

Abstract
Aim:
To investigate the effect of ginsenoside Rb1 on voltage-gated calcium currents in cultured rat hippocampal neurons and the modulatory mechanism.
Methods:
Cultured hippocampal neurons were prepared from Sprague Dawley rat embryos. Whole-cell configuration of the patch-clamp technique was used to record the voltage-gated calcium currents (VGCCs) from the hippocampal neurons,and the effect of Rb1 was examined.
Results:
Rb1 (2–100 μmol/L) inhibited VGCCs in a concentration-dependent manner, and the current was mostly recovered upon wash-out. The specific L-type Ca2+ channel inhibitor nifedipine (10 μmol/L) occluded Rb1-induced inhibition on VGCCs. Neither the selective N-type Ca2+ channel blocker ω-conotoxin-GVIA (1 μmol/L), nor the selective P/Q-type Ca2+ channel blocker ω-agatoxin IVA (30 nmol/L) diminished Rb1-sensitive VGCCs. Rb1 induced a leftward shift of the steady-state inactivation curve of ICa to a negative potential without affecting its activation kinetics or reversal potential in the I–V curve. The inhibitory effect of Rb1 was neither abolished by the adenylyl cyclase activator forskolin (10 μmol/L), nor by the PKA inhibitor H-89 (10 μmol/L).
Conclusion:
Ginsenoside Rb1 selectively inhibits the activity of L-type voltage-gated calcium channels, without affecting the N-type or P/Q-type Ca2+ channels in hippocampal neurons. cAMP-PKA signaling pathway is not involved in this effect.
Keywords: ginsenoside Rb1, L-type Ca2+ channel, nifedipine, ω-conotoxin-GVIA, ω-agatoxin IVA, patch-clamp technique, hippocampus, cAMP-PKA signaling pathway
Introduction
Ca2+, an important regulator of neuronal activity, controls membrane excitability, triggers the release of neurotransmitters and mediates activity-dependent changes in gene expression. Voltage-gated Ca2+ channels play a key role in the control of free cytosolic Ca2+ 1. According to their pharmacological and electrophysiological properties, at least 5 distinct types of voltage-gated Ca2+ channels have been identified and designated as L, N, P/Q, R, and T2, 3. L-, N-, and P/Q-type Ca2+ channels are pharmacologically identified and characterized by their specific blockers. Nifedipine is an L-type Ca2+ channel inhibitor, ω-conotoxin GVIA is an N-type Ca2+ channel inhibitor, and ω-agatoxin IVA is a P/Q-type Ca2+ channel inhibitor4, 5, 6, 7, 8, 9. Because the elevation of intracellular Ca2+ levels ([Ca2+]i) caused by the excessive stimulation of Ca2+ channels plays a key role in the excitotoxic damage of neurons, agents blocking the elevation of [Ca2+]i by regulating Ca2+ channels might have neuroprotective effects10, 11, 12.
Ginseng, the root of Panax ginseng CA Meyer, has been used worldwide as an herbal medicine for the alleviation of many ailments, particularly those associated with aging and memory deterioration. Ginsenoside Rb1, a protopanaxadiol type saponin, is one of the most important active compounds of ginseng. Recently, ginsenoside Rb1 has been reported to effectively protect neurons from glutamate-induced toxicity and β-amyloid (Aβ)-induced toxicity by reducing intracellular Ca2+ levels13, 14, 15, 16, 17, 18. However, how Rb1 decreases [Ca2+]i by regulating Ca2+ channels remains unclear. Although studies have found that Rb1 inhibits voltage-gated Ca2+ channels in other cell types and that its mechanisms of action vary by cell type, there have been no research studies to date that have investigated the action of ginsenoside Rb1 on voltage-gated Ca2+ channels in hippocampal neurons. In this paper, we analyzed the effect of ginsenoside Rb1 on voltage-gated Ca2+channels in hippocampal neurons and the possible mechanism for this modulation.
Materials and methods
Materials
Ginsenoside Rb1 was obtained from the Department of Organic Chemistry at Jilin University (Changchun, China) with a purity >98%. Stock solutions of the Ca2+ channel antagonists, ω-conotoxin GVIA (Alomone Labs, UK), nifedipine (Sigma, UK) ω-agatoxin IVA (Alomone Labs, UK) and adenylyl cyclase agonist Forskolin (Sigma, UK), were prepared with the appropriate amounts of deionized water or dimethyl sulfoxide (DMSO) and frozen at −20 °C before appropriate dilution in the recording medium. H-89 was dissolved in the pipette solution (described below) and stored at −20 °C. After the whole-cell configuration was obtained, H-89 was dialyzed into the cell through the pipette.
Hippocampal neuron cultures
Chemical media and culture media were obtained from Sigma unless otherwise noted. The care and use of animals followed the guidelines of the Shanghai Institutes for Biological Sciences Animal Research Advisory Committee. The hippocampal neuron cultures were prepared as described previously19 with some modifications. Briefly, whole brains were isolated from 18-day-old SD rat embryos, and the hippocampi were dissected and treated with 0.125% trypsin at 37 °C for 12 min. The cells were suspended with Dulbecco's Modified Eagle's Medium (DMEM) (GIBCO) containing 10% fetal bovine serum (HyClone, Logan, UT, USA) and 10% F-12 (GIBCO) and were plated at a density of 60 000 cells/mL on poly D-lysine-coated 35 mm dishes (Costar). Twenty-four hours after plating, half of the medium was changed to serum-free Neurobasal (NB) medium with 2% B27 supplement (GIBCO) and 1% glutamine. Thereafter, half of the changed medium was replaced twice a week with NB medium containing 2% B27 supplement and 0.25% glutamine. After 7 d in vitro, glial cell proliferation was inhibited by exposure to 2–4 mmol/L cytosine arabinoside. All the recordings were made with cells between d 6 and 8.
Electrophysiological recordings
Single patch recordings of Ca2+ channels from cultured hippocampal neurons at 6–8 d in vitro were made at room temperature using an EPC-9 patch-clamp amplifier and its corresponding Patchmaster software (Heka Electroniks, Germany) or an Axopatch-200B amplifier (Axon Instruments) with pCLAMP acquisition software. The gain was set to 1, filtered at 1 kHz, stored on videotape after digitization with a PCM processor, and displayed with a thermal pen recorder. The membrane capacitance and series resistance compensation were optimized.
Patch pipettes were fabricated from borosilicate glass capillaries (outer diameter 1.2 mm, inner diameter 0.69 mm, length 7.5 cm; B-120-69-15, Sutter Instruments) on a horizontal puller (Sutter Instruments). The microelectrodes had tip diameters of 2–3 μm and resistances of 3–6 MΩ. The pipettes were filled with an intracellular solution containing 80 mmol/L Cs-methanesulfonate, 20 mmol/L tetraethylammonium chloride (TEA-Cl), 1 mmol/L CaCl2, 5 mmol/L MgCl2, 11 mmol/L ethylene glycole-bis-(2-aminoethyl)-tetraacetic acid (EGTA), 10 mmol/L N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES), and 10 mmol/L Na2ATP. The chemicals were obtained from Sigma. CsOH was used to adjust the pH to 7.2–7.3. The osmolarity of the pipette solution was adjusted to 300 mOsm with sucrose. As suggested by Smirnov20, the replacement of 1.5 mmol/L Ca2+ with 5 mmol/L Ba2+ was used to augment the amplitude of the inward current through Ca2+ channels. The potential dependency of activation and in-activation with 5 mmol/L Ba2+ was very similar to the results observed in 1.5 mmol/L Ca2+. To isolate the Ba2+ current (IBa), the following reagents were used for the external solution: 115 mmol/L choline-Cl, 25 mmol/L TEA-Cl, 5 mmol/L 4-aminopyridine (4-AP), 5 mmol/L BaCl2, 10 mmol/L glucose, 10 mmol/L HEPES, and 0.0005 mmol/L tetrodotoxin (TTX). Tris was used to adjust the pH to 7.4. The osmolarity of the extracellular solution was adjusted to 300 mOsm with sucrose. 4-AP and TEA were used to eliminate outward K+ currents. TTX was used to eliminate inward Na+ currents.
We recorded the voltage-gated calcium channel Ba2+ current (IBa) and used the following stimulating programs: an activation procedure and a drug application program. For the activation procedure, the cells were held at a potential of −60 mV and depolarized to potentials ranging from −70 mV to +70 mV with 10 mV as a step for a duration of 150 ms, and the steps were repeated every 10 s. For the drug application program, the cells were held at a potential of −60 mV and depolarized to 0 mV (0–+20 mV) for a duration of 200 ms, and the steps were repeated every 10 s.
Experimental drug application and treatment
The “U-tube” solution exchange method21, 22 was used to apply the drugs. The whole cell measurements were initiated 5 min after break-in. Little run-down was observed during the 15 min necessary to collect the data. The current amplitudes of the cell before and after the experiment and the current densities of the cells of the different groups were compared.
Analysis of the electrophysiological recordings
The current recordings were analyzed using Clampfit 8.0 software (Axon, USA). Further analyses were performed using Microsoft Excel 2003 and Microcal Origin 8.0. All the data were described as the mean±the standard error of the mean. All the current recordings were normalized according to the whole cell capacitance to give the current density. A repeated measures ANOVA with Tukey-Kramer's post-test was used to compare the differences among entire current-voltage (I–V) relationships, and an unpaired Student's t-test was used to compare points on different curves that were activated by stepping to the same potential. P values less than 0.05 were considered significant. The peak current was measured as the maximal current observed during the depolarizing step.
The calcium current steady-state activation curve was fitted to a Boltzmann equation of the following form:
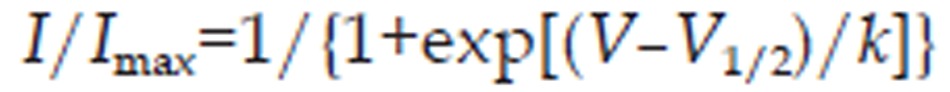 |
where I is the voltage-dependent current amplitude, V is the membrane potential for activation, V1/2 is the voltage at which activation is half maximal, and k is the slope factor.
Results
Voltage-gated calcium channel currents in hippocampal neurons
We recorded the whole-cell membrane currents from the somatic region of the neurons (Figure 1A) and identified a potent Ca2+ channel antagonist by its sensitivity to cadmium. We successively discriminated N-, P/Q-, and L-type Ca2+ channels by their specific blockers. ω-conotoxin GVIA and ω-agatoxin IVA showed irreversible blocking effects, while nifedipine exerted a partially reversible blocking effect (Figure 1B).
Figure 1.

(A) Phase-contrast image showing a single patch recording from 7-d cultured hippocampal neurons for the recording of the VGCCs. Scale bar, 10 μm. (B) Pharmacological separation of the VGCC subtypes in hippocampal neurons. Upper panel, inward Ca2+ channel Ba2+ currents evoked by pulses from −60 mV to 0 mV at the times indicated in the lower panel. Lower panel, time course of effects of ω-conotoxin GVIA (1 μmol/L), ω-agatoxin IVA (30 nmol/L) and nifedipine (10 μmol/L) on the Ba2+ current amplitude.
Effect of ginsenoside Rb1 on VGCCs in hippocampal neurons
The method of extracellular micro-perfusion was used to study the effects of Rb1 on VGCCs. In these experiments with Rb1 treatment, only the peak currents were selected for comparison, and the current was evoked by a pulse of 200 ms duration from −60 mV to 0 mV (0–+20 mV). The inhibition rate of the IBa peak current was calculated as follows: [(maximum current value before administration – maximum current value after administration)/maximum current value before administration×100%]. The control group was treated with the extracellular solution, and the experimental groups were treated with 1, 2, 5, 10, and 100 μmol/L Rb1. Both the control group and the 1 μmol/L Rb1 group showed no inhibitory effects on the IBa (Figure 2A, 2B). However, the other experimental groups with 2, 5, 10, and 100 μmol/L Rb1 demonstrated inhibitory effects on the IBa. The inhibition rates were 2%±0.87% (n=6), 5%±1.78% (n=4), 20%±3.96% (n=9), and 40%±6.71% (n=5), respectively. The IBa peak current inhibition rate of each group was significantly higher than that of the previous dose group (P<0.01) (Table 1). The effects of Rb1 on the IBa were partially reversible after wash-out with the bathing solution (Figure 2C, 2D).
Figure 2.

Effect of the extracellular solution, Rb1 (1 μmol/L) and Rb1 (10 μmol/L), on the IBa (A, B, and C, respectively). (1) Before drug application; (2) drug application; (3) washout. (D) represents the time course of the experiment corresponding to (C).
Table 1. Effects of Ginsenoside Rb1 at different concentrations (1, 2, 5, 10, and 100 μmol/L) on the amplitude of IBa. Mean±SD. cP<0.01 compared with the previous group.
| Groups | IBa inhibition (%) |
|---|---|
| Control | 0±0.74 |
| 1 μmol/L Rb1 | 0±1.12 |
| 2 μmol/L Rb1 | 2±0.87c |
| 5 μmol/L Rb1 | 5±1.78c |
| 10 μmol/L Rb1 | 20±3.96c |
| 100 μmol/L Rb1 | 40±6.71c |
Mechanism of action of ginsenoside Rb1 on the VGCCs in hippocampal neurons
The IBa was still elicited by depolarizing from −60 mV to 0 mV (0–+20 mV) and recorded continuously every 10 s. Under the maximum activated voltage, the currents achieved stability after recording 5 to 6 times. As shown in Figure 3A, 10 μmol/L ginsenoside Rb1 inhibited the IBa by 21.53%±2.81% (n=5). The inhibitory effect was eliminated after the application of nifedipine, a selective blocker of L-type Ca2+ channels.
Figure 3.

Rb1 inhibited the IBa in hippocampal neurons, and this inhibitory effect was eliminated after the application of nifedipine (A). Neither ω-conotoxin-GVIA nor ω-agatoxin IVA diminished the Rb1-sensitive IBa (B and C, respectively). Upper panel, pairs of the inward currents evoked by pulses from −60 to +0 mV (0–+20 mV) at the times indicated in the lower panel. Lower panel, time course of the effects of 10 μmol/L Rb1 on the IBa amplitude before and after application of the Ca2+ channel antagonists (10 μmol/L nifedipine, 1 μmol/L ω-conotoxin GVIA and 30 nmol/L ω-agatoxin IVA). The bar graphs for Rb1 inhibition (mean±SEM, n=5 for Rb1) on the IBa in cells untreated or treated with Ca2+ channel antagonists. cP<0.01 compared with Rb1 treatment alone.
As shown in Figure 3B, 10 μmol/L Rb1 inhibited the IBa by 20.19%±2.98% (n=5) before ω-conotoxin-GVIA treatment and inhibited the IBa by 20.51%±3.15% (n=5) in the presence of ω-conotoxin-GVIA (P>0.05 compared to Rb1 treatment alone). As shown in Figure 3C, 10 μmol/L Rb1 inhibited the IBa by 19.80%±3.21% (n=5) before ω-agatoxin IVA treatment and inhibited the IBa by 20.34%±2.58% (n=5) in the presence of ω-agatoxin IVA (P>0.05 compared to Rb1 treatment alone). Thus, neither ω-conotoxin-GVIA nor ω-agatoxin IVA could diminish the Rb1-sensitive VGCCs.
To gain a better understanding of the action of Rb1 on the IBa, we explored its action on the I–V curve and the steady-state inactivation curve of the IBa. A dose of 10 μmol/L ginsenoside Rb1 inhibited the IBa at the maximum amplitude (control: IBa=752.56±48.42 pA, Rb1: IBa=600±40.70 pA, P<0.01, n=6), but had no effect on the activation threshold potential or the reversal potential of the IBa in the I–V relationship (Figure 4). Furthermore, 10 μmol/L Rb1 shifted the steady-state inactivation curve of the IBa to a hyperpolarizing voltage (Figure 5) (control: V1/2=-17.70±0.40 mV, k=6.26±0.41; Rb1:V1/2=-25.53±0.53 mV, k=8.24±0.47; P<0.05, n=6).
Figure 4.

The I-V relationships of the IBa showed the inhibitory effects of 10 μmol/L Rb1 on the VGCCs (n=6). The holding potential was −60 mV, and the test potentials ranged from −70 mV to +70 mV in 10 mV increments.
Figure 5.

The effects of 10 μmol/L Rb1 on the voltage-dependence of the steady-state of IBa inactivation (n=6). The data were fitted to the Boltzmann equation.
A continuous recording with 10 s intervals was used after the application of Rb1, and the inhibitory effects of Rb1 on the IBa were observed during the first 10 s interval in hippocampal neurons. The results indicated that Rb1 inhibited the IBa within 10 s. To determine if phosphorylation was involved in the inhibition of the IBa by ginsenoside Rb1, the adenylyl cyclase (AC) agonist forskolin and the protein kinase A (PKA) antagonist H-89 were used. The percentage of IBa inhibitory action by Rb1 was 20.15%±3.96% (n=9), while that with the bath application of forskolin (10 μmol/L) and Rb1 was 22.5%±2.95% (n=11). Forskolin did not offset the inhibitory effect of Rb1. In the presence of H-89 (10 μmol/L), the percent inhibitory action by Rb1 was reduced to 20.85%±3.78% (n=12), a value with no statistical significance compared with that of Rb1 alone (P>0.05), demonstrating that H-89 did not affect the inhibition of the IBa caused by Rb1 (Figure 6).
Figure 6.

(A) The percentage of inhibitory action by 10 μmol/L Rb1 and Rb1 co-administered with forskolin. (B) The percentage of inhibitory action by 10 μmol/L Rb1 in the presence and absence of H-89.
Discussion
Ginsenosides, which are the pharmacologically active ingredients of Panax ginseng, produce reversible and selective inhibitory effects on voltage-dependent and ligand-gated ion channels23, 24, 25, 26. Studies have also found that the mechanisms of action of saponins vary due to their types or the cell types they act on. For example, ginsenosides activate Gαq/11, a protein coupled to PLC, leading to IP3-dependent endoplasmic reticulum calcium release in Xenopus oocytes. However, this effect does not occur in neurons27. Although previous studies have reported the diversity of voltage-dependent Ca2+ channels in hippocampal neurons, and this diversity was also confirmed in the present experiment, none of the research literature mentions the action of the ginsenoside Rb1 on VGCCs in hippocampal neurons. We found that Rb1 at a concentration range of 1 to 100 μmol/L inhibited the calcium channel currents of hippocampal neurons in a dose-dependent manner. The ICa peak current inhibition rate paralleled the increase of Rb1 concentration, and the inhibitory effect was mostly reversible.
Most studies support the effect of Rb1 in preventing neuronal death linked to neurodegenerative diseases13, 15, 28, 29. Its perturbed neuronal Ca2+ homeostasis is implicated in aging and age-related cognitive impairments12. Chen also showed that it could decrease the Aβ-induced elevation of intracellular calcium and stabilize microtubule integrity18. It is widely known that postsynaptic [Ca2+]i and L-type voltage-gated calcium channel currents are upregulated in the hippocampus during aging, despite a significant decrease of cell density30. Elevated postsynaptic [Ca2+]i and L-type voltage-gated calcium channel activity contribute to impaired synaptic plasticity31 and working memory32 in aged hippocampal neurons. The increase of L-type voltage-gated calcium channel currents also enhances the susceptibility of aging neurons for apoptosis. Fu has shown that the cholinesterase inhibitor tacrine can reduce Aβ-induced neuronal apoptosis by regulating L-type voltage-gated calcium channel activity33. Our results show that the ginsenoside Rb1 selectively targets L-type calcium channels by inhibiting voltage-gated calcium channels. Different calcium channels have distinct electrophysiological characteristics and are closely related to different cell functions. For example, ginsenoside selectively acts on the non-L-type calcium channels of chromaffin cells, which are related to the regulation of the secretion of catecholamines34. Additionally, ginsenoside Rf selectively acts on the N-type calcium channels of sensory neurons, which are related to the inhibition of neurotransmitter release following painful stimuli35. Therefore, we infer that the selective action of Rb1 on the L-type calcium channels of hippocampal neurons may be the cellular basis of its pharmacological effects in preventing neuronal death linked to neurodegenerative diseases.
In this study, Rb1 induced a leftward shift of the steady-state inactivation curves of the IBa to a negative potential without affecting its activation kinetics or reversal potential in the I–V curve, indicating that Rb1 may alter the biophysical nature of the calcium channel and inhibit channel activity by accelerating channel activity access to the inactivation state without affecting its activation characteristics. These results suggest that Rb1 regulates the activity of calcium channels by altering their time dependence.
Protein phosphorylation modulates the function of VGCCs, and the AC-cAMP-PKA system plays a key role in this phosphorylation36, 37. Therefore, we used forskolin and H-89 to investigate whether the action of Rb1 on the ICa is involved in this mechanism. We found that co-application of forskolin and Rb1 did not affect the reduction caused by Rb1. Additionally, the action of Rb1 was not affected by H-89, indicating that the cAMP-PKA system might not be involved in the mechanism by which Rb1 reduces the ICa. These results confirmed our previous speculation that it is difficult to achieve Ca2+ channels phosphorylation within 10 s.
In summary, this study provides electrophysiological evidence that Rb1 induces calcium current inhibition by inhibiting the activity of the L-type Ca2+ channels in hippocampal neurons. This finding raises the possibilities that Rb1 may be useful and potentially therapentic choices in the treatment of neurological disorders.
Author contribution
Xiao-chun CHEN designed the research; Zhi-ying LIN, Li-min CHEN, Jing ZHANG, and Xiao-dong PAN performed the experiments; Yuan-gui ZHU, Qin-yong YE, and Hua-pin HUANG contributed new analytical tools and performed the data analysis; Zhi-ying LIN wrote the paper.
Acknowledgments
We thank Prof Shu-min DUAN (Shanghai Institute of Neuroscience, Shanghai Institute of Biological Sciences, Chinese Academy of Sciences, Shanghai, China) for technical support. This work was supported by grants from the National Natural Science Foundation of China (No 30772555).
References
- Rosenthal W, Hescheler J, Trautwein W, Schultz G. Control of voltage-dependent Ca2+ channels by G protein-coupled receptors. FASEB J. 1988;2:2784–90. doi: 10.1096/fasebj.2.12.2457531. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–38. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Ahlijanian MK, Westenbroek RE, Catterall WA. Subunit structure and localization of dihydropyridine-sensitive calcium channels in mammalian brain, spinal cord, and retina. Neuron. 1990;4:819–32. doi: 10.1016/0896-6273(90)90135-3. [DOI] [PubMed] [Google Scholar]
- Chin H, Smith MA, Kim HL, Kim H. Expression of dihydropyridine-sensitive brain calcium channels in the rat central nervous system. FEBS Lett. 1992;299:69–74. doi: 10.1016/0014-5793(92)80103-n. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Arnot MI, Doering CJ, Zamponi GW. Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gbeta isoforms. J Biol Chem. 2001;276:45051–8. doi: 10.1074/jbc.M107784200. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Hashimotodani Y, Ano M, Takeda S, Tsubokawa H, Kano M. Endocannabinoid signalling triggered by NMDA receptor-mediated calcium entry into rat hippocampal neurons. J Physiol. 2007;584:407–18. doi: 10.1113/jphysiol.2007.137505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ, Morris NP, Fyffe RE, Robertson B. The Cav2.1/alpha1A (P/Q-type) voltage-dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur J Neurosci. 2001;13:1902–12. doi: 10.1046/j.0953-816x.2001.01566.x. [DOI] [PubMed] [Google Scholar]
- Yarotskyy V, Elmslie KS. Omega-conotoxin GVIA alters gating charge movement of N-type (CaV2.2) calcium channels. J Neurophysiol. 2009;101:332–40. doi: 10.1152/jn.91064.2008. [DOI] [PubMed] [Google Scholar]
- Menne J, Park JK, Agrawal R, Lindschau C, Kielstein JT, Kirsch T. Cellular and molecular mechanisms of tissue protection by lipophilic calcium channel blockers. FASEB J. 2006;20:994–6. doi: 10.1096/fj.05-4087fje. [DOI] [PubMed] [Google Scholar]
- Nikonenko I, Bancila M, Bloc A, Muller D, Bijlenga P. Inhibition of T-type calcium channels protects neurons from delayed ischemia-induced damage. Mol Pharmacol. 2005;68:84–9. doi: 10.1124/mol.104.010066. [DOI] [PubMed] [Google Scholar]
- Nah SY, Kim DH, Rhim H. Ginsenosides: are any of them candidates for drugs acting on the central nervous system. CNS Drug Rev. 2007;13:381–404. doi: 10.1111/j.1527-3458.2007.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Hakuba N, Hata R, Morizane I, Yoshida T, Shudou M. Ginsenoside Rb1 protects against damage to the spiral ganglion cells after cochlear ischemia. Neurosci Lett. 2007;415:113–17. doi: 10.1016/j.neulet.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Liu M, Zhang J. Effects of ginsenoside Rb1 and Rg1 on synaptosomal free calcium level, ATPase and calmodulin in rat hippocampus. Chin Med J. 1995;108:544–7. [PubMed] [Google Scholar]
- Radad K, Gille G, Moldzio R, Saito H, Rausch WD. Ginsenosides Rb1 and Rg1 effects on mesencephalic dopaminergic cells stressed with glutamate. Brain Res. 2004;1021:41–53. doi: 10.1016/j.brainres.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Sakanaka M, Zhu P, Zhang B, Wen TC, Cao F, Ma YJ. Intravenous infusion of dihydroginsenoside Rb1 prevents compressive spinal cord injury and ischemic brain damage through upregulation of VEGF and Bcl-XL. J Neurotrauma. 2007;24:1037–54. doi: 10.1089/neu.2006.0182. [DOI] [PubMed] [Google Scholar]
- Yuan QL, Yang CX, Xu P, Gao XQ, Deng L, Chen P. Neuroprotective effects of ginsenoside Rb1 on transient cerebral ischemia in rats. Brain Res. 2007;1167:1–12. doi: 10.1016/j.brainres.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Chen X, Huang T, Zhang J, Song J, Chen L, Zhu Y. Involvement of calpain and p25 of CDK5 pathway in ginsenoside Rb1's attenuation of beta-amyloid peptide25–35-induced tau hyperphosphorylation in cortical neurons. Brain Res. 2008;1200:99–106. doi: 10.1016/j.brainres.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–82. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Aaronson PI. Ca2+ currents in single myocytes from human mesenteric arteries: evidence for a physiological role of L-type channels. J Physiol. 1992;457:455–75. doi: 10.1113/jphysiol.1992.sp019387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretschneider F, Markwardt F. Drug-dependent ion channel gating by application of concentration jumps using U-tube technique. Methods Enzymol. 1999;294:180–9. doi: 10.1016/s0076-6879(99)94011-9. [DOI] [PubMed] [Google Scholar]
- Duan S, Cooke IM. Selective inhibition of transient K+ current by La3+ in crab peptide-secretory neurons. J Neurophysiol. 1999;81:1848–55. doi: 10.1152/jn.1999.81.4.1848. [DOI] [PubMed] [Google Scholar]
- Choi S, Jung SY, Lee JH, Sala F, Criado M, Mulet J. Effects of ginsenosides, active components of ginseng, on nicotinic acetylcholine receptors expressed in Xenopus oocytes. Eur J Pharmacol. 2002;442:37–45. doi: 10.1016/s0014-2999(02)01508-x. [DOI] [PubMed] [Google Scholar]
- Choi S, Lee JH, Oh S, Rhim H, Lee SM, Nah SY. Effects of ginsenoside Rg2 on the 5-HT3A receptor-mediated ion current in Xenopus oocytes. Mol Cells. 2003;15:108–13. [PubMed] [Google Scholar]
- Sala F, Mulet J, Choi S, Jung SY, Nah SY, Rhim H. Effects of ginsenoside Rg2 on human neuronal nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2002;301:1052–9. doi: 10.1124/jpet.301.3.1052. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–63. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SM, Lee JH, Kim S, Rhim H, Lee BH, Kim JH. Ginseng saponins induce store-operated calcium entry in Xenopus oocytes. Br J Pharmacol. 2004;142:585–93. doi: 10.1038/sj.bjp.0705797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook-Jung I, Hong HS, Boo JH, Lee KH, Yun SH, Cheong MY. Ginsenoside Rb1 and Rg1 improve spatial learning and increase hippocampal synaptophysin level in mice. J Neurosci Res. 2001;63:509–15. doi: 10.1002/jnr.1045. [DOI] [PubMed] [Google Scholar]
- Liao B, Newmark H, Zhou R. Neuroprotective effects of ginseng total saponin and insenosides Rb1 and Rg1 on spinal cord neurons in vitro. Exp Neurol. 2002;173:224–34. doi: 10.1006/exnr.2001.7841. [DOI] [PubMed] [Google Scholar]
- Coon AL, Wallace DR, Mactutus CF, Booze RM. L-type calcium channels in the hippocampus and cerebellum of Alzheimer's disease brain tissue. Neurobiol Aging. 1999;20:597–603. doi: 10.1016/s0197-4580(99)00068-8. [DOI] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, Browning MD. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res. 2003;110:193–202. doi: 10.1016/s0169-328x(02)00643-5. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–56. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK. Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem. 2006;98:1400–10. doi: 10.1111/j.1471-4159.2006.03960.x. [DOI] [PubMed] [Google Scholar]
- Choi S, Jung S, Kim C, Kim H, Rhim H, Kim S. Effect of ginsenosides on voltage-dependent Ca(2+) channel subtypes in bovine chromaffin cells. J Ethnopharmacol. 2001;74:75–81. doi: 10.1016/s0378-8741(00)00353-6. [DOI] [PubMed] [Google Scholar]
- Nah SY, Park HJ, McCleskey EW. A trace component of ginseng that inhibits Ca2+ channels through a pertussis toxin-sensitive G protein. Proc Natl Acad Sci U S A. 1995;92:8739–43. doi: 10.1073/pnas.92.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. J Neurosci. 1997;17:5334–48. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Munoz I, Sanchez-Franco F, Vallejo M, Fernández A, Palacios N, Fernández M. Activity-dependent somatostatin gene expression is regulated by cAMP-dependent protein kinase and Ca2+-calmodulin kinase pathways. J Neurosci Res. 2010;88:825–36. doi: 10.1002/jnr.22264. [DOI] [PubMed] [Google Scholar]