

Abstract
In this review, we discuss the genetic factors in both the aetiology and treatment of ischaemic stroke. We discuss candidate gene association studies, family linkage studies and the more recent whole genome association studies and whole genome expression studies. We also briefly discuss genetic testing for stroke risk and genetic analysis of treatment complications.
Introduction
Ischaemic stroke (IS) is a common neurological disease and a leading cause of severe disability and death in western countries [1]. About 85–90% of strokes are ischaemic [2, 3]. In the majority of cases, stroke is believed to be a multifactorial disorder or complex trait for which it is not possible to demonstrate classical patterns of inheritance; however, twin, family and animal model studies have consistently suggested a genetic influence on stroke risk and prognosis [4–6].
Genetic factors could act at various levels: by predisposing to conventional risk factors, by modelling the effects of such risk factors on the end organs, or, alternatively, by a direct independent effect on stroke risk, infarct size and outcome [5]. Similarly, interactions between genes and the environment factors could occur at various levels. In addition, to make it more complex, genetic predisposition could differ depending on IS subtype.
Genetic validity of subtypes
Many investigators have approached IS as a complex phenotype, dividing the IS population into distinct subtypes. A systematic review in 2002 of research into the genetics of human IS showed that when investigators subtyped IS, they typically used either nonstandard classification systems or systems of undetermined reliability [7]. Important methodological issues, including blinding and prespecification of the classification system, were rarely reported. Since then, there have been modest improvements in research methodology regarding subtyping [8]. Furthermore, the scientific rationale for looking for subtype-specific genetic risk factors continues to mount. Family history of stroke is more frequent in large- and small-vessel stroke than in cardioembolic stroke (CES) [9].
In the multigenerational linkage study performed in Iceland, no obvious pattern of inheritance emerged for IS grouped according to presumed aetiology [10]. A family study was performed based on a pooled analysis of two cohorts of male and female adult sibling pairs with symptomatic IS [11]. Subtype diagnoses were based on Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria [12]. Agreement for subtype diagnoses within families was poor (kappa = 0.17 ± 0.04). Occurrence of one IS subtype in a proband was not associated with a greater likelihood of that subtype being the qualifying stroke subtype in the sibling. Comparable poor levels of agreement were seen when restricting the analysis to stroke in same-sex sibling pairs and when restricting the analysis to young stroke.
Genetic approaches for IS as an entity
Candidate genes
Historically the two most common approaches to finding genes involved in disease have been linkage and candidate gene association studies. Association studies have become an increasingly common approach to mapping variants that affect IS, especially as a result of lower power in family studies and advances in high-throughput genotyping. However, although large numbers of candidate genes for IS have been investigated, few associations have been consistently replicated. The potential reasons that have been discussed for this lack of reproducibility are false-positive associations that correctly were not replicated, false-negative association in the replication study, methodological differences such as study design and/or differences in genetic or environmental background [13]. Strong evidence suggests that most likely, many alleles with small effect sizes are contributing to IS and to be reliably detected large sample sizes in the order of 1000 patients or more are required. In the case of stroke, most studies that have identified polymorphisms related to IS were underpowered studies with small sample size that did not achieve that number.
There is a long list of candidate gene pathways and genes that have been studied. Most of them involved in inflammation, lipid metabolism, nitric oxide release, coagulation and renin-angiotensin-aldosterone systems, haemostasis, etc. Here we focus on meta-analysis or recent studies with larger sample size. Until recently the larger-scale studies came from meta-analysis. Casas et al. performed a meta-analyses of 120 stroke candidate gene case–control studies and a total of 51 polymorphisms in 32 genes. Statistically significant associations with IS were identified for factor V Leiden Arg506Gln (FVL), methylenetetrahydrofolate reductase C677T (MTHFR), prothrombin G20210A and angiotensin-converting enzyme insertion/deletion (ACE I/D) [14]. Other meta-analyses restricted to one or more common variant from one gene reported association with IS for glycoprotein Ib alpha (GP1BA) 13 or nonassociation for plasminogen activator inhibitor (PAI)-1, tumour necrosis factor-α and integrin, alpha 2 (ITGA2) [15–17].
Although meta-analyses facilitate the overall interpretation of association, they also need to be interpreted with caution. Some meta-analyses do not include stroke risk factors as covariates and sample sizes remain small when correctly taking into account differences in ethnicity and inclusion study criteria [inclusion of children and adults, patients with transient ischaemic attack (TIA), etc.].
Berger et al. [18] investigated a total of 106 single nucleotide polymorphisms (SNPs) located in 63 candidate genes for potential associations with IS in two independent case–control studies from a German population. All genes tested were related to pathways important in the pathophysiology of cardiovascular and inflammatory diseases. Only the glu298asp polymorphism in the nitric oxide synthase-3 gene was reported to be replicated in the second study. The association was independent of age, gender, hypertension, diabetes and hypercholesterolaemia in both studies. Wang et al. [19] evaluated the association between 105 polymorphisms in 64 inflammatory and cardiovascular system-related genes and IS. None of these SNPs remained statistically significant after false discovery rate (FDR) correction. Only when the data were stratified on hypertension status did two polymorphisms in lymphotoxin-alpha (LTA) remain significantly associated with IS in nonhypertensive subjects. The data were not adjusted for other stroke risk factors such as diabetes or heart disease. Review of the literature suggests that stroke risk for the majority of these SNPs is likely to be weak or perhaps restricted to specific populations or IS subtypes.
Combining genome-wide linkage analysis and association approaches in IS
Phosphodiesterase 4D (PDE4D)
Investigators at deCODE (http://www.decode.com) performed a genome-wide linkage scan for stroke susceptibility genes in the Icelandic population [20]. Evidence for linkage with stroke was found in a region of human chromosome 5q12 that included the PDE4D gene. A risk haplotype, comprising a microsatellite (AC008818-1) and an SNP (SNP45), conferred a 1.5-fold increased risk for IS in the Icelandic population [21].
PDE4D gene belongs to a superfamily of phosphodiesterases (PDE4 family) involved in the degradation of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate. PDE4D degrades second-messenger cAMP, which might be playing an important role in the proliferation of smooth muscle cells and macrophages, and therefore possibly in atherosclerosis and plaque stability [22–24]. PDE4D protein has been thus hypothesized to be involved in the cause of stroke risk through an atherosclerotic pathway.
Numerous replication studies have attempted to confirm these findings in other cohorts and populations with variable results. In some populations the PDE4D gene seems to be a risk factor for stroke [25–29] whilst others have found no association [30–34]. Moreover in these positive studies no SNP seems to be consistent, suggesting that the causal genetic variant still needs to be identified. A meta-analysis of nine case–control studies of 3808 stroke cases and 4377 controls confirmed a significant association between IS and SNP 87, SNP 83 and SNP 41. However, heterogeneity amongst the studies in the direction of association between individual SNPs and stroke suggests that the SNPs tested are in linkage disequilibrium (LD) with the causal allele(s) [28, 29]. A recent meta-analysis comprising 16 studies, 5216 cases and 6615 controls revealed that any of the PDE4D SNPs analysed (all PDE4D variants previously associated with IS) was associated with IS [35].
Lipoxygenase activating protein (ALOX5AP)
In a separate study deCODE reported a susceptibility locus predisposing to stroke and myocardial infarction (MI) was mapped to 13q12–13 9 [36]. Within this region, a candidate gene, ALOX5AP, previously implicated in risk for atherosclerosis, was proposed as a stroke susceptibility locus. A haplotype of ALOX5AP (Hap A), defined by four SNPs that span the first four exons, was strongly associated with risk for stroke and MI. In the Icelandic population, Hap A conferred a 1.8-fold increased risk for MI and a 1.67-fold increased risk for stroke. Hap A was also found to be a risk factor for stroke in a Scottish population. A different haplotype of ALOX5AP (Hap B) was positively associated with stroke in a British population [37].
Replication studies produce conflicting results with some reporting association [30, 36] whereas others did not [25, 38, 39]. Recently, Zintzaras et al. [40] performed a meta-analysis of all studies with ALOX5AP genotyping (5194 stroke cases and 4566 controls) showing significant heterogeneity amongst studies and nonsignificant association between the Hap A (OR: 1.13; 95% CI: 0.88–1.45) or Hap B (OR: 1.03; 95% CI: 0.77–1.37) and stroke risk.
Genome-wide linkage studies in IS
Recently, Nilsson-Ardnor et al. [41] reported data for a whole genome linkage scan of 56 pedigrees from a relatively genetically homogeneous region of northern Sweden. They used three different disease models: including all stroke cases [excluding TIA but including subarachnoid haemorrhage (SAH)], ischaemic and intracerebral haemorrhagic stroke, and IS only. The initial linkage findings were refined in two steps. Fine-mapping in the first step detected moderate linkage on chromosomes 5q.10, 9q, 13q and 18p. In the second step, 53 additional families from the same region were included and linkage was still detected in all of these regions apart from chromosome 9q. In the combined analysis including all 109 families, the highest allele-sharing LOD scores were obtained on chromosome 18p (LOD = 1.53 at marker D18S59) when stroke was considered alone. The study did not identify any new major loci for IS.
Genome-wide association studies
The completion of the International Haplotype Map Project (http://www.hapmap.org), coupled with the availability of high-throughput genotyping methodologies, has allowed the application of association testing in a genome-wide manner. Genome-wide association (GWA) scans can identify novel genetic loci, and do not require a prespecified hypothesis implicating a particular candidate pathway. To date, variants or regions associated with disease with nearly 40 complex diseases or traits have been identified and replicated in diverse population samples [42]. However, few GWAS have been performed in the context of IS.
As a first phase of a GWAS in stroke we performed a GWAS using more than 400 000 unique SNPs in a cohort of 249 patients with IS and 268 neurologically normal controls, white non-Hispanics from North America [43]. As expected, the screening approach yielded hundreds of nominally statistically significant associated markers, none significant after correction for multiple testing. Some of the most significant SNPs were within or near interesting candidate loci and the individual risk estimates provided by SNPs were moderate-to-high with odds ratios (OR) ranging from 0.40 (0.26–0.62) to 0.54 (0.41–0.74) and 1.9 (1.42–2.65) to 8.0 (2.85–22.33). The published pilot study was underpowered in isolation to detect genetic loci that exert a moderate risk for stroke; however, the study was useful to suggest that there is not a single common genetic variant conferring a major risk on IS and generate publicly available genome-wide genotypes that could allow other investigators to independently pursue follow-up. The data generated were published with public release of genotype data in all controls and 88% of the stroke patients.
Recently, Kubo et al. [44] performed a GWA study using 1112 Japanese cases with IS and 1112 age- and gender-matched controls. First, they genotyped 188 cases and 188 age- and sex-matched controls using 52 608 tag SNPs selected from the Japanese SNP (JSNP) database. In a second stage, they genotyped the remaining samples for the 1098 SNPs showing P-values below 0.01. They also performed analysis by stroke subtypes. A nonsynonymous SNP (1425 G/A) in a member of protein kinase C (PKC) family, PRKCH, was significantly associated with lacunar infarction (P = 4.73 × 10−6). The same group replicated this association with an independent cohort of 1137 cases with lacunar infarction and 1875 controls selected from the BioBank Japan project. Combined analysis of two case–control samples showed that the A allele was significantly associated with lacunar infarction under a dominant model, with an OR of 1.40 (95% C.I: 1.23–1.59; P = 5.1 × 10−7).
Protein kinase C is a serine-threonine kinase that regulates a wide variety of important cellular functions including proliferation, differentiation and apoptosis. Functional analysis showed that PKC was expressed mainly in vascular endothelial cells and foamy macrophages in human atherosclerotic lesions, and its expression increased as the lesion type progressed. The study supports a role for PRKCH in the pathogenesis of IS, likely specific to Asian populations through atherosclerosis [44].
Recently, two SNPs rs11833579 and rs12425791 located at chromosome 12p13 have been reported to be associated with stroke and IS in a community-based GWAS, formed for four white cohort studies and replicated in an African American cohort and a white-control sample [45]. The study suggested that each copy of a minor allele at these two loci increased the hazards ratio (HR) for IS by 1.39–1.41 (95% CI: 1.27–1.56) and for total stroke by 1.31–1.32 (95% CI: 1.19–1.44). The association of these two SNPs with atherothrombotic subtype stroke in whites was stronger.
The SNPs are located closed to the gene Ninjurin2 (NINJ2) and WNK1. Nerve injury-induced protein 2 (NINJ2) is a cell surface adhesion molecule, and its expression is constitutive and abundant in almost all the sensory ganglion neurones and enteric neurones but weak in peripheral glial cells and neurones in the autonomic system [46]. Ninjurin2 is also highly expressed in hematopoietic and lymphatic tissues. Ninjurin2 is upregulated in Schwann cells in the distal nerve segment after peripheral nerve injury, and it promotes neurite outgrowth [47, 48]. The authors suggested that level of expression of Ninjurin2 could affect how the brain tolerates ischaemic insults.
WNK1, a serine-threonine kinase regulating numerous ion channels involved in sodium and potassium transport [49], is ubiquitously expressed, with particularly high levels of expression in the kidney and cardiovascular system. Mutations in WNK1 have been linked to Pseudohypoaldosteronism type 2 (PHA2), a rare autosomal dominant disorder primarily characterized by early-onset hypertension and hyperkalemia [50]. Heterozygous knock-out mice that lack WNK1 expression have low blood pressure (BP) [51] and multiple rare and common WNK1 variants have been reported to contribute to BP variation and hypertension [52]. Replication in independent cohorts remains necessary to confirm or refute this as an unequivocal risk factor for stroke.
Atrial fibrillation and chromosome 4q25
Nearly 6% of those over 65 years of age have atrial fibrillation (AF) [53]. The excess 5-year risk of stroke imparted by AF varies from about 2% to 16%, depending on clinical and demographic factors [54]. Early identification of AF is an important clinical objective because warfarin in patients with AF reduces the risk of stroke by about 60% [55].
Mutations in potassium-channel genes cause some cases of familial AF, but this constitutes only a small fraction of all AF cases. A GWAS, followed by replication studies in three European-descent populations and a Chinese population found an association between two sequence variants on chromosome 4q25 and sporadic AF [56]. About 35% of individuals of European descent have at least one of the variants. The risk of AF increases by 1.72 and 1.39 per copy. Association with the stronger variant was replicated in the Chinese population, where it is carried by 75% of individuals and the risk of AF is increased by 1.42 per copy. Both variants are adjacent to Pitx2, which has a role in the development of left–right cardiac asymmetry.
A GWAS subsequently demonstrated that risk variants for AF on chromosome 4q25 were also risk factors for IS. A total of 1661 Icelandic subjects with IS and 10 815 control subjects were genotyped using the Infinium HumanHap300 chip [57]. The most significant signals were replicated in two European populations (2224 cases and 2583 control subjects). Two SNPs, rs2200733 and rs10033464, were tested in additional European populations (2327 patients and 16 760 control subjects). In the Icelandic samples and the two replication sets combined, rs2200733 associated significantly with CES (OR: 1.54). The rs2200733 SNP associated significantly with IS in all sets combined (OR: 1.26). Both the rs2200733 SNP and the adjacent rs10033464 associated strongly with CES. The rs2200733 also significantly associated with non-CES. One possible explanation for this is that amongst the cases classified as noncardioembolic there may be a substantial subset of subjects with cryptogenic cardioembolism [58]. This raises the question whether patients who present with IS but not AF should undergo gene testing for AF risk gene variants. Those who test positive may have a high likelihood of detecting intermittent AF with cardiac monitoring, potentially justifying more intensive monitoring. More intensive cardiac rhythm monitoring has a higher yield of detecting AF [59, 60]. Relatively unselective use of warfarin for secondary stroke prophylaxis is not superior to aspirin [61, 62]. However, it is possible that patients with stroke who test positive for AF variants are at such high risk of developing AF as to justify long-term anticoagulation.
Myocardial infarction, coronary disease, IS and chromosome 9p21
There is growing evidence that a risk locus for coronary artery disease (CAD) resides on chromosome 9p21. A GWAS identified a 58-kb interval on chromosome 9p21 that consistently associated with CAD in six independent samples (>23 000 participants) from four populations of European descent [63]. This interval is located near the CDKN2A and CDKN2B genes. Up to 20–25% of people of European descent are homozygous for the risk allele and have about a 30–40% increased risk of CAD. A separate joint analysis of two GWAS of CAD was performed [64]. Chromosomal loci strongly associated with CAD were first identified in the Wellcome Trust Case Control Consortium (WTCCC) study (1926 CAD cases; 2938 controls) and replication was sought in the German MI Family Study (875 cases with MI; 1644 controls). Data on other SNPs that were significantly associated with CAD in either study (P < 0.001) were then combined to identify additional loci. The chromosome 9p21.3 locus had the strongest association with CAD in both the WTCCC and German studies. A third GWAS of 4587 cases and 12 767 controls identified a variant adjacent to CDKN2A and CDKN2B associated with CAD [65]. About 21% of individuals in the population were homozygous for this variant, and their estimated risk of suffering MI was 1.64 times as great as that of noncarriers.
A meta-analysis found that the risk allele (C) of the lead SNP, rs1333049, representing the 9p21.3 locus, was uniformly associated with CAD in seven case–control studies [66]. In a pooled analysis, the OR per copy of the risk allele was 1.29 (95% CI: 1.22–1.37; P = 0.0001). The meta-analysis of the rs1333049 SNP in 12 004 cases and 28 949 controls increased the overall level of evidence for association with CAD to P = 6.04 × 10−10 (OR: 1.24; 95% CI: 1.20–1.29). Genotyping of 31 additional SNPs in the region identified several that significantly associated with CAD, but none was more predictive than rs1333049.
Analysis of the 9p21 region showed that the same locus that was a risk factor for CAD was also a risk factor for IS [67]. This analysis took advantage of a prior pilot IS GWAS [43]. A larger study using multiple case series confirmed the association [68].
The SNP rs10757278-G, which tags 9p21, has been associated with CAD, abdominal aortic aneurysm (AAA; OR = 1.31; P = 1.2 × 10−12) and intracranial aneurysm (OR = 1.29; P = 2.5 × 10−6) [69]. It is not surprising that there would be a common risk factor for MI and AAA formation, as both share atherosclerosis pathologically. However, it is less clear why intracranial aneurysms should be associated with this group, as atherosclerotic pathology is not a major feature of intracranial aneurysms. It is possible that the 9q21 variant acts through an interaction with tobacco smoking, which is a risk factor for all phenotypes as well.
Copy number variants and IS
In addition to SNP data, the genotyping assay use in GWA studies also generates metrics that permit detection of copy number variants (CNVs): segments of the genome that may be deleted or duplicated. We investigated whether CNVs may modulate risk for IS. They examined CNV in 263 cases with IS and each identified CNV was compared with changes identified in neurologically normal control populations. The analysis identified 247 CNVs, corresponding to 185 insertions (76%) and 60 deletions (24%) ranging in size from 1.7 kb to 2.1 Mb. Most alterations (81%) were the same as, or overlapped with CNVs previously reported in healthy individuals and the reminder was not recurrent in more than one IS sample. The study did not detect any common genomic structural variation unequivocally linked to IS, although the authors did not exclude that smaller CNVs or CNVs in genomic regions poorly covered by this methodology may confer risk for IS [70].
Limitations of the GWAS approach
The GWA approach has its limitations. Inconsistent or poorly defined measurements of the phenotype, population stratification, insufficient genome coverage, low power and failure to control the rate of false discoveries are potential reasons for missing genuine associations. In the context of a multifactorial disease such as stroke, care in the selection of the samples having identical straightforward definitions in the diagnosis, classification and the different variables analysed in stroke and control samples, such as stroke risk factors would help to minimize errors and find true associations.
However, low power could be our major issue if multiple different genes with really small effects are influencing risk for IS and different studies and approaches suggest that is the case. In fact, all susceptibility alleles discovered so far explain only a small fraction of disease risk, with OR typically in the range of 1.2–1.5 [42, 71]. As these are high-cost studies and very large numbers of subjects will be needed, public registration of protocol (helping to obviate biases) and databases (making possible to join different parties) or multinational studies such as the Wellcome Trust or the International Stroke Genetics Consortium could be a solution to arrive at solid conclusions that otherwise would be impossible for a single research group working alone.
Other factors to take into account is that the GWA approach is unable to detect susceptibility loci that harbour numerous individually rare (recent) polymorphisms and also a proportion of the genome is comprised of recombination hotspots that are not amenable to LD-based approaches.
Moreover, some genetic determinants that may be relatively common in Caucasian population could be uncommon in other nonstudied populations. Larger number of tag SNPs is likely to be required in African populations (or those with very recent origins in Africa) because these populations generally contain more variations and less LD. A resequencing approach will be needed to identify rare alleles.
Commercially available risk assessment tests
There are commercially available tests for stroke risk factor loci on chromosome regions 4q25 and 9p21. In both instances, clinical validation studies are warranted to see how these tests could best be integrated into clinical practice.
Atrial fibrillation and chromosome 4q25
As discussed above, a GWA for IF and subsequently replication in European sets found two SNPs on Chr 4p25 that double the risk of AF. deCODE is offering risk assessment testing based on this finding.
Chromosome 9p21
deCODE is also offering a test for the identification of subjects with increased risk for MI, through the genotyping of the SNPs in 9p21 region that were associated with the disease. However, it would seem that the reported OR for the SNP do not warrant such testing.
Pharmacogenetics applied to IS management
Response to clopidogrel
Clopidogrel requires transformation into an active metabolite by cytochrome P-450 (CYP) enzymes for its antiplatelet effect. The genes encoding CYP enzymes are polymorphic, with common alleles conferring reduced function. The association between functional genetic variants in CYP genes, plasma concentrations of active drug metabolite and platelet inhibition in response to clopidogrel was tested in 162 healthy subjects [72]. The association was then examined between these genetic variants and cardiovascular outcomes in a separate cohort of 1477 subjects with acute coronary syndromes who were treated with clopidogrel in the TRITON-TIMI 38 trial. In healthy subjects treated with clopidogrel, carriers of at least one CYP2C19 reduced-function allele (about 30% of the study population) had a relative reduction of 32.4% in plasma exposure to the active metabolite of clopidogrel, compared with noncarriers (P < 0.001). Carriers also had an absolute reduction in maximal platelet aggregation in response to clopidogrel that was 9% less than that seen in noncarriers (P < 0.001). Amongst clopidogrel-treated subjects in TRITON-TIMI 38, carriers had a relative increase of 53% in the composite primary efficacy outcome of the risk of stroke, MI or cardiovascular death, compared with noncarriers [12.1% vs. 8.0%; HR for carriers, 1.53; 95% confidence interval (CI): 1.07–2.19; P = 0.01] and an increase by a factor of 3 in the risk of stent thrombosis (2.6% vs. 0.8%; HR: 3.09; 95% CI: 1.19–8.00; P = 0.02).
In a separate study involving a nationwide French registry, 2208 patients presenting with an acute MI and receiving clopidogrel therapy were enrolled [73]. Investigators assessed the relation of allelic variants of genes modulating clopidogrel absorption (ABCB1), metabolic activation (CYP3A5 and CYP2C19) and biological activity (P2RY12 and ITGB3) to the risk of stroke, MI or all-cause mortality within the first year of follow-up. None of the selected SNPs in CYP3A5, P2RY12 or ITGB3 was associated with a risk of an adverse outcome. Patients with two variant alleles of ABCB1 (TT at nucleotide 3435) had a higher rate of cardiovascular events at 1 year than those with the ABCB1 wild-type genotype (CC at nucleotide 3435) (15.5% vs. 10.7%; adjusted HR: 1.72; 95% CI: 1.20–2.47). Patients carrying any two CYP2C19 loss-of-function alleles (*2, *3, *4 or *5), had a higher event rate than patients with none (21.5% vs. 13.3%; adjusted HR: 1.98; 95% CI: 1.10–3.58). Amongst the 1535 patients who underwent percutaneous coronary intervention during hospitalization, the rate of cardiovascular events amongst patients with two CYP2C19 loss-of-function alleles was 3.58 times the rate amongst those with none (95% CI: 1.71–7.51).
The CYP2C19 variants appear relevant to young individuals with coronary atherosclerosis as well. In a study of 259 young patients (age <45 years) who survived a first MI and were exposed to clopidogrel treatment for at least a month, the primary end-point of MI, urgent coronary revascularization and death occurred more frequently in carriers of the CYP2C19*2 variant than in noncarriers (15 vs. 11 events; HR: 3.69; 95% CI: 1.69–8.05;P = 0.0005) [74]. The same was true for in-stent thrombosis (8 vs. 4 events; HR 6.02; 95% CI: 1.81–20.04;P = 0.0009). The detrimental effect of the CYP2C19*2 genetic variant persisted from 6 months after clopidogrel initiation to the end of follow-up (HR 3.00; 95% CI: 1.27–7.10; P = 0.009). After multivariable analysis, the CYP2C19*2 genetic variant was the only independent predictor of cardiovascular events (HR 4.04; 95% CI: 1.81–9.02; P = 0.0006).
Statin-associated myopathy
In rare cases, myopathy occurs in association with statin therapy, especially when the statins are administered at higher doses and with certain other medications.
Organic anion transporting polypeptide 1B1 (OATP1B1) is an uptake transporter located at the sinusoidal membrane of human hepatocytes encoded by the gene SLO1B1. Four healthy volunteers with the homozygous SLCO1B1 c.521CC genotype, 12 with the heterozygous c.521TC genotype and 16 with the homozygous c.521TT genotype (controls) were recruited in a study to investigate the effects of genetic polymorphism in the SLCO1B1 gene on the pharmacokinetics of simvastatin [75]. Each participant ingested a single 40 mg dose of simvastatin. Plasma concentrations of simvastatin (inactive lactone) and its active metabolite simvastatin acid were measured for 12 h. The AUC0–∞ of simvastatin acid was 120% and 221% higher in participants with the SLCO1B1 c.521CC genotype than in those with the c.521TC and c.521TT genotypes respectively (P < 0.001). TheCmax of simvastatin acid was 162% and 200% higher in participants with the c.521CC genotype than in those with the c.521TC and c.521TT genotypes (P < 0.001). The Cmax of simvastatin acid occurred earlier in participants with the c.521CC and c.521TC genotypes than in those with the c.521TT genotype (P < 0.05). Raised plasma concentrations of simvastatin acid in patients carrying the SLCO1B1 c.521C variant allele may enhance the risk of systemic adverse effects during simvastatin treatment.
A GWAS was carried out using about 300 000 markers (and additional fine-mapping) in 85 subjects with definite or incipient myopathy and 90 controls, all of whom were taking 80 mg of simvastatin daily as part of a trial involving 12 000 participants [76]. Replication was tested in a trial of 40 mg of simvastatin daily involving 20 000 participants. The genome-wide scan yielded a single strong association of myopathy with the rs4363657 SNP located within SLCO1B1 on chromosome 12 (P = 4 × 10−9). The noncoding rs4363657 SNP was in near-complete LD with the nonsynonymous rs4149056 SNP (r2 = 0.97), which has been linked to statin metabolism. The prevalence of the rs4149056 C allele in the population was 15%. The OR for myopathy was 4.5 (95% CI: 2.6–7.7) per copy of the C allele, and 16.9 (95% CI: 4.7–61.1) in CC compared with TT homozygotes. More than 60% of these myopathy cases could be attributed to the C variant. The association of rs4149056 with myopathy was replicated in the trial of 40 mg of simvastatin daily, which also showed an association between rs4149056 and the cholesterol-lowering effects of simvastatin.
Response to warfarin
Warfarin is the most effective means of preventing CES in the setting of AF. High variability in drug response and a narrow therapeutic index complicate warfarin therapy initiation (Fig. 1). The principal enzyme involved in warfarin metabolism is cytochrome P450 (CYP) 2C9 (CYP2C9), and the pharmacologic target of warfarin is vitamin K epoxide reductase (VKORC1). Variation in these genes leads to variable response to warfarin.
Figure 1.
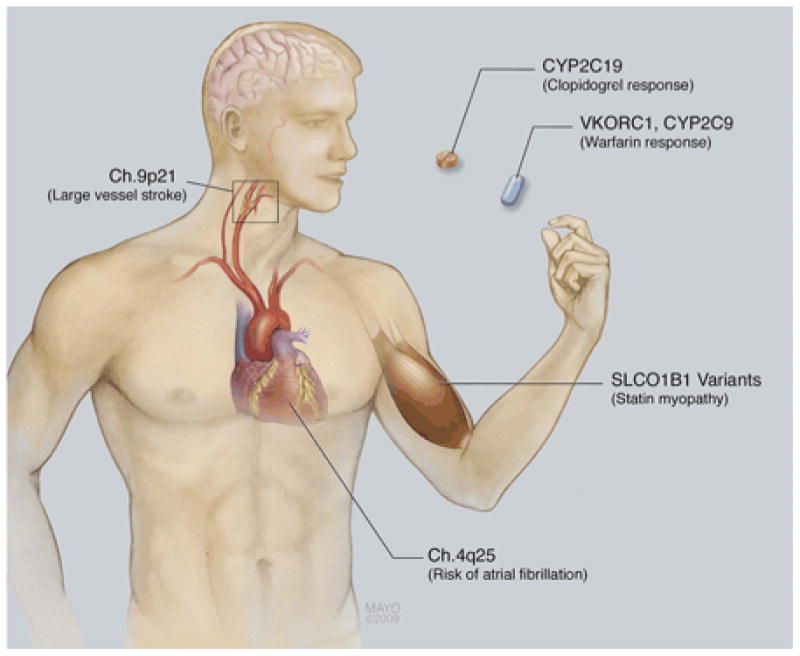
Potential pharmacogenetic considerations in optimizing stroke prevention. Genetic testing could be used to stratify risk of future large vessel atherosclerotic stroke or cardioembolic stroke. Genetic testing could also be used to help safely initiate warfarin anticoagulation or identify individuals not likely to respond to clopidogrel. It could also be used to identify individuals at risk of myositis associated with statin use.
Polymorphisms in the CYP2C9 and VKORC1 genes were genotyped in 92 patients undergoing total hip or knee replacement [77]. A model for refining the warfarin dose after the third warfarin dose was developed that explained four-fifths of the variability in therapeutic dose. Significant (P > .05) predictors were international normalized ratio (INR) value after three doses, first warfarin dose, CYP2C9*3 and CYP2C9*2 genotype, estimated blood loss, smoking status and VKORC1.
A study of patients receiving long-term warfarin maintenance therapy determined VKORC1 haplotype frequencies in a multiracial population, and VKORC1 messenger RNA (mRNA) expression in human liver samples [78]. Ten common noncoding VKORC1 SNPs were identified. A low-dose haplotype group (A) and a high-dose haplotype group (B) were identified. Mean maintenance dose of warfarin differed significantly amongst haplotype groups. VKORC1 haplotype groups A and B explained about 25% of the variance in dose. Asian Americans had a higher proportion of group A haplotypes and African Americans a higher proportion of group B haplotypes. VKORC1 mRNA levels varied according to haplotype combination, suggesting that VKORC1-related warfarin response variability is regulated at the transcriptional level.
A retrospective cohort study of 200 patients receiving long-term warfarin therapy for various indications was conducted at two anticoagulation clinics [79]. Amongst 185 patients with analysable data, 58 (31.4%) had at least one variant CYP2C9 allele and 127 (68.6%) had the wild-type (*1/*1) genotype. Mean maintenance dose varied significantly amongst the six genotype groups. Patients with at least one variant allele had an increased risk of above-range INRs. The variant group also required more time to achieve stable dosing. Subjects with a variant genotype also had a significantly increased risk of a serious bleeding event (HR: 2.39; 95% CI: 1.18–4.86).
Schelleman et al. [80] studied whether warfarin dosing algorithms developed for Caucasians and African Americans on the basis of clinical, environmental and genetic factors performed better than an empirical starting dose of 5 mg day−1. From April 2002 to December 2005, 259 subjects (Caucasians and African Americans) who started using warfarin were prospectively followed up until they reached maintenance dose. The Caucasian algorithm included 11 variables (R2 = 0.43). This model predicted 51% of the doses to within 1 mg of the observed dose. This was better than the 5 mg day−1 fixed-dose approach, which predicted 29% of the doses to within 5 ± 1 mg. The African American algorithm included 10 variables (R2 = 0.28). This model predicted 37% of the doses to within 1 mg of the observed dose, representing a small improvement compared with 5 mg day−1 (which predicted 34% of the doses to within 1 mg of 5 mg day−1). These results were similar to the results we obtained from testing other published algorithms. The algorithms performed only marginally better for African Americans when compared with giving 5 mg empirically.
A retrospective GWAS was designed to identify polymorphisms that could explain a large fraction of the dose variance [81]. White patients from an index warfarin population (n = 181) and two independent replication patient populations (n = 374) were studied. From the approximately 550 000 polymorphisms tested, the most significant independent effect was associated with VKORC1 polymorphisms (P = 6.2 × 10−13) in the index patients. CYP2C9 (rs1057910, CYP2C9*3 and rs4917639) was associated with dose at moderate significance levels (P approximately 10−4). Replication polymorphisms (355 SNPs) from the index study did not show any significant effects in the replication patient sets. The investigators concluded that common SNPs with large effects on warfarin dose are unlikely to be discovered outside the CYP2C9 and VKORC1 genes.
A study of 297 patients starting warfarin therapy assessed CYP2C9 genotypes (CYP2C9 *1, *2 and *3), VKORC1 haplotypes (designated A and non-A), clinical characteristics, response to therapy (as determined by INR) and bleeding events [82]. Compared to patients with the non-A/non-A haplotype, patients with the A/A haplotype of VKORC1 had a decreased time to the first INR within the therapeutic range and to the first INR >4. The CYP2C9 genotype was a significant predictor of time to the first INR >4 (P = 0.03). Both the CYP2C9 genotype and VKORC1 haplotype had a significant influence on the required warfarin dose after the first 2 weeks of therapy.
Several commercial platforms for pharmacogenetic dosing are available. The assays typically take 3–8 h to complete and the accuracies range from 99% to 100% [83].
A total of 206 participants being initiated on warfarin were randomized to pharmacogenetic-guided or standard dosing [84]. DNA was genotyped for CYP2C9 *2, CYP2C9 *3 and VKORC1C1173T. Standard dosing followed an empirical protocol, whereas pharmacogenetic-guided dosing followed a regression equation including the three genetic variants and age, sex and weight. Prothrombin time INR was measured on days 0, 3, 5, 8, 21, 60 and 90. A research pharmacist un-blinded to treatment strategy managed dose adjustments. Patients were followed for up to 3 months. Pharmacogenetic-guided predicted doses more accurately approximated stable doses (P < 0.001), resulting in smaller (P = 0.002) and fewer (P = 0.03) dosing changes and INRs (P = 0.06). However, per cent out-of-range INRs (pharmacogenetic = 30.7%, standard = 33.1%), the primary end-point, did not differ significantly between arms. Multiple variant allele carriers were at increased risk of an INR of ≥4 (P = 0.03).
Gene expression and IS
As mentioned previously, there are some interesting results with novel polymorphisms which have been linked to increased risk of stroke or other common complex diseases. In these cases, as is likely in most complex diseases, the risk variants are noncoding. This would suggest that genetic variability in expression and/or splicing is critical in disease, and different approaches have all shown that genetic variability is an important component in the regulation of gene expression [85]. Notably the work describing the association between variability at PDE4D and risk of IS showed that this variability affected PDE4D gene expression [21]. The model of locus identification followed by investigation into the consequences of such variation on expression is clearly useful; however, ad hoc investigation into such effects is resource hungry and uninformative with respect to distal or trans-effects.
ALOX5P and the gene coding for the counter-inflammatory cytokine IL-1ra (ILRN) are other examples of direct evidence of connection between genetic variability and in expression and predisposition for disease and/or vulnerability to complications. Helgadottir et al. [36] found a greater production of LTB4, one of the key products of 5-LO, in ionomycin-stimulated neutrophils from male carriers of the at-risk haplotype, suggesting that the disease-associated variants increase the response of five-lipoxygenase activating protein (FLAP) to factors that stimulate inflammatory cells.
ILRN has been found to have a variable number tandem repeat polymorphism in intron 2. Homozygotes for alelle 2 (less common 2 repeat) have a two- to threefold lower IL-1ra production than homozygotes for allele 1 (common 4 repeat polymorphism) [86]. Allele 2 has been found increased in coronary [87] and carotid atherosclerosis [88].
Gene expression profiling
Gene expression profiling involves the study of mRNA levels in a tissue sample to determine the expression levels of genes that are expressed or transcribed from genomic DNA. For example, gene expression profiling in the study of cancer tissue samples has facilitated the identification and refinement of tumour subtypes [89], distinction between good-prognosis and poor-prognosis tumours [90] and the prediction of response to treatment [91, 92].
In stroke, peripheral blood mononuclear cells (PBMCs) and whole blood have been used as a source of mRNA to study gene expression. Moore et al. [93] compared gene expression levels on 20 patients with IS to 20 referent controls and, using prediction analysis for microarrays, found that a panel of 22 genes classified stroke in a validation cohort with a sensitivity of 78% and a specificity of 80%. Tang et al. [94] reported that a large number of genes change expression in the peripheral blood of humans as early as 3 h after IS. Little overlap in the set of genes between both studies was probably due to differences in methodology (whole blood versus PBMCs) and blood sample times.
Recently, some studies have been trying to identify gene expression profiles for IS patients with specific phenotypes [95, 96]. For example, Xu et al. [96] reported that gene expression profiles in blood can differ between different aetiologies of IS. Seventy-three genes differentiated CES from large-vessel atherosclerotic stroke with at least 95.2% specificity and 95.2% sensitivity for each.
The small percentage of patients who actually receive recombinant tissue-type plasminogen activator (rt-PA) (3–5%), the only approved therapy for the acute phase of stroke and the large numbers of patients who leave the hospital with either a diagnosis of TIA or stroke of undetermined cause confirms the need to identify additional means of stroke diagnosis. Peripheral blood is easily accessible in all clinical situations and therefore provides greater clinical utility. Gene expression profiling could provide new insight into the molecular pathways involved in brain recovery and health and elucidate complex genomic interactions that may play a role in outcome. A greater understanding of the molecular response to acute stroke could allow for a more accurate selection of pathways to target novel stroke therapeutics.
Limitations of gene expression work
Although these findings need to be confirmed in larger cohorts, these studies provide a novel and sophisticated approach to delineate the pathogenesis of stroke. However, there are some shortcomings of previous studies: (i) Most of the studies do not use mRNA from ischaemic target tissues such as brain or arteries and many diseases manifest their phenotype in certain tissues and not in others; (ii) They only examined segments of the entire sequence of interest rather than the entire architecture of the risk locus; (iii) They used small sample sizes (n ≤ 20); (iv) Many studies used stroke animal models that may not entirely reflect the pathophysiological process through which stroke evolves in humans. The species difference is one of the main reasons accounting for the lack of success of ‘bench to bedside translation’ in the area of stroke and (v) Remain difficult to distinguish between causal and reactive effect. Still it will be needed to know if the genes that showed different profile are stroke risk genes or genes that respond differently to the ischaemic event.
Gene expression in human brain
Brain ischaemia is thought to be regulated by more genes than any other condition possibly because ischaemia damages all cellular elements including neurones, glia, axons/white matter and the vessels. Clearly, expression profiling has the potential to divine complex patterns of expression such as these.
Promising advancement in this direction can already be found in the literature. Myers et al. [97] reported genetic variants influencing normal human cortical expression.
The effects of genetic variability on gene expression in brains obtained from stroke cases have not been examined in detail, but two pilot studies have investigated the dynamic changes in gene expression in brain samples from patients with various times of survival following stroke.
Vikman and Edvinsson [98] investigated gene expression in 11 samples of human brain after thromboembolic stroke in the middle cerebral artery (MCA) using samples 7–10 days poststroke and 2–3 days postmortem. Thirteen subjects who had died from extracranial events, cardiogenic insufficiency or MI (age: 36–91 years) were used as controls. They focused mainly on mRNA expression of receptors. Analysis of the different expression patterns resulted in 82 significantly upregulated and 17 downregulated genes. Between the pathways found, they chose seven genes (RhoA, LY64, ELK3, POU3F4, MetallothioninIG, actinα2 and smoothelin) for validation with RT-PCR, and six were consistent with the array.
Mitsios et al. [99] investigated gene expression in brain tissue taken from infarcts of 12 patients who died from acute IS and had large MCA strokes confirmed by computed tomography or magnetic resonance imaging. They established three mRNA expression profiles according to time poststroke: between 2–6, 9–20 and 26–37 days after stroke in human patients. Seventy-seven, 92 and 15 genes were de-regulated in stroke-affected regions in these three patient survival groups respectively. Thus the number of deregulated genes seems to decline 26–37 days after stroke, indicating that dynamic changes in gene expression occur during the first days to few weeks in the human postischaemic brain. The authors examined in more detail a small subset of genes with no prior reported role in stroke (PAK1, MMP11 and INI1). The temporal expression patterns of these genes following RT-PCR supported the validity of the data obtained from microarrays.
Gene expression in the arterial circle of Willis
Atherosclerosis of the cerebral and precerebral vessels remains a major cause of stroke despite the recognition and treatment of atherosclerotic risk factors. Atherosclerosis is both a systemic disease and an organ-specific disease. At the systemic level, atherosclerosis in one vascular bed predicts atherosclerosis in other vessels. Vascular territories follow a somewhat predictable time course of plaque development. At the individual level, dramatic variation exists in the vulnerability of specific vessels to atherosclerosis. This is also seen at a population level with Asians and African Americans having higher risk of intracranial atherosclerosis and Caucasians having greater susceptibility to large vessel extracranial atherosclerosis [100, 101]. New data from the Warfarin-Aspirin Symptomatic Intracranial Disease study underscores the tremendous burden of intracranial disease with stroke rates of ∼20% in both treatment arms [102]. Patients with this disease are at extremely high risk and the lack of a good animal model has severely impeded efforts to better understand the pathophysiology and to develop treatments for this disease.
In autopsy studies performed in the 1960s it was found that the arterial circle of Willis in infarcted brains had a higher number of hypoplastic vessel segments than brains without such infarctions [103, 104]. There are also studies that show significant associations between stroke severity and the functioning of collateral pathways in the arterial circle of Willis [104–108]. Thus some studies indicate that in patients with severe internal carotid artery stenosis or occlusion, a well-functioning circle of Willis can protect the brain against ischaemia [109, 110].
From another perspective, a recent study has assessed the role of an important haplotype of the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene as a possible risk factor for IS mediated by the severity of intracranial atherosclerosis. The E607G SNP of PCSK9 was associated significantly with risk of LAAS stroke subtype. The same allele also tended to be associated with increased atherosclerosis of the large intracranial arteries forming the circle of Willis in an independent Finnish autopsy study with a gene dose effect [111].
Thus the study of common genetic variants and expression profiling in tissues such as the brain and the arterial circle of Willis could provide clues to identify genes and subsequent pathways involved in this type of stroke and their interaction with environmental factors.
The future
The coming months and years will see GWA studies of stroke and related phenotypes performed in multiple population samples, integrating analysis of SNPs, haplotypes and CNVs. As the sample sizes become larger the data will become more reliable and genuine and reproducible risk variants will be discovered. A subsequent challenge will be to move from these associated SNPs to a deeper understanding of the biology of risk for stroke. Many of the discovered susceptibility polymorphisms fall in noncoding regions and they are probably only tagging the real functional variants [112]. In future it will be necessary to move from association signal to causal variant, and from causal variant to the molecular and cellular mechanisms involved in generating phenotypic effects. There is a need for methodologies that allow both the interpretation of functional consequences of variants and the description of functionally important variants [113]. Linkage and association studies coupling expression with genetic variability data have started to reveal the genetics underlying part of this variation, including complex allele-specific interactions and its relatively high level of heritability [97, 113–116].
Integrating expression with genetic variability in human stroke targeting organs such as the brain and cerebral vessels could tell us about the causal variants of IS and how they lead to disease. Finally the next-generation whole-sequencing technologies will be able to explore genome that right now is inaccessible or unassayed, providing a better profile of genomic differences between cases and controls. Already commercialization of the meagre replicated findings is assuring, and whilst this is probably premature, it will undoubtedly continue to increase. Responsible use of the genetic data that comes from these risk analyses will pose a challenge both to the medical profession and to the public if we are to achieve the goal of well-organized personal medicine.
Acknowledgments
This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Center's funding scheme. Work in our laboratories is sponsored by the MRC. This work was also supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services (project Z01 AG000958-06). Our stroke sample collection was supported by NIH/NINDS grants to J.M. Ancillary
Footnotes
Conflict of interest statement: No conflict of interest was declared.
References
- 1.Bonita R. Epidemiology of stroke. Lancet. 1992;339:342–4. doi: 10.1016/0140-6736(92)91658-u. [DOI] [PubMed] [Google Scholar]
- 2.Rothwell PM, Coull AJ, Giles MF, et al. Change in stroke incidence, mortality, case-fatality, severity, and risk factors in Oxfordshire, UK from 1981 to 2004 (Oxford Vascular Study) Lancet. 2004;363:1925–33. doi: 10.1016/S0140-6736(04)16405-2. [DOI] [PubMed] [Google Scholar]
- 3.Brown RD, Whisnant JP, Sicks JD, O'Fallon WM, Wiebers DO. Stroke incidence, prevalence, and survival: secular trends in Rochester, Minnesota, through 1989. Stroke. 1996;27:373–80. [PubMed] [Google Scholar]
- 4.Joutel A, Corpechot C, Ducros A, et al. Notch 3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–10. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 5.Hassan A, Markus HS. Genetics and ischaemic stroke. Brain. 2000;123:1784–812. doi: 10.1093/brain/123.9.1784. [DOI] [PubMed] [Google Scholar]
- 6.Tournier-Lasserve E. New players in the genetics of stroke. N Engl J Med. 2002;347:1711–2. doi: 10.1056/NEJMcibr022035. see comment. [DOI] [PubMed] [Google Scholar]
- 7.Meschia JF. Addressing the heterogeneity of the ischaemic stroke phenotype in human genetics research. Stroke. 2002;33:2770–4. doi: 10.1161/01.str.0000035261.28528.c8. [DOI] [PubMed] [Google Scholar]
- 8.Dichgans M, Markus HS. Genetic association studies in stroke: methodological issues and proposed standard criteria. Stroke. 2005;36:2027–31. doi: 10.1161/01.STR.0000177498.21594.9e. [DOI] [PubMed] [Google Scholar]
- 9.Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischaemic stroke. Stroke. 2004;35:212–27. doi: 10.1161/01.STR.0000107187.84390.AA. [DOI] [PubMed] [Google Scholar]
- 10.Gretarsdottir S, Sveinbjornsdottir S, Jonsson HH, et al. Localization of a susceptibility gene for common forms of stroke to 5q12. Am J Hum Genet. 2002;70:593–603. doi: 10.1086/339252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiklund PG, Brown WM, Brott TG, et al. Lack of aggregation of ischaemic stroke subtypes within affected sibling pairs. Neurology. 2007;68:427–31. doi: 10.1212/01.wnl.0000252955.17126.6a. [DOI] [PubMed] [Google Scholar]
- 12.Adams HP, Bendixen BH, Kappelle LJ, et al. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment Stroke. 1993;24:35–41. doi: 10.1161/01.str.24.1.35. [DOI] [PubMed] [Google Scholar]
- 13.Newton-Cheh C, Hirschhorn J. Genetic association studies of complex traits: design and analysis issues. Mutat Res. 2005;573:54–69. doi: 10.1016/j.mrfmmm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Casas JP, Hingorani AD, Bautista LE, Sharma P. Meta-analysis of genetic studies in ischaemic stroke: thirty-two genes involving approximately 18,000 cases and 58,000 controls. Arch Neurol. 2004;61:1652–61. doi: 10.1001/archneur.61.11.1652. [DOI] [PubMed] [Google Scholar]
- 15.Nikolopoulos GK, Tsantes AE, Bagos PG, Travlou A, Vaiopoulos G. Integrin, alpha 2 gene C807T polymorphism and risk of ischaemic stroke: a meta-analysis. Thromb Res. 2007;119:501–10. doi: 10.1016/j.thromres.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Pereira TV, Rudnicki M, Franco RF, Pereira AC, Krieger JE. Effect of the G-308A polymorphism of the tumor necrosis factor alpha gene on the risk of ischaemic heart disease and ischaemic stroke: a meta-analysis. Am Heart J. 2007;153:821–30. doi: 10.1016/j.ahj.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 17.Tsantes AE, Nikolopoulos GK, Bagos PG, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism and risk of ischaemic stroke: a meta-analysis. Blood Coagul Fibrinolysis. 2007;18:497–504. doi: 10.1097/MBC.0b013e3281ec4eee. [DOI] [PubMed] [Google Scholar]
- 18.Berger K, Stogbauer F, Stoll M, et al. The glu298asp polymorphism in the nitric oxide synthase 3 gene is associated with the risk of ischaemic stroke in two large independent case–control studies. Hum Genet. 2007;121:169–78. doi: 10.1007/s00439-006-0302-2. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Cheng S, Brophy VH, et al. A meta-analysis of candidate gene polymorphisms and ischaemic stroke in 6 study populations: association of lymphotoxin-alpha in nonhypertensive patients. Stroke. 2009;40:683–95. doi: 10.1161/STROKEAHA.108.524587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gretarsdottir S, Sveinbjornsdottir S, Jonsson HH, et al. Localization of a susceptibility gene for common forms of stroke to 5q12. Am J Hum Genet. 2002;70:593–603. doi: 10.1086/339252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gretarsdottir S, Thorleifsson G, Reynisdottir ST, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131–8. doi: 10.1038/ng1245. [DOI] [PubMed] [Google Scholar]
- 22.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Deng C, Bugaj-Gaweda B, et al. Cloning and characterization of novel PDE4D isoforms PDE4D6 and PDE4D7. Cell Signal. 2003;15:883–91. doi: 10.1016/s0898-6568(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 24.Gulcher JR, Gretarsdottir S, Helgadottir A, Stefansson K. Genes contributing to risk for common forms of stroke. Trends Mol Med. 2005;11:217–24. doi: 10.1016/j.molmed.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Meschia JF, Brott TG, Brown RD, Jr, et al. Phosphodiesterase 4D and 5-lipoxygenase activating protein in ischaemic stroke. Ann Neurol. 2005;58:351–61. doi: 10.1002/ana.20585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saleheen D, Bukhari S, Haider SR, et al. Association of phosphodiesterase 4D gene with ischaemic stroke in a Pakistani population. Stroke. 2005;36:2275–7. doi: 10.1161/01.STR.0000182242.59466.ee. [DOI] [PubMed] [Google Scholar]
- 27.Woo D, Kaushal R, Kissela B, et al. Association of phosphodiesterase 4D with ischaemic stroke: a population-based case–control study. Stroke. 2006;37:371–6. doi: 10.1161/01.STR.0000198843.72824.0a. [DOI] [PubMed] [Google Scholar]
- 28.Zee RY, Brophy VH, Cheng S, Hegener HH, Erlich HA, Ridker PM. Polymorphisms of the phosphodiesterase 4D, cAMP-specific (PDE4D) gene and risk of ischaemic stroke: a prospective, nested case-control evaluation. Stroke. 2006;37:2012–7. doi: 10.1161/01.STR.0000230608.56048.38. [DOI] [PubMed] [Google Scholar]
- 29.Staton JM, Sayer MS, Hankey GJ, et al. Association between phosphodiesterase 4D gene and ischaemic stroke. J Neurol Neurosurg Psychiatry. 2006;77:1067–9. doi: 10.1136/jnnp.2006.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohmussaar E, Gschwendtner A, Mueller JC, et al. ALOX5AP gene and the PDE4D gene in a central European population of stroke patients. Stroke. 2005;36:731–6. doi: 10.1161/01.STR.0000157587.59821.87. [DOI] [PubMed] [Google Scholar]
- 31.Rosand J, Bayley N, Rost N, De Bakker PI. Many hypotheses but no replication for the association between PDE4D and stroke. Nat Genet. 2006;38:1091–2. doi: 10.1038/ng1006-1091. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama T, Asai S, Sato N, Soma M. Genotype and haplotype association study of the STRK1 region on 5q12 among Japanese: a case–control study. Stroke. 2006;37:69–76. doi: 10.1161/01.STR.0000194961.17292.33. [DOI] [PubMed] [Google Scholar]
- 33.Kuhlenbaumer G, Berger K, Huge A, et al. Evaluation of single nucleotide polymorphisms in the phosphodiesterase 4D gene (PDE4D) and their association with ischaemic stroke in a large German cohort. J Neurol Neurosurg Psychiatry. 2006;77:521–4. doi: 10.1136/jnnp.2005.073577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brophy VH, Ro SK, Rhees BK, et al. Association of phosphodiesterase 4D polymorphisms with ischaemic stroke in a US population stratified by hypertension status. Stroke. 2006;37:1385–90. doi: 10.1161/01.STR.0000221788.10723.66. [DOI] [PubMed] [Google Scholar]
- 35.Bevan S, Dichgans M, Gschwendtner A, Kuhlenbaumer G, Ringelstein EB, Markus HS. Variation in the PDE4D gene and ischaemic stroke risk: a systematic review and meta-analysis on 5200 cases and 6600 controls. Stroke. 2008;39:1966–71. doi: 10.1161/STROKEAHA.107.509992. [DOI] [PubMed] [Google Scholar]
- 36.Helgadottir A, Manolescu A, Thorleifsson G, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–9. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 37.Helgadottir A, Gretarsdottir S, St Clair D, et al. Association between the gene encoding 5-lipoxygenase-activating protein and stroke replicated in a Scottish population. Am J Hum Genet. 2005;76:505–9. doi: 10.1086/428066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zee RY, Cheng S, Hegener HH, Erlich HA, Ridker PM. Genetic variants of arachidonate 5-lipoxygenase-activating protein, and risk of incident myocardial infarction and ischaemic stroke: a nested case–control approach. Stroke. 2006;37:2007–11. doi: 10.1161/01.STR.0000229905.25080.01. [DOI] [PubMed] [Google Scholar]
- 39.Kostulas K, Gretarsdottir S, Kostulas V, et al. PDE4D and ALOX5AP genetic variants and risk for ischaemic cerebrovascular disease in Sweden. J Neurol Sci. 2007;263:113–7. doi: 10.1016/j.jns.2007.06.042. [DOI] [PubMed] [Google Scholar]
- 40.Zintzaras E, Rodopoulou P, Sakellaridis N. Variants of the arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene and risk of stroke: a HuGE Gene-Disease Association review and meta-analysis. Am J Epidemiol. 2009;169:523–32. doi: 10.1093/aje/kwn368. [DOI] [PubMed] [Google Scholar]
- 41.Nilsson-Ardnor S, Janunger T, Wiklund PG, et al. Genome-wide linkage scan of common stroke in families from northern Sweden. Stroke. 2007;38:34–40. doi: 10.1161/01.STR.0000251643.37454.16. [DOI] [PubMed] [Google Scholar]
- 42.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matarin M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol. 2007;6:414–20. doi: 10.1016/S1474-4422(07)70081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubo M, Hata J, Ninomiya T, et al. A nonsynonymous SNP in PRKCH (protein kinase C eta) increases the risk of cerebral infarction. Nat Genet. 2007;39:212–7. doi: 10.1038/ng1945. [DOI] [PubMed] [Google Scholar]
- 45.Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. N Engl J Med. 2009;360:1718–28. doi: 10.1056/NEJMoa0900094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araki T, Milbrandt J. Ninjurin2, a novel homophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J Neurosci. 2000;20:187–95. doi: 10.1523/JNEUROSCI.20-01-00187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seilheimer B, Schachner M. Studies of adhesion molecules mediating interactions between cells of peripheral nervous system indicate a major role for L1 in mediating sensory neuron growth on Schwann cells in culture. J Cell Biol. 1988;107:341–51. doi: 10.1083/jcb.107.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lemmon V, Farr KL, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 49.Naray-Fejes-Toth A, Snyder PM, Fejes-Toth G. The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na+ transport. Proc Natl Acad Sci USA. 2004;101:17434–9. doi: 10.1073/pnas.0408146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cope G, Golbang A, O'Shaughnessy KM. WNK kinases and the control of blood pressure. Pharmacol Ther. 2005;106:221–31. doi: 10.1016/j.pharmthera.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Zambrowicz BP, Abuin A, Ramirez-Solis R, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA. 2003;100:14109–14. doi: 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newhouse S, Farrall M, Wallace C, et al. Polymorphisms in the WNK1 gene are associated with blood pressure variation and urinary potassium excretion. PLoS ONE. 2009;4:e5003. doi: 10.1371/journal.pone.0005003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feinberg WM, Albers GW, Barnett HJ, et al. Guidelines for the management of transient ischaemic attacks. From the Ad Hoc Committee on Guidelines for the Management of Transient Ischaemic Attacks of the Stroke Council of the American Heart Association. Circulation. 1994;89:2950–65. doi: 10.1161/01.cir.89.6.2950. [DOI] [PubMed] [Google Scholar]
- 54.Rietbrock S, Heeley E, Plumb J, Van Staa T. Chronic atrial fibrillation: incidence, prevalence, and prediction of stroke using the Congestive heart failure, Hypertension, Age >75, Diabetes mellitus, and prior Stroke or transient ischaemic attack (CHADS2) risk stratification scheme. Am Heart J. 2008;156:57–64. doi: 10.1016/j.ahj.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med. 2007;146:857–67. doi: 10.7326/0003-4819-146-12-200706190-00007. [DOI] [PubMed] [Google Scholar]
- 56.Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–7. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 57.Gretarsdottir S, Gudmar T, Manolescu A, et al. Risk variants for atrial fibrillation on 4q25 associate with ischaemic stroke. Ann Neurol. 2008;64:402–9. doi: 10.1002/ana.21480. [DOI] [PubMed] [Google Scholar]
- 58.Meschia JF. Decoding cryptogenic cardioembolism. Ann Neurol. 2008;64:364–6. doi: 10.1002/ana.21470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jabaudon D, Sztajzel J, Sievert K, Landis T, Sztajzel R. Usefulness of ambulatory 7-day ECG monitoring for the detection of atrial fibrillation and flutter after acute stroke and transient ischaemic attack. Stroke. 2004;35:1647–51. doi: 10.1161/01.STR.0000131269.69502.d9. [DOI] [PubMed] [Google Scholar]
- 60.Douen AG, Pageau N, Medic S. Serial electrocardiographic assessments significantly improve detection of atrial fibrillation 2.6-fold in patients with acute stroke. Stroke. 2008;39:480–2. doi: 10.1161/STROKEAHA.107.492595. [DOI] [PubMed] [Google Scholar]
- 61.Mohr JP, Thompson JL, Lazar RM, et al. A comparison of warfarin and aspirin for the prevention of recurrent ischaemic stroke. N Engl J Med. 2001;345:1444–51. doi: 10.1056/NEJMoa011258. [DOI] [PubMed] [Google Scholar]
- 62.A randomized trial of anticoagulants versus aspirin after cerebral ischemia of presumed arterial origin. The Stroke Prevention in Reversible Ischemia Trial (SPIRIT) Study Group. Ann Neurol. 1997;42:857–65. doi: 10.1002/ana.410420606. [DOI] [PubMed] [Google Scholar]
- 63.McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–3. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 66.Schunkert H, Gotz A, Braund P, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation. 2008;117:1675–84. doi: 10.1161/CIRCULATIONAHA.107.730614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matarin M, Brown WM, Singleton A, Hardy JA, Meschia JF. Whole genome analyses suggest ischaemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke. 2008;39:1586–9. doi: 10.1161/STROKEAHA.107.502963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gschwendtner A, Bevan S, Cole JW, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol. 2009;65:531–9. doi: 10.1002/ana.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 70.Matarin M, Simon-Sanchez J, Fung HC, et al. Structural genomic variation in ischaemic stroke. Neurogenetics. 2008;9:101–8. doi: 10.1007/s10048-008-0119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009;360:354–62. doi: 10.1056/NEJMoa0809171. see comment. [DOI] [PubMed] [Google Scholar]
- 73.Simon T, Verstuyft C, Mary-Krause M, et al. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360:363–75. doi: 10.1056/NEJMoa0808227. see comment. [DOI] [PubMed] [Google Scholar]
- 74.Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet. 2009;373:309–17. doi: 10.1016/S0140-6736(08)61845-0. see comment. [DOI] [PubMed] [Google Scholar]
- 75.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–9. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- 76.Group SC. Link E, Parish S, et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med. 2008;359:789–99. doi: 10.1056/NEJMoa0801936. see comment. [DOI] [PubMed] [Google Scholar]
- 77.Millican EA, Lenzini PA, Milligan PE, et al. Genetic-based dosing in orthopedic patients beginning warfarin therapy. Blood. 2007;110:1511–5. doi: 10.1182/blood-2007-01-069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005;352:2285–93. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- 79.Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287:1690–8. doi: 10.1001/jama.287.13.1690. [DOI] [PubMed] [Google Scholar]
- 80.Schelleman H, Chen J, Chen Z, et al. Dosing algorithms to predict warfarin maintenance dose in Caucasians and African Americans. Clin Pharmacol Ther. 2008;84:332–9. doi: 10.1038/clpt.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cooper GM, Johnson JA, Langaee TY, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112:1022–7. doi: 10.1182/blood-2008-01-134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med. 2008;358:999–1008. doi: 10.1056/NEJMoa0708078. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.King CR, Porche-Sorbet RM, Gage BF, et al. Performance of commercial platforms for rapid genotyping of polymorphisms affecting warfarin dose. Am J Clin Pathol. 2008;129:876–83. doi: 10.1309/1E34UAPR06PJ6HML. [DOI] [PubMed] [Google Scholar]
- 84.Anderson JL, Horne BD, Stevens SM, et al. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation. 2007;116:2563–70. doi: 10.1161/CIRCULATIONAHA.107.737312. see comment. [DOI] [PubMed] [Google Scholar]
- 85.Wang D, Sadee W. Searching for polymorphisms that affect gene expression and mRNA processing: example ABCB1 (MDR1) AAPS J. 2006;8:E515–20. doi: 10.1208/aapsj080361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dewberry R, Holden H, Crossman D, et al. Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2394–400. doi: 10.1161/01.atv.20.11.2394. [DOI] [PubMed] [Google Scholar]
- 87.Francis SE, Camp NJ, Dewberry RM, et al. Interleukin-1 receptor antagonist gene polymorphism and coronary artery disease. Circulation. 1999;99:861–6. doi: 10.1161/01.cir.99.7.861. [DOI] [PubMed] [Google Scholar]
- 88.Worrall BB, Azhar S, Nyquist PA, et al. Interleukin-1 receptor antagonist gene polymorphisms in carotid atherosclerosis. Stroke. 2003;34:790–3. doi: 10.1161/01.STR.0000057815.79289.EC. [DOI] [PubMed] [Google Scholar]
- 89.Bittner M, Meltzer P, Chen Y, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–40. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 90.Jones MH, Virtanen C, Honjoh D, et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet. 2004;363:775–81. doi: 10.1016/S0140-6736(04)15693-6. [DOI] [PubMed] [Google Scholar]
- 91.Sims A, Bartlett J. Approaches towards expression profiling the response to treatment. Breast Cancer Res. 2008;10:115. doi: 10.1186/bcr2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang JC, Wooten EC, Tsimelzon A, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–9. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- 93.Moore DF, Li H, Jeffries N, et al. Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischaemic stroke: a pilot investigation. Circulation. 2005;111:212–21. doi: 10.1161/01.CIR.0000152105.79665.C6. [DOI] [PubMed] [Google Scholar]
- 94.Tang Y, Xu H, Du X, et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischaemic stroke in humans: a microarray study. J Cereb Blood Flow Metab. 2006;26:1089–102. doi: 10.1038/sj.jcbfm.9600264. [DOI] [PubMed] [Google Scholar]
- 95.Grond-Ginsbach C, Hummel M, Wiest T, et al. Gene expression in human peripheral blood mononuclear cells upon acute ischaemic stroke. J Neurol. 2008;255:723–31. doi: 10.1007/s00415-008-0784-z. [DOI] [PubMed] [Google Scholar]
- 96.Xu H, Tang Y, Liu DZ, et al. Gene expression in peripheral blood differs after cardioembolic compared with large-vessel atherosclerotic stroke: biomarkers for the etiology of ischaemic stroke. J Cereb Blood Flow Metab. 2008;28:1320–8. doi: 10.1038/jcbfm.2008.22. [DOI] [PubMed] [Google Scholar]
- 97.Myers AJ, Gibbs JR, Webster JA, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–9. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 98.Vikman P, Edvinsson L. Gene expression profiling in the human middle cerebral artery after cerebral ischemia. Eur J Neurol. 2006;13:1324–32. doi: 10.1111/j.1468-1331.2006.01496.x. [DOI] [PubMed] [Google Scholar]
- 99.Mitsios N, Saka M, Krupinski J, et al. A microarray study of gene and protein regulation in human and rat brain following middle cerebral artery occlusion. Arterioscler Thromb Vasc Biol. 2007;8:93. doi: 10.1186/1471-2202-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Solberg LA, McGarry PA, Moossy J, et al. Distribution of cerebral atherosclerosis by geographic location, race, and sex. Lab Invest. 1968;18:604–12. [PubMed] [Google Scholar]
- 101.McGarry P, Solberg LA, Guzman MA, Strong JP. Cerebral atherosclerosis in New Orleans. Comparisons of lesions by age, sex, and race. Lab Invest. 1985;52:533–9. [PubMed] [Google Scholar]
- 102.Chimowitz M, Lynn M, Howlett-Smith H, et al. Warfarin-Aspirin Symptomatic Intracranial Disease (WASID) Trial: final results. Stroke. 2004;35:235. Abstract. [Google Scholar]
- 103.Battacharji S, Hutchinson E, McCall A. The circle of Willis: the incidence of developmental abnormalities in normal and infracted brains. Brain. 1967;90:747–58. doi: 10.1093/brain/90.4.747. [DOI] [PubMed] [Google Scholar]
- 104.Ringelstein EB, Weiller C, Weckesser M, Weckesser S. Cerebral vasomotor reactivity is significantly reduced in low-flow as compared to thromboembolic infarctions: the key role of the circle of Willis. J Neurol Sci. 1994;121:103–9. doi: 10.1016/0022-510x(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 105.Schomer DF, Marks MP, Steinberg GK, et al. The anatomy of the posterior communicating artery as a risk factor for ischaemic cerebral infarction. N Engl J Med. 1994;330:1565–70. doi: 10.1056/NEJM199406023302204. see comment. [DOI] [PubMed] [Google Scholar]
- 106.Hedera P, Bujdakova J, Traubner P. Effect of collateral flow patterns on outcome of carotid occlusion. Eur Neurol. 1995;35:212–6. doi: 10.1159/000117130. [DOI] [PubMed] [Google Scholar]
- 107.Henderson RD, Eliasziw M, Fox AJ, Rothwell PM, Barnett HJ. Angiographically defined collateral circulation and risk of stroke in patients with severe carotid artery stenosis. North American Symptomatic Carotid Endarterectomy Trial (NASCET) Group. Stroke. 2000;31:128–32. doi: 10.1161/01.str.31.1.128. [DOI] [PubMed] [Google Scholar]
- 108.Vernieri F, Pasqualetti P, Matteis M, et al. Effect of collateral blood flow and cerebral vasomotor reactivity on the outcome of carotid artery occlusion. Stroke. 2001;32:1552–8. doi: 10.1161/01.str.32.7.1552. [DOI] [PubMed] [Google Scholar]
- 109.Silvestrini M, Vernieri F, Troisi E, et al. Cerebrovascular reactivity in carotid artery occlusion: possible implications for surgical management of selected groups of patients. Acta Neurol Scand. 1999;99:187–91. doi: 10.1111/j.1600-0404.1999.tb07342.x. [DOI] [PubMed] [Google Scholar]
- 110.Hoksbergen AW, Legemate DA, Csiba L, Csati G, Siro P, Fulesdi B. Absent collateral function of the circle of Willis as risk factor for ischaemic stroke. Cerebrovasc Dis. 2003;16:191–8. doi: 10.1159/000071115. see comment. [DOI] [PubMed] [Google Scholar]
- 111.Abboud S, Karhunen PJ, Lutjohann D, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) gene is a risk factor of large-vessel atherosclerosis stroke. PLoS ONE. 2007;2:e1043. doi: 10.1371/journal.pone.0001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nica AC, Dermitzakis ET. Using gene expression to investigate the genetic basis of complex disorders. Hum Mol Genet. 2008;17:R129–34. doi: 10.1093/hmg/ddn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stranger BE, Nica AC, Forrest MS, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–24. doi: 10.1038/ng2142. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–9. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dixon AL, Liang L, Moffatt MF, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–7. doi: 10.1038/ng2109. see comment. [DOI] [PubMed] [Google Scholar]
- 116.Goring HH, Curran JE, Johnson MP, et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet. 2007;39:1208–16. doi: 10.1038/ng2119. see comment. [DOI] [PubMed] [Google Scholar]