The myogenic differentiation of MuSCs—alone or after coculture with FAPs—was assessed by immunostaining for MYHC (green). Nuclei were counterstained with DAPI (blue). (D) Graph showing the fusion index of the experiment represented in C. We measured the percentage of nuclei that were in MYHC− or MYHC+ mononucleated myotubes (n < 2), the percentage of nuclei that were inside myotubes containing between two and five nuclei (2 < n < 5), and the percentage of nuclei inside myotubes containing more than five nuclei (n > 5). (E) FAPs’ myogenic and adipogenic potential after TSA treatment or BAF60 variant down-regulation was assessed by immunofluorescence for MYHC (green) and PERILIPIN (red). Nuclei were counterstained with DAPI (blue). (F) Graph showing the quantification of the myogenic differentiation of FAPs in the experiment represented in E. We measured the percentage of nuclei in MYHC-positive cells. (G) Graph showing the quantification of the adipogenic differentiation of FAPs in the experiment represented in E. We measured the percentage of the PERILIPIN-positive cells per field. Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01.
These results support the concept that HDAC-regulated expression of myomiRs controls the mutually exclusive incorporation of BAF60 variants into distinct SWI/SNF complexes that can alternatively direct adipogenic or myogenic programs in FAPs from young mdx mice. Interestingly, the resistance to TSA of FAPs from muscles of mdx mice at late stages of disease progression, when the regeneration potential is exhausted, suggests that FAPs’ response to HDACis is influenced by regeneration signals. We therefore tested whether induction of regeneration by cardiotoxin (CTX)-mediated muscle injury could enable FAPs of young wild-type mice to adopt the myogenic lineage in response to TSA. Supplemental Figure 6 shows that TSA does not affect the phenotype of FAPs from uninjured muscles of wild-type mice but does promote myogenesis at the expense of adipogenesis in FAPs from CTX-injured regenerating muscles (Supplemental Fig. S6A–E), an effect reminiscent of that observed in young mdx mice. Consistently, FAPs from injured muscles exposed to TSA showed an increased ability to support the myogenic potential of MuSCs (Supplemental Fig. S6F,G). Moreover, ectopic expression of each of the myomiRs (miR-1.2, miR-133a, and miR-206) individually promoted myogenesis and inhibited adipogenesis only in FAPs from CTX-injured wild-type muscles (Supplemental Fig. S7A–D). Likewise, knocking down Baf60 variants Baf60a and Baf60b, but not Baf60c, promoted myogenesis and inhibited adipogenesis only in FAPs from CTX-injured wild-type muscles (Supplemental Fig. S7E–H).
Collectively, these results indicate that regeneration signals—such as those released by injured wild-type muscles and dystrophic muscles at the early, compensatory stages of disease progression—instruct FAPs to adopt the myogenic lineage in response to HDACis. This evidence explains the reported stage-dependent beneficial effect of HDACis in mdx mice (Mozzetta et al. 2013).
BAF60C is an essential component of the chromatin machinery that directs the promyogenic phenotype of FAPs in response to TSA treatment
The data shown in Figure 6 point to the key role of BAF60C in directing the promyogenic phenotype of FAPs. To conclusively demonstrate the role of BAF60C in directing the phenotype of FAPs from dystrophic muscles, we crossed Baf60cfl/fl mice with mdx mice to generate mdx;Baf60cfl/fl mice. FAPs were isolated from these mice as previously described (Mozzetta et al. 2013), and Baf60c was heritably deleted by infection with adenovirus expressing CRE (AdCRE) followed by culture in adipogenic conditions (see scheme in Fig. 7A). Mdx;Baf60cfl/fl FAPs infected with control adenovirus (AdCTR) displayed the expected phenotype, yielding occasional MYHC-positive mononucleated cells (Fig. 7B,D) within a predominant population of Oil red O-positive adipocytes (Fig. 7E,G). In vitro treatment with TSA increased the number of MYHC-positive myotubes, with formation of multinucleated myotubes (Fig. 7B,D), albeit to a reduced extent as compared with FAPs from in vivo treated young mdx mice (cf. Fig. 1). This difference is possibly due to the negative, nonspecific effect of adenoviral infection on the myogenic potential of FAPs. TSA also inhibited the formation of Oil red O-positive adipocytes (Fig. 7E,G). Adenoviral (AdCRE)-mediated deletion of Baf60c (Fig. 7C) reduced the basal number of MYHC-positive cells and abrogated TSA-mediated induction of myogenesis (Fig. 7B,D). However, while BAF60C deficiency increased the number of Oil red O-positive adipocytes, it did not effectively counter the ability of TSA to prevent the activation of the adipogenic program in FAPs (Fig. 7E,G). This was also confirmed by qRT–PCR analysis of gene expression, as the TSA-mediated repression of adipogenic genes, such as Pparγ (PPARγ) and Cebpα (CEBPα), was observed in either control (AdCTR) or Baf60c-deficient (AdCRE) FAPs derived from mdx;Baf60cfl/fl mice (Fig. 7F).
Figure 7.

Genetic ablation of BAF60C in FAPs prevents the HDACi-mediated myogenic phenotype in FAPs but not the repression of the adipogenic program. (A) Schematic representation of the experimental setting. Cells were isolated from young Baf60cfl/fl mdx mice and, after 3 d in growth medium (GM), were infected with VQAd Cre/GFP virus (AdCRE) (Viraquest, Inc.) or VQAd CMV eGFP virus (AdCTR) (Viraquest, Inc.). Twenty-four hours after infection, cells were treated with TSA. After treatment, cells where switched to differentiation medium (DM). (B) FAPs’ myogenic potential was assessed by immunofluorescence for MYHC (green). Nuclei were counterstained with DAPI (blue). (C) Relative expression of Baf60c transcript in FAPs infected with AdCTR or AdCRE and treated or not with TSA. (D) Graph showing the quantification of the myogenic differentiation of FAPs in the experiment represented in B. We measured the percentage of nuclei in MYHC-positive cells. (E) FAPs’ adipogenic differentiation was assessed by Oil red O staining. (F) qRT–PCR of Pparγ and Cebpα. (G) Relative quantification of the Oil red O area measured as pixels squared per field. Data are represented as average ± SEM of three different experiments (n = 3); (*) P < 0.05; (**) P < 0.01.
These results support the conclusion that BAF60C is an essential component of the chromatin-modifying machinery that promotes the promyogenic phenotype in the FAPs of young mdx mice exposed to HDACis. However, while the absence of BAF60C is sufficient to enhance the adipogenic potential of FAPs from mdx mice, BAF60C does not appear to be required for HDACi-mediated repression of the adipogenic program in FAPs.
Discussion
Our study has identified an HDAC-regulated network that controls the “functional” phenotype of FAPs from dystrophic muscles. This network consists of myomiRs that target two alternative variants of the SWI/SNF chromatin remodeling complex, BAF60A and BAF60B, thereby favoring the formation of a BAF60C-based SWI/SNF complex that confers on MYOD the ability to activate the myogenic program (Forcales et al. 2012; Albini et al. 2013).
HDAC-mediated repression of myomiRs has been previously reported in dystrophic muscles (Cacchiarelli et al. 2010). Furthermore, myomiRs have been extensively reported to regulate skeletal myogenesis by targeting individual regulatory components of the myogenic network (Braun and Gautel 2011). Interestingly, treatment with HDACis (Minetti et al. 2006) or up-regulation of miR-206 (Liu et al. 2012) invariably exerts therapeutic effects in muscles of mdx mice. Our results reveal a previously unrecognized link between HDACs, myomiRs, and chromatin remodeling that underlies the transition of FAPs from the “promyogenic” phenotype that supports compensatory regeneration at early stages of DMD to a “pathological” phenotype typical of the late stages of the disease. We show that reversible HDAC-mediated repression of myomiRs and of the two central components of the transcriptional machinery that activates muscle gene expression—BAF60C and MYOD—underlies the latent myogenic phenotype in FAPs observed in young mdx mice. This potential appears to be a specific feature of FAPs from regenerating dystrophic muscles, as it was not observed in wild-type mice or old mdx mice. As such, this observation reveals an unanticipated lineage “plasticity” of FAPs that correlates with the highly regenerative environment typical of dystrophic muscles at the early stages of the disease. In contrast, FAPs isolated from old mdx mice, in which the regeneration potential declines, showed no myogenic potential. Thus, we speculate that the latent myogenic phenotype of FAPs from regenerating muscles of mdx young mice might reflect the acquisition of a bipotency that is likely adopted in response to regenerative cues and is resolved into the myogenic phenotype by HDAC inhibition. In contrast, an impairment of the reciprocal interactions between environmental signals, FAPs, and MuSCs contributes to the progressive impairment of the regeneration niche in dystrophic muscles. In this regard, it should be noted that Guttridge and colleagues (He et al. 2013) have recently shown that muscle interstitial cells expressing the same surface markers that define FAP identity (SCA1pos, PDGF-Rαpos, and CD34pos) adopt the myogenic lineage upon conditions that trigger cachexia. However, these cells fail to fully differentiate into myotubes because of an adverse environment imposed by cachexia (He et al. 2013).
The constitutive fibro-adipogenic phenotype of FAPs from old mdx mice and the related resistance to HDACis appears to be epigenetically determined, as suggested by the striking changes in chromatin accessibility revealed by NA-seq in FAPs isolated from old mdx mice, as compared with FAPs from young mdx mice. Our results indicate that the relative levels of BAF60 variants is a key determinant of the changes in the epigenetic landscape that ultimately directs FAP lineage commitment and that myomiRs are essential upstream regulators of the individual BAF60 variant levels. Previous reports have linked miRs with alternative lineage determination (Yin et al. 2013b; for review, see Lessard and Crabtree 2010). In particular, work from Crabtree and collaborators (Lessard et al. 2007; Yoo et al. 2011) has elucidated a miR-mediated regulation of BAF53 and BAF45 variants that controls sequential stages of neurogenic differentiation and can even mediate conversion of fibroblasts into neurons. These data are in analogy with our observation that a myomiR–BAF60 variant axis controls lineage potency of FAPs (Figs. 5, 6) and collectively support the notion that miR-regulated composition of SWI/SNF chromatin remodeling complexes determines the epigenetic landscape that controls the cell identity of tissue progenitors (Puri and Mercola 2012).
Our data clearly indicate that the propensity of FAPs to adopt a myogenic phenotype invariably coincides with the activation of the MYOD–BAF60C axis and myomiRs. Although the role of MYOD and BAF60C in the myogenic conversion of somatic and embryonic stem cells has been reported (Davis et al. 1987; Weintraub et al. 1989; Albini et al. 2013), the myogenic conversion of FAPs by myomiRs (Fig. 5) is unprecedented. Interestingly, myomiR-mediated conversion of FAPs from dystrophic muscles is highly reminiscent of miR-mediated conversion of cardiac fibroblasts (Jayawardena et al. 2012). In particular, the dependence of myomiR function on signals from regenerating young dystrophic muscle (this study) mirrors the emerging view that miR-based lineage conversion in multiple contexts is influenced by signals released by injured tissues, such as acute ischemic myocardium or regenerating skeletal muscle. This is consistent with an essential role of regeneration cues in generating an epigenetic landscape that is permissive for lineage conversion. Our data suggest that when FAPs are exposed to these instructive signals, HDAC-mediated repression of the Myod1–myomiR–BAF60 network maintains a “lineage plasticity,” which can be experimentally resolved by perturbations such as HDAC blockade, myomiR overexpression, or BAF60A and BAF60B knockdown that bias the network toward the formation of the MYOD/BAF60C-based SWI/SNF complex. Signal-dependent activation of this complex by regeneration cues, such as those that activate the p38 pathway, has been previously reported (Simone et al. 2004; Forcales et al. 2012). As myomiR expression is induced by the combined activity of MYOD and the SWI/SNF complex (Mallappa et al. 2010) and myomiRs favor the composition of the promyogenic BAF60C–SWI/SNF complex, by targeting the alternative BAF60A and BAF60B variants, FAP reprogramming into the myogenic phenotype appears to rely on a HDAC-supervised feed-forward circuitry composed by external signals, MYOD, myomiR, and selection of specific BAF60 variants. This is consistent with the robust fibroblast reprogramming into cardiac cells induced by the combined activity of cardiogenic factors and miRs (Nam et al. 2013).
Our data show that FAPs from mdx mice exposed to HDACis adopt the myogenic lineage and have an enhanced ability to support MuSC activity, an effect collectively referred as to induction of the promyogenic phenotype. While the relationship between these two effects is not conclusively established by the present study, it is important to note that both are dependent on the up-regulation of BAF60C and myomiR-mediated down-regulation of BAF60A and BAF60B. However, given the reported evidence that FAPs from injured muscles of wild-type mice do not adopt the myogenic lineage but retain the ability to support MuSC-mediated regeneration (Joe et al. 2010), it is likely that these two events could be separately activated. Likewise, the ability of HDACis to promote the myogenic phenotype and repress the fibro-adipogenic lineage seems to rely on distinct mechanisms, as the genetic ablation of BAF60C impairs HDACi-mediated activation of the myogenic phenotype but not HDACis’ ability to repress the fibro-adipogenic program in FAPs. Given the heterogeneity of FAPs (Pannérec et al. 2013), it will be interesting to determine whether different fates adopted by these cells rely on the transition from one subpopulation to another.
Overall, we reported the first evidence of an HDAC–myomiR–BAF60 network that controls, in concert with environmental cues, the identity and activity of a cell type (FAPs) that plays a key role in muscle regeneration and is implicated in the pathogenesis of MDs. Future studies will shed further light on the pharmacological potential to reprogram the phenotype of these cells by epigenetic drugs that can be used in the treatment of DMD.
Materials and methods
Animals and in vivo treatments
Wild-type C57/BL10 and C57Bl10 mdx mice were purchased from Jackson Laboratories. Baf60Cfl/fl C57/BL10 mice were crossed with mdx C57/BL10 mice to generate the Baf60cfl/fl mdx mice. Further details on treatments are provided in the Supplemental Material.
Cell preparation and FACS isolation
MuSCs were isolated as TER119−/CD45−/CD31−/α7INTEGRIN+/SCA-1− cells; FAP cells were isolated as TER119−/CD45−/CD31−/α7INTEGRIN−/SCA-1+ cells, as described in Mozzetta et al. (2013). Further details on FACS sorting and culture conditions are provided in the Supplemental Material.
Luciferase assay
HEK293 and C2C12 cells were transfected with psiCheck 2 Renilla luciferase reporter vectors containing BAF60A, BAF60B, or BAF60C 3′ UTR fragments. Further details on construct preparation and luciferase analysis are provided in the Supplemental Material.
Western blot
Western blot was performed using antibodies against the following proteins: BAF60A (BD), BAF60B (Santa Cruz Biotechnology), BAF60C (Forcales et al. 2012), MYOD (Santa Cruz Biotechnology), MYHC (MF20, Developmental Studies Hybridoma Bank [DSHB]), PERILIPIN (Sigma-Aldrich), and TUBULIN (Cell Signaling). Further experimental details are provided in the Supplemental Material.
Immunofluorescence
Primary antibodies used for immunofluorescence were against MYOD (1:20; Santa Cruz Biotechnology), MYHC (1:20; MF-20, DSHB), PERILIPIN A/B (1:100; Sigma-Aldrich), Sca-1-FITC (1:100; eBioscience), and α7-Integrin-PE (1:100; eBioscence). Oil red O was used to stain lipids, as described in Mozzetta et al. (2013). Further experimental details are provided in the Supplemental Material.
qRT–PCR
Total RNA was extracted with Trizol, and 0.5–1 µg was retrotranscribed using the TaqMan reverse transcription kit (Applied Biosystems). Real-time qPCR was performed using primers and conditions provided in the Supplemental Material.
FAIRE
Chromatin for FAIRE analysis was prepared as previously described in Albini et al. (2013). Further experimental details and primers used are provided in the Supplemental Material.
ChIP
ChIP assay was performed as previously described in Albini et al. (2013). The following antibodies were used: anti-acetylated Histone 3 (AcH3; Millipore) and normal rabbit IgG (IgG; Santa Cruz Biotechnology). Further experimental details and primers for RT–PCR are provided in the Supplemental Material.
miR HTS
For miR screening, HEK293 cells were cotransfected with 10 nM Ambion pre-miR miR precursor human version 2.0 miR library and 100 ng of SMARCD 3′ UTR target sensor plasmids (Fig. 3A) per well in white 384-well plates (Greiner) in triplicate using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Further experimental details are provided in the Supplemental Material.
Gene expression analysis
RNA from 1 × 105 freshly sorted cells was extracted using the RNeasy kit (Qiagen) following the manufacturer’s protocol. We analyzed Agilent mouse GE 4x44K version 2 (single-color) arrays using the manufacturer’s reader and collected primary data using the supplied scanner software. Further experimental details on data analysis are provided in the Supplemental Material.
NA-seq
The library for NA-seq was prepared as described in Gargiulo et al. (2009). Further details on data analysis are provided in the Supplemental Material.
Small RNA-seq
C57Bl10 mdx mice (1.5 mo old) were treated for 15 d with TSA. RNA was harvested from freshly isolated FAPs using Trizol reagent, and small RNA libraries were prepared for sequencing using standard Illumina protocols. Further experimental details are provided in the Supplemental Material.
Statistical analysis
Data are presented as mean ± SEM. Comparisons were made using the Student’s t-test assuming a two-tailed distribution, with significance being defined as P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Acknowledgments
P.L.P. is an investigator of Sanford Children’s Health Research Center. This work has been supported by the following grants to P.L.P.: R01AR056712, R01AR052779, and P30 AR061303 from the National Institute of Health (NIH)/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), EPIGEN, and the Muscular Dystrophy Association (MDA). This work has benefited from research funding from the European Community’s Seventh Framework Programme in the project FP7-Health-2009 ENDOSTEM 241440 (activation of vasculature-associated stem cells and MuSCs for the repair and maintenance of muscle tissue). S.M. was supported by EPIGEN. C.M. was supported by AFM fellowship. T.R. is supported by an American Heart Association fellowship. M.M. was supported by NIH RO1HL113601, R01HL108176, and R33HL088266 and Fondation Leducq.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.234468.113.
References
- Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV, Puri PL 2013. Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep. 3: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Rando TA 2012. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 10: 504–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T, Gautel M 2011. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol 12: 349–361 [DOI] [PubMed] [Google Scholar]
- Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, et al. 2010. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 12: 341–351 [DOI] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. 2010. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18: 662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ 2006. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, Ragone G, Pescatori M, Zaccagnini G, Antonini A, et al. 2008. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci 105: 19183–19187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consalvi S, Saccone V, Giordani L, Minetti G, Mozzetta C, Puri PL 2011. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med 19: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, Rocchetti M, Leoni F, Monzani V, Mascagni P, et al. 2013. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 20: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkilic I, Kunkel LM 2003. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 13: 231–238 [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB 1987. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987–1000 [DOI] [PubMed] [Google Scholar]
- Eisenberg I, Alexander MS, Kunkel LM 2009. miRNAS in normal and diseased skeletal muscle. J Cell Mol Med 13: 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, Coutinho P, Saccone V, Consalvi S, Williams R, et al. 2012. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J 31: 301–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo G, Levy S, Bucci G, Romanenghi M, Fornasari L, Beeson KY, Goldberg SM, Cesaroni M, Ballarini M, Santoro F, et al. 2009. NA-seq: a discovery tool for the analysis of chromatin structure and dynamics during differentiation. Dev Cell 16: 466–481 [DOI] [PubMed] [Google Scholar]
- Giordani L, Puri PL 2013. Epigenetic control of skeletal muscle regeneration: integrating genetic determinants and environmental changes. FEBS J 280: 4014–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goljanek-Whysall K, Pais H, Rathjen T, Sweetman D, Dalmay T, Munsterberg A 2012. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J Cell Sci 125: 3590–3600 [DOI] [PubMed] [Google Scholar]
- Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, Cardani R, Perbellini R, Isaia E, Sale P, et al. 2009. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 23: 3335–3346 [DOI] [PubMed] [Google Scholar]
- He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, et al. 2013. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A 2013. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153: 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ 2012. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res 110: 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM 2010. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 2: 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RN, Zhang RH, Rossi FM 2013. Tissue-resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J 280: 4100–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A 2006. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 174: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard JA, Crabtree GR 2010. Chromatin regulatory mechanisms in pluripotency. Annu Rev Cell Dev Biol 26: 503–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR 2007. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55: 201–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG 2004. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 432: 107–112 [DOI] [PubMed] [Google Scholar]
- Liu N, Williams AH, Maxeiner JM, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN 2012. MicroRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest 122: 2054–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallappa C, Nasipak BT, Etheridge L, Androphy EJ, Jones SN, Sagerström CG, Ohkawa Y, Imbalzano AN 2010. Myogenic microRNA expression requires ATP-dependent chromatin remodeling enzyme function. Mol Cell Biol 30: 3176–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, et al. 2006. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med 12: 1147–1150 [DOI] [PubMed] [Google Scholar]
- Mozzetta C, Minetti G, Puri PL 2009. Regenerative pharmacology in the treatment of genetic diseases: the paradigm of muscular dystrophy. Int J Biochem Cell Biol 41: 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchell KJ, Marazzi G, Borsellino G, Battistini L, Sassoon D, et al. 2013. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med. 5: 626–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, et al. 2013. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci 110: 5588–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannérec A, Formicola L, Besson V, Marazzi G, Sassoon DA 2013. Defining skeletal muscle resident progenitors and their cell fate potentials. Development 140: 2879–2891 [DOI] [PubMed] [Google Scholar]
- Puri PL, Mercola M 2012. BAF60 A, B, and Cs of muscle determination and renewal. Genes Dev 26: 2673–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Muñoz-Cánoves P 2011. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol 96: 167–201 [DOI] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL 2004. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 36: 738–743 [DOI] [PubMed] [Google Scholar]
- Sokol NS, Ambros V 2005. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev 19: 2343–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Bruneau BG 2009. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 459: 708–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K 2010. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 2: 143–152 [DOI] [PubMed] [Google Scholar]
- Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe-Suzuki Y, et al. 2011. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 124: 3654–3664 [DOI] [PubMed] [Google Scholar]
- von Maltzahn J, Chang NC, Bentzinger CF, Rudnicki MA 2012. Wnt signaling in myogenesis. Trends Cell Biol 22: 602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD 1989. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci 86: 5434–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AH, Liu N, van Rooij E, Olson EN 2009. MicroRNA control of muscle development and disease. Curr Opin Cell Biol 21: 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Price F, Rudnicki MA 2013a. Satellite cells and the muscle stem cell niche. Physiol Rev 93: 23–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Pasut A, Soleimani VD, Bentzinger CF, Antoun G, Thorn S, Seale P, Fernando P, van Ijcken W, Grosveld F, et al. 2013b. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab 17: 210–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR 2011. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476: 228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]