

Abstract
Context
Pyruvate dehydrogenase complex (PDC) deficiency is a genetic mitochondrial disorder commonly associated with lactic acidosis, progressive neurological and neuromuscular degeneration and, usually, death during childhood. There has been no recent comprehensive analysis of the natural history and clinical course of this disease.
Objective
We reviewed 371 cases of PDC deficiency, published between 1970 and 2010, that involved defects in subunits E1α and E1β and components E1, E2, E3 and the E3 binding protein of the complex.
Data sources and extraction
English language peer-reviewed publications were identified, primarily by using PubMed and Google Scholar search engines.
Results
Neurodevelopmental delay and hypotonia were the commonest clinical signs of PDC deficiency. Structural brain abnormalities frequently included ventriculomegaly, dysgenesis of the corpus callosum and neuroimaging findings typical of Leigh syndrome. Neither gender nor any clinical or neuroimaging feature differentiated the various biochemical etiologies of the disease. Patients who died were younger, presented clinically earlier and had higher blood lactate levels and lower residual enzyme activities than subjects who were still alive at the time of reporting. Survival bore no relationship to the underlying biochemical or genetic abnormality or to gender.
Conclusions
Although the clinical spectrum of PDC deficiency is broad, the dominant clinical phenotype includes presentation during the first year of life; neurological and neuromuscular degeneration; structural lesions revealed by neuroimaging; lactic acidosis and a blood lactate:pyruvate ratio≤20.
Keywords: Congenital lactic acidosis, Dichloroacetate, Neuroimaging, Ketogenic diet, Pyruvate dehydrogenase complex, Thiamine
1. Introduction
The mitochondrial pyruvate dehydrogenase complex (PDC) catalyzes the rate-limiting step in the aerobic glucose oxidation and is thus integral to cellular energetics [1,2] (Fig. 1). It comprises multiple copies of three enzymatic subunits: pyruvate dehydrogenase (E1), dihydrolipoamide transacetylase (E2) and dihydrolipoamide dehydrogenase (E3), as well as an E3 binding protein (BP). The E1 component is a heterotetramer of 2 alpha and 2 beta subunits. The gene for the E1α subunit is located on the X chromosome and all proteins of the complex are nuclear encoded. Rapid regulation of the complex is regulated mainly by reversible phosphorylation of the E1α subunit [3] that is mediated by a family of PDC kinases (PDK) and phosphatases (PDP) [4,5].
Fig. 1.
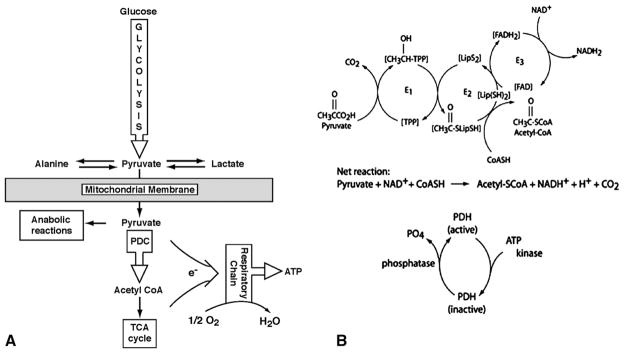
A. Central role of PDC in cellular energetics. B. PDC multi-enzyme complex. Abbreviations: TCA, tricarboxylic acid; e−, electrons; E1, pyruvate decarboxylase; E2, dihydrolipoamide transactylase; E3, dihydrolipoamide dehydrogenase; TPP, thiamine pyrophosphate; Lip, lipoate (reduced and oxidized).
Although over 40 years have elapsed since the first description of a congenital deficiency of the PDC [6], the incidence and prevalence of this life-threatening condition remain unknown. Several excellent earlier reviews exist of the clinical and biochemical characteristics or of the molecular genetic etiologies of PDC deficiency [2,7–9]. However, there has been no recent comprehensive analysis of the natural history of the disease, nor attempts to discern phenotypic differences or predictable outcomes based on biochemical defects or mutations in specific components of the complex. Here we summarize the clinical, biochemical and genetic findings contained in published reports and personal experience of 371 individual cases of PDC deficiency. We sought associations among various pathological indices that could provide new insight into the pathobiology and clinical course of this disease.
2. Material and methods
2.1. Source material
We reviewed all English language publications under “pyruvate dehydrogenase deficiency” and deficiency of each individual subunit and component of the complex listed in PubMed and Google Scholar from 1970 through December, 2010 [6,10–163]. Additional publications were found by reviewing the references included in articles not identified by the search engines. We also included information on a previously unpublished case of PDC E1α deficiency.
2.2. Definition of PDC deficiency
To be included a report had to provide a quantitative assessment of overall PDC activity and/or of the activities of its component parts. In most cases, PDC specific activity was determined by measuring the decarboxylation of 1-14C-pyruvate to 14CO2 and was expressed as a unit of 14CO2 production per tissue mass per unit of time. No case was included unless a “normal” control mean value or range of control values was given. For cases in which only the range of normal values was provided, we used the minimum value of the range. For cases in which the values for enzyme activity were listed for more than one cell type, the default choice was: fresh or frozen skeletal muscle, then primary cultures of skin fibroblasts, then freshly isolated lymphocytes or transformed lymphoblasts, then other tissues (e.g., liver). This order was adopted because skeletal muscle is highly dependent upon mitochondrial oxidative metabolism and is thus a target tissue in PDC deficiency and because a skin biopsy was the most common source of cells used in making the biochemical diagnosis. If serial measurements were made in different tissue specimens, we used the lowest reported value, applying the same default strategy described above. If a publication described a patient with a clinical presentation consistent with PDC deficiency (e.g., psychomotor retardation, hypotonia, neonatal lactic acidosis) who was found to have a pathological mutation in a subunit or component enzyme of the PDC or who had a first degree relative who possessed a pathological mutation involving the complex, we accepted the patient as having PDC deficiency, even without confirmation by standard enzymological criteria. Thus, the total number of PDC cases exceeds the number of cases in which a measureable enzymological defect was obtained.
2.3. Clinical features
Age of clinical disease onset was defined as the earliest reported clinical or biochemical sign or symptom consistent with a genetic mitochondrial disease. Quantification of intellectual, neuromuscular or peripheral nerve function or other clinical signs was seldom reported; therefore, we included them only as descriptive variables.
2.4. Metabolites
We combined data on whole blood, serum and plasma concentrations for lactate and pyruvate and chose the highest value reported for an individual case. We also used the highest reported values for cerebrospinal fluid (CSF) lactate and pyruvate. Circulating levels of alanine, which is in equilibrium with pyruvate and lactate (Fig. 1A), and other metabolites were frequently described as “elevated” but often were not quantified; consequently, we expressed the overall frequency of reported abnormalities in these molecules descriptively.
2.5. Neuroimaging
Findings derived from computerized tomography or magnetic resonance imaging of the brain, with or without the use of contrast material, were included in this category. “Leigh syndrome” (subacute necrotizing encephalomyopathy) was applied to cases in which patients had neuroimaging findings that included focal, bilaterally symmetric lesions in one or more parts of the basal ganglia, thalamus and brainstem.
2.6. Statistical analysis
Although this paper is mainly descriptive, we did conduct a number of comparisons. Qualitative associations were conducted by Fisher’s exact test. Comparisons between two groups for quantitative measures were conducted by the Satterthwaite t-test, which corrects for unequal population standard deviations. Associations between quantitative variables were conducted by the Pearson’s Correlation Coefficient. Readers are cautioned that, because a large number of statistical tests were conducted, some associations may be spurious.
3. Results
We found 159 full-length, peer-reviewed publications of 392 case reports of patients with PDC deficiency from the first reported case in 1970 [6]. A few papers reported complimentary details of the same patient [6,21,28,34,36,37,41,43,45,62,65,71,73,76,79,80,92,93,99,101,108, 110,124]. Twenty-one cases were omitted from our analysis, owing to lack of explicit enzymological or molecular genetic confirmation of disease [10–20]. Hence, this review summarizes the findings of 371 patients [6,12,21–163] (see Suppl. Table 1).
3.1. Clinical signs
Table 1 summarizes the most frequently reported clinical signs of PDC deficiency. We combined 211 cases in which patients were described as having “developmental delay”, “mental retardation” or “psychomotor retardation” because of the ambiguity of these terms and the considerable overlap in their application to patients. This category was the most frequently reported clinical sign and was present in 57% of cases. Hypotonia and/or hypertonia (169 cases) were the next most frequently reported signs; most of these patients were hypotonic, whereas hypertonia was reported in a few instances. Ataxia was reported in 72 patients and was more common in males (p=0.012). Other frequently reported signs of motor dysfunction included spasticity, ptosis, and choreoathetoid movements. Reports of seizure activity frequently did not discriminate between focal and generalized convulsions, so this category includes both presentations. Microcephaly was usually reported without reference to a measured head circumference or in relation to a normative value. Respiratory distress typically occurred in the setting of acute acid–base decompensation and lactic acidemia, particularly in the neonatal period. Facial dysmorphism was reported to affect girls more frequently than boys and included such features as narrow head, frontal bossing, prominent philtrum and wide nasal bridge. Ocular manifestations of disease included optic atrophy, nystagmus and strabismus. The term “peripheral neuropathy” was used descriptively in most cases, even when formal nerve conduction testing was apparently undertaken during a patient’s evaluation, and was reported more frequently in males (p=0.004).
Table 1.
Clinical signs associated with PDC deficiency.
| Sign | No. of cases (% of total) | % Males |
|---|---|---|
| DD/MR/PMR | 211 (57) | 45 |
| Hypotonia/hypertonia | 169 (46) | 51 |
| Seizures | 95 (26) | 47 |
| Microcephaly | 83 (22) | 23 |
| Ataxia | 72 (19) | 78 |
| Respiratory distress | 51 (14) | 59 |
| Facial dysmorphism | 44 (11) | 34 |
| Spasticity | 38 (10) | 34 |
| Peripheral neuropathy | 26 (7) | 88 |
| Optic atrophy | 15 (4) | 60 |
| Nystagmus | 11 (3) | 64 |
| Ptosis | 9 (2.5) | 89 |
| Choreoathetoid movements | 8 (2) | 88 |
| Strabismus | 6 (1.6) | 100 |
3.2. Biochemical features
Among all patients, 326 (89%) reports provided enzymological data and, in most cases, additional biochemical investigation that identified the affected subunit (Table 2). The mean±SD residual enzyme activity among cases was 37.0±34.6% of the normative data recorded in these reports. Most patients with PDC deficiency presented with clinical signs consistent with the disease (Table 1) before 1 year of age, regardless of the affected subunit or component. Seventy-six percent of patients had PDC deficiency due to a deficit in the α subunit of E1. Males slightly outnumbered females and consanguinity accounted for about 7% of all cases. A majority of patients with a deficiency of E1β [132,151,157], E2 [136] or E3BP [12,58,78,126,128,140,143,171] were products of consanguinity, in which the disease often occurred in more than one family member.
Table 2.
Enzymological classification and age of clinical presentation in PDC deficiency.
| Patients (%) | Males (%) | Consanguinity (%) | PDC activity (% of nl) | Age of clinical onset (mos) | |
|---|---|---|---|---|---|
| Total | 371 (100) | 52.1 | 6.8 | 37±34.6 | 8.0±15.8 |
| E1 | 65 (17.5) | 58.4 | 0 | 38.8±25.9 | 7.3±15.7 |
| E1α | 210 (56.6) | 50.4 | 1 | 41.7±39.2 | 8.9±17.3 |
| E1β | 9 (2.4) | 55.6 | 55.6 | 24.2±22.2 | 3.1±6.2 |
| E2 | 4 (1.1) | 50 | 66.7 | 48.5±16 | 9.0±8.6 |
| E3 | 20 (5.1) | 66.7 | 13.3 | 48.5±21.2 | 5.9±9.5 |
| E3BP | 23 (6.2) | 60.8 | 60.8 | 18.1±13.2 | 5±10.2 |
| Unclassified | 40 (10.8) | 46.9 | 0 | 42.3±30.2 | 9.3±16.6 |
Cases included as E1 deficiency were those in which a defect in the alpha or beta subunit was not described.
Table 3 summarizes the maximum reported lactate and pyruvate concentrations in blood and CSF and the blood lactate:pyruvate ratio. In some cases, only single values were reported; in others, serial blood levels were documented, although the time sequences varied greatly. Blood lactate was the most frequently reported value. The blood lactate: pyruvate ratio was calculated for those cases in which these measurements were obtained simultaneously. Virtually no temporal associations were described between measurements of blood and CSF chemistries; thus, we made no associations between blood and CSF lactate or pyruvate levels. These values were similar between genders and among the various biochemical diagnostic categories.
Table 3.
Blood and CSF lactate and pyruvate concentrations in patients with PDC deficiency.
| Blood lactate (mmol/L) | Blood pyruvate (mmol/L) | Blood L:P ratio | CSF lactate (mmol/L) | CSF pyruvate (mmol/L) | |
|---|---|---|---|---|---|
| Total | 7.8±5.6 (216) | 0.76±2.3 (118) | 19.6±22 (45) | 6.1±3.04 (123) | 0.47±0.27 (43) |
| E1 | 9.9±8.2 (16) | 0.67±0.99 (15) | 18.6±7.2 (5) | 5.90±4.5 (9) | 0.57±0.4 (4) |
| E1α | 7.02±5.8 (115) | 0.56±0.83 (66) | 20.1±25.6 (32) | 6.07±3 (74) | 0.48±0.27 (26) |
| E1β | 11.8±6.6 (8) | 20.2±0.28 (2) | |||
| E2 | 2.58±2.1 (3) | 0.137 (1) | 1.4±0.39 (2) | ||
| E3 | 11.3±4.6 (15) | 4.37±8.6 (7) | 3.01±1.47 (4) | ||
| E3BP | 10.3±6.9 (21) | 0.56±0.43 (10) | 13.4±4.7 (5) | 6.03±2.3 (6) | 0.53±0.06 (2) |
| Unclassified | 6.0±2.6 (38) | 0.37±0.19 (19) | 17.6 (1) | 6.9±2.8 (28) | 0.40±0.26 (11) |
3.3. Neuroimaging findings
One hundred eighty-six patients had one or more imaging studies of the brain (Fig. 2). Seven subjects were reported to have normal imaging findings; all had E1α deficiency [80,110,125,137,145,147,156,163]. Ventriculomegaly was the most frequent abnormality, affecting 65 patients (35%) in whom an abnormal brain imaging finding was reported. Leigh syndrome was described in 50 patients (27%); autopsy confirmation of this condition was reported very rarely. The basal ganglia, brain stem and cerebellum are anatomical areas frequently reported in association with Leigh syndrome and were identified as isolated abnormalities in some patients. Hypogenesis or agenesis of the corpus callosum was reported in 57 patients (31%) and in slightly over one-third of E1α deficient individuals.
Fig. 2.

Brain imaging findings in 186 patients with PDC deficiency. Abbreviation: BS, brain stem.
3.4. Molecular genetics
There were 237 cases (66%) that reported a mutation in a PDC component gene (Supplemental Table 1). Mutational data are listed chronologically and in relation to patient gender, residual PDC enzyme activity and age of clinical onset of disease. Missense mutations were more prevalent than frameshift mutations and occurred more often in males; most females harbored frameshift mutations (Table 4). Although patients possessing missense mutations were most likely to present clinically later in life (7.73±11.37 months vs. 3.60±7.64 months for frameshift mutations), there were no significant differences between these mutational classes in residual enzyme activity, maximum blood lactate concentration or survival at the time of case reporting. Not surprisingly, most mutations (84%) were located in the PDHA1 gene. Among E1α deficient individuals, most missense mutations localized to exons 1–9 (77%), whereas most frameshift mutations localized to exons 10 and 11 (86.5%). Associations among PDHA1 missense mutations, the amino acid sequence of the E1α subunit and residual PDC enzyme activities are described in the Supplemental Text and Supplemental Figs. 1–5.
Table 4.
Mutational classification in relation to various clinical indices.
| Mutation | % Males | % Females | Enzyme activity | Age of clinical onset | Max. blood lactate | % Alive | % Deceased |
|---|---|---|---|---|---|---|---|
| Missense | 62.7 (96) | 36.3 (57) | 37.8±35.20 (142) | 7.78±12.90 (111) | 7.81±5.70 (95) | 65.6 (86) | 34.3 (45) |
| Frameshift | 38.6 (27) | 61.4 (43) | 32.7±29.30 (64) | 3.60±7.70 (47) | 7.41±4.20 (35) | 63.5 (33) | 36.5 (19) |
The number of cases is in parentheses.
3.5. Clinical outcome
We examined the cross-sectional relationship between PDC deficiency, based on biochemical diagnosis, and survival (Fig. 3). There were 294 cases (79% of total) in which the age of the patient at death was reported or in which the patient was said to be alive at the time the case was described. Table 5 shows that 108 patients (36.6% of total) died at the time of reporting, with a mean age at death of 2.70±4.66 years. The remaining 187 patients were alive at the time of reporting, with a mean age of 7.10±6.15 years. There were 243 patients alive at 0.5 years of age, but only 10 patients were alive at age 20 years (Fig. 3). These statistics were closely matched by the data from the E1 and E1α patient subgroups. The only other category in which similar results could be compared was the group of E3BP deficient subjects. However, as noted in Table 2, over 60% of these patients were from consanguineous relationships. Patients who died before case reporting were 8 months younger at the time of clinical presentation, compared to those who were alive at the time of reporting (2.9±6.56 months vs. 11.3±19.8 months; p=0.001). Deceased patients also had higher maximum blood lactate concentrations (11.1±7.3 mmol/L vs. 6.2±3.8 mmol/L; p<0.001) and lower residual PDC activity (26.6±23.6% vs. 37.7±33.4%; p<0.002). However, these three variables were only weakly correlated with each other (range of r: 0.235 to 0.281).
Fig. 3.
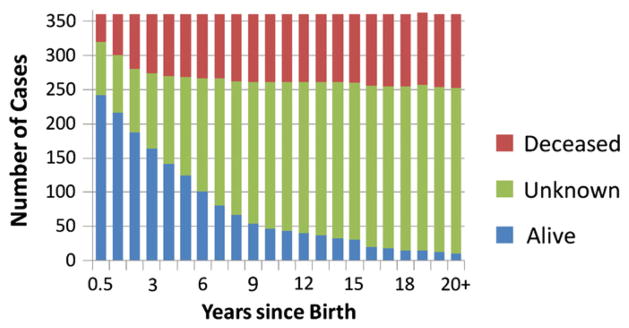
Outcome of 371 patients with PDC deficiency. There were 294 patients in whom death or survival was documented at the time of reporting and 77 patients whose outcome was unknown at the time of reporting.
Table 5.
Outcome of 294 patients with PDC deficiency.
| Subunit (no. of cases) | Deceased |
Alive |
||||
|---|---|---|---|---|---|---|
| Percent | Mean age in years | Range in years | Percent | Mean age in years | Range in years | |
| Total (294) | 36.2 (108) | 2.71±4.53 (107) | 0.1–35 | 63.8 (186) | 7.1±6.11 (167) | 0–21 |
| E1 (57) | 42.1 (24) | 0.98±1.69 (24) | 0–8 | 57.9 (33) | 6.65±5.17 (31) | 0.1–22 |
| E1α (163) | 36.8 (60) | 3.22±5.22 (60) | 0–21 | 63.2 (103) | 7.5±6.43 (92) | 0.2–35 |
| E1β (9) | 44.4 (4) | 0.65±0.48 (4) | 0.1–1.1 | 55.5 (5) | 9.14±10.3 (5) | 0.3–27 |
| E2 (4) | 0 | 100 (4) | 7.5±4.77 (3) | 3.5–11 | ||
| E3 (13) | 50.0 (9) | 2.40±2.16 (8) | 0.3–5.5 | 50.0 (9) | 4.01±2.86 (8) | 0.8–5 |
| E3BP (22) | 22.7 (5) | 4.88±6.67 (5) | 0.1–16 | 77.2 (17) | 7.8±7.08 (17) | 0.9–24 |
| Unclassified (21) | 28.6 (6) | 4.5±6.58 (6) | 0–16 | 71.4 (15) | 5.78±4.72 (14) | 0.8–15 |
Because E1α deficient patients represented the largest single subgroup in PDC deficiency, we undertook a similar cross-sectional comparison of age versus outcome in those E1α deficient patients in whom a mutation in the PDHA1 gene was identified (Fig. 4). Of 187 patients with PDHA1 mutations, the most frequently reported were at amino acid positions 302 (15 cases), 263 (15 cases) and 378 (14 cases); 35 were missense mutations. Note that a mutation affecting amino acid position 378 may be particularly lethal. Of those individuals with a mutation affecting this position, seven were dead within the first 2 years of life. In contrast, patients with an amino acid substitution at either position 263 or 302 appeared to have a less truncated life span. Males accounted for 29 (83%) of these common mutations.
Fig. 4.
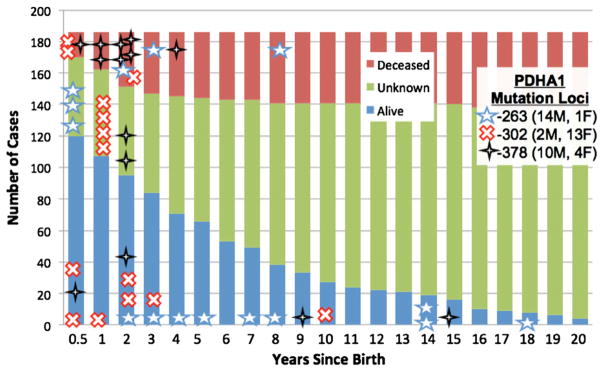
Outcome of 187 patients with PDC deficiency due to an identified biochemical and/or molecular genetic defect in E1α.
We next examined the effect of gender on the outcome of all cases with PDC deficiency or E1α deficiency (Table 6). The proportion of males who died was greater than that of females in both groups. Unexpectedly, we also found that females who died presented earlier in life and died at a younger age than males, yet had higher residual enzyme activity. The reason for this apparent discrepancy is unclear.
Table 6.
Gender differences in clinical presentation and course.
| Gender | Number of deceased (%) | Age of onset (mos) | Age of death (years) | PDC activity (% nl) |
|---|---|---|---|---|
| Male | 62 (64)/37 (64) | 3.5/4.1 | 3.2/3.8 | 23.5/26.1 |
| Female | 38 (36)/21 (36) | 1.1/0.9 | 2.1/2.4 | 32.7/41 |
Standard font represents total number of cases, while italicized text represents E1α cases.
3.6. Other associations
We found no statistically significant associations between genders regarding the following variables: survival outcome at the time of case reporting; mental retardation/developmental delay/psychomotor retardation; hypotonia/hypertonia; microcephaly; facial dysmorphism; corpus callosum hypogenesis or agenesis; Leigh syndrome; basal ganglia involvement; or brainstem and cerebellar involvement. There was also no significant difference between the individual enzyme component deficiencies and the categories above and no significant association between corpus callosum agenesis or hypogenesis and Leigh syndrome, or between microcephaly and ventriculomegaly.
3.7. Treatment
Finally, we sought information relating to the clinical management of PDC deficiency. Alkali, usually as sodium bicarbonate, was administered often but sporadically, usually as a temporary treatment for acute acid–base decompensation. The most common interventions used chronically were thiamine (vitamin B1; 73 cases), a ketogenic diet, comprising at least 65% of total calories (19 cases), and dichloroacetate (DCA; 15 cases). Thiamine doses varied, from a few milligrams per day to doses exceeding 1 g/day. These latter cases typically involved patients in whom PDC deficiency was considered due to a mutation affecting the interaction of thiamine pyrophosphate with the E1α subunit. The frequency with which ketogenic diets were employed may be underrepresented because many reports failed to specify the proportion of fat calories. Some patients received intravenous DCA for acute treatment of lactic acidosis but the majority of patients received DCA for chronic oral treatment at doses usually of 25 mg/kg/d or more. No intervention for PDC deficiency has been evaluated in a randomized controlled trial.
4. Discussion
PDC deficiency has been considered one of the most common biochemically proven causes of congenital lactic acidosis [2]. Our review of 371 cases of biochemically and/or genetically established PDC deficiency supports this notion. Regardless of which subunit or component of the complex is defective, most patients present within the first few months of life with both clinical and biochemical evidence of disease. The dominant presenting phenotype of PDC deficiency includes impairment of neurological and motor function; structural abnormalities of the brain, particularly ventriculomegaly, collossal dysgenesis and findings consistent with Leigh syndrome; hyperlactatemia; and a circulating lactate:pyruvate ratio≤20. Nevertheless, the clinical spectrum of PDC deficiency is broad, ranging from neonatal death with overwhelming lactic acidosis to a relatively benign course early in life with few overt signs.
Inhibition of PDC activity impairs the mitochondrial oxidation of pyruvate and promotes its cytoplasmic reduction to lactate or transamination to alanine. Diminished flux through the PDC also decreases the availability of acetyl CoA for the TCA cycle. In turn, reduction of both PDC and TCA cycle activities decreases the generation of reducing equivalents (as NADH and FADH2) that donate electrons to the respiratory chain to complete the process of oxidative phosphorylation. Consequently, potentially any congenital or acquired defect in any PDC component may give rise to lactic acidosis and to cellular energy failure, the latter most commonly expressed as progressive neurological and neuromuscular deterioration. Thus, it is not surprising that we found no clinical, neuroimaging or biochemical signs that distinguish patients with the most commonly reported E1α deficiency from patients with defects in E1β, E2, E3 or the E3BP of the complex. In general, those who died were younger, had earlier clinical onset and had lower PDC activity.
The natural history of PDC deficiency appears generally very similar between genders. However, boys may die proportionately more often than girls, yet at an older age, a phenomenon that may reflect the impact of lyonization of the PDHA1 allele. Although significant gender differences were observed for ataxia and peripheral neuropathy, we caution against overinterpreting these findings. Ataxia has been reported in response to carbohydrate feeding in a subset of males who exhibited an otherwise mild phenotype during the first 2 decades of life [57,111]. However, gross motor abnormalities occur in affected patients of both sexes (Table 1) and it is difficult to rationalize why one of these features should necessarily afflict one gender more than another. Electrical evidence of peripheral neuropathy, when sought, is a very common finding in patients with genetic mitochondrial diseases, including PDC deficiency [164–168], as would be expected for a tissue reliant mainly on oxidative metabolism for energy. Most of the published reports in our series that described the presence of peripheral neuropathy provided little or no objective documentation of sensory or motor nerve dysfunction and subtle abnormalities probably were either not sought or missed in many other patients. Thus, we tentatively conclude that the apparent gender discrepancy for this clinical sign is probably spurious and will not be confirmed by future studies in which formal peripheral nerve conduction testing is undertaken.
Facial dysmorphism is both an uncommon and curious finding in PDC deficiency, affecting only 11% of patients (mostly girls) in this review. Intriguing comparisons have been made between the dysmorphic features in PDC deficiency and fetal alcohol syndrome [reviewed in 2], given that the latter condition can potentially inhibit PDC activity and disrupt energy metabolism. However, it is likely that newborns with congenital PDC deficiency have suffered a degree of cellular energy failure at least as protracted and severe as children born to mothers who consume alcohol during pregnancy. Therefore, if PDC deficiency is the dominant biochemical mechanism underlying facial dysmorphism in both disorders, it is surprising that its prevalence is not higher in our series.
Structural brain lesions have long been noted as a common feature of PDC deficiency, but none of the findings reported here is unique to the disease. Moreover, their true frequencies are probably underestimated because only 186 (51%) of the cases contained evaluable neuroimaging details. In addition, given the high prevalence of ventricular enlargement in our series, it is probable that the true prevalence of cerebral atrophy is underreported.
Mosaicism in the PDHA1 gene due to variability in the pattern of X chromosome inactivation may make it difficult to diagnose PDC deficiency in females [148] and a few cases of somatic mosaicism have also been reported in males [144,169]. Consequently, cells obtained by biopsy for enzymological diagnosis could falsely overestimate global residual PDC activity in E1α deficient girls. It might be assumed that the amount of residual E1α activity measured in cells would correlate more closely with clinical phenotype in males who are heterozygous for a mutation than in females, in whom the biochemical expression of the defect depends on the degree of lyonization. However, as we and others [2] have found, there is generally a poor correlation between measured enzyme activity and clinical presentation and course, a conclusion applicable to PDC deficiency in general, regardless of the underlying component defect. This discrepancy between enzyme activity and the clinical phenotype of PDC deficiency is at least partly explained by both random X inactivation (in cases of E1α deficiency) and the dependency of the tissue used to diagnose the disease on PDC activity for supporting its energy needs.
PDHA1 gene mutations have been subdivided broadly into 1) missense point mutations, with loss of PDC catalytic activity only; 2) deletions or exon skipping mutations, resulting in loss of E1α mRNA and protein; and 3) deletions, missense or nonsense mutations, resulting in loss of E1α protein without loss of E1α mRNA (unstable E1α) [10,23,138,147,154,170,172,178]. Despite the variable nature of these mutations, we found it difficult to draw sharp distinctions between specific mutations of the PDC and the natural history of the disease, although mutations affecting position 378 in PDHA1 may be particularly lethal. As a whole, missense mutations occur more frequently in boys, whereas frameshift mutations are more common in girls. However, this difference does not appear to be translated into biochemically or clinically meaningful differences.
Treatment of most patients with PDC deficiency has been disappointing and no intervention specific to this disease has been evaluated in a randomized controlled trial [173]. Nutritional mixtures of various cofactors and vitamins are commonly provided to patients with PDC deficiency, usually, with limited biochemical rationale. In the majority of cases we reviewed, the doses of these modalities were omitted from the reports. Thiamine doses ranged from a few mg/d to >1 g/d. Thiamine pyrophosphate is an obligate cofactor for the E1 component of PDC, and most cases in which higher thiamine doses were administered involved cases in which a pathological mutation affecting the thiamine pyrophosphate binding site of E1 was assumed or established.
Patients with PDC deficiency do not oxidize carbohydrates efficiently; hence, the pyruvate derived from glycolysis is more likely to be reduced in the cytoplasm to lactate. This has led to the widespread use of high fat diets, typically with a caloric ratio of 3–4 fats to 1 carbohydrate plus protein, which induce ketosis and provide an alternative source of acetyl CoA, particularly for the nervous system. Reports of a few children with PDC deficiency whose clinical course improved dramatically while following a high fat diet [23,111] are consistent with this postulate and have resulted in the incorporation of such diets in the routine care of many patients with PDC deficiency [111,174]. However, most of the reports reviewed here described the administration of a “ketogenic,” “high fat” or “low carbohydrate” diet without providing information on the caloric distribution of metabolizable fuels. Therefore, we included under the category of ketogenic diet only those cases in which fat comprised at least 65% of total calories. No controlled trials have prospectively evaluated any type of “ketogenic diet” in the treatment of PDC deficiency.
DCA increases PDC activity by inhibiting the activity of PDKs in virtually all tissues, thereby maintaining E1α in its phosphorylated, catalytically active, state [175,176] and by stabilizing the complex and decreasing its rate of turnover [176]. When DCA was administered to PDC deficient patients, doses frequently varied over time in a given patient and, among subjects, in duration of treatment. The drug has been evaluated in randomized controlled trials in patients with various genetic mitochondrial diseases [177–179] in which a few children with PDC deficiency participated. These and other open label reports [10] have suggested that chronic DCA may benefit patients with PDC deficiency.
4.1. Conclusions
In summary, our analysis of 371 cases of proven PDC deficiency found that neither gender nor any biochemical or clinical feature differentiates the various enzymological or molecular genetic etiologies of the disease. The clinical spectrum of PDC deficiency is broad, but typically presents in early childhood with neurodevelopmental and neuromuscular compromise. Hyperlactatemia is a frequent, but not universal, finding early in the disease process, is associated with a blood lactate:pyruvate ratio≤20 and often portends an early demise when present during the neonatal period. Boys and girls may differ in a few clinical characteristics and in the type of PDC mutation they harbor, but otherwise share a fairly common clinical course. Although many cases in our series described the administration of thiamine, a ketogenic diet and DCA, singly or in combination, such uncontrolled studies involving one or a few subjects provide no new insight into the potential safety and efficacy of these interventions. Rigorous, prospective evaluation of promising therapies is needed for PDC deficiency. Such controlled trials should incorporate a standardized approach to the longitudinal study of the natural history of this devastating disease.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R34HD065991-01, UL1RR025208 and U54RR025208-01 and by the University of Florida’s Clinical and Translational Science Institute. We thank Ms. Kathryn St. Croix for editorial assistance.
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.ymgme.2012.03.017.
References
- 1.Reed LJ. Multienzyme complexes. Acc Chem Res. 1974;7:40–46. [Google Scholar]
- 2.Robinson BH. Lactic academia (disorders of pyruvate carboxylase, pyruvate dehydrogenase) In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 1995. pp. 1479–1499. [Google Scholar]
- 3.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141:761–774. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagliarini DR, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64:830–849. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blass JP, Avigan J, Uhlendorf BW. A defect in pyruvate decarboxylase deficiency in a child with an intermittent movement disorder. J Clin Invest. 1970;49:423–432. doi: 10.1172/JCI106251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerr DS, Schmotzer C. Variability of human pyruvate dehydrogenase complex deficiency. In: Patel MS, Jordan F, editors. Thiamine: Catalytic Mechanisms and Role in Normal and Disease States. Marcel Dekker Inc; New York: 2004. pp. 471–483. [Google Scholar]
- 8.DeBrosse SD, Okajima K, Schmotzer CL, Frohnapfel MB, Kerr DS. Spectrum of clinical outcomes in pyruvate dehydrogenase deficiency. United Mitochondrial Disease Foundation Annual Symposium; June 15–19, 2011; Chicago, IL. 2011. [Google Scholar]
- 9.Stacpoole PW, Gilbert LR. Pyruvate dehydrogenase complex deficiency. In: Glew RH, Rosenthal MD, editors. Clinical Cases in Medical Biochemistry. Oxford University Press; New York: 2006. pp. 77–88. [Google Scholar]
- 10.Berendzen K, Theriaque DS, Shuster J, Stacpoole PW. Therapeutic potential of dichloroacetate for pyruvate dehydrogenase complex deficiency. Mitochondrion. 2006;6:126–135. doi: 10.1016/j.mito.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Livingstone IR, Gardner-Medwin D, Pennington RJT. Familial intermittent ataxia with possible X-linked recessive inheritance. J Neurol Sci. 1984;64:89–97. doi: 10.1016/0022-510x(84)90059-5. [DOI] [PubMed] [Google Scholar]
- 12.Ramadan DG, Head RA, Al-Tawari A, Habeeb Y, Zaki M, Al-Ruqum F, et al. Lactic acidosis and developmental delay due to deficiency of E3 binding protein (protein X) of the pyruvate dehydrogenase complex. J Inherit Metab Dis. 2004;27:477–485. doi: 10.1023/B:BOLI.0000037336.91549.44. [DOI] [PubMed] [Google Scholar]
- 13.Brown R, Head RA, Brown G. Pyruvate dehydrogenase E3 binding protein deficiency. Hum Genet. 2002;110:187–191. doi: 10.1007/s00439-001-0665-3. [DOI] [PubMed] [Google Scholar]
- 14.Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Kuroda Y. Thiamine-responsive lactic acidaemia: role of pyruvate dehydrogenase complex. Eur J Pediatr. 1998;157:648–652. doi: 10.1007/s004310050903. [DOI] [PubMed] [Google Scholar]
- 15.Arai Y, Miyasato Y, Koide H. Characteristic changes on brain CT in a case of Leigh encephalopathy with deficiency of pyruvate dehydrogenase. Brain Dev. 1991;13:457–458. doi: 10.1016/s0387-7604(12)80050-7. [DOI] [PubMed] [Google Scholar]
- 16.Medina L, Chi TL, DeVivo DC, Hilal SK. MR findings in patients with subacute necrotizing encephalomyelopathy (Leigh syndrome): correlation with the biochemical defect. Am J Neuroradiol. 1990;11:379–384. [PMC free article] [PubMed] [Google Scholar]
- 17.Chow CW, Anderson RM, Kenny GC. Neuropathology in cerebral lactic acidosis. Acta Neuropathol. 1987;74:393–396. doi: 10.1007/BF00687218. [DOI] [PubMed] [Google Scholar]
- 18.Cederbaum SD, Blass JP, Minkoff N, Brown WJ, Cotton ME, Harris SH. Sensitivity to carbohydrate in a patient with familial intermittent lactic acidosis and pyruvate dehydrogenase deficiency. Pediatr Res. 1976;10:713–720. doi: 10.1203/00006450-197608000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Haworth JC, Perry TL, Blass JP, Hansen S, Urquhart N. Lactic acidosis in three sibs due to defects in both pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase complexes. Pediatrics. 1976;58:564–572. [PubMed] [Google Scholar]
- 20.Stromme JH, Borud O, Moe PJ. Fatal lactic acidosis in a newborn attributable to a congenital defect of pyruvate dehydrogenase. Pediatr Res. 1976;10:62–66. doi: 10.1203/00006450-197601000-00012. [DOI] [PubMed] [Google Scholar]
- 21.Blass JP, Kark AP, Engel WK. Clinical studies of a patient with pyruvate decarboxylase deficiency. Arch Neurol. 1971;25:449–460. doi: 10.1001/archneur.1971.00490050083007. [DOI] [PubMed] [Google Scholar]
- 22.Farrell DF, Clark AF, Scott CR, Wennberg RP. Absence of pyruvate decarboxylase activity in man: a cause of congenital lactic acidosis. Science. 1975;187:1082–1084. doi: 10.1126/science.803713. [DOI] [PubMed] [Google Scholar]
- 23.Falk RE, Cederbaum SD, Blass JP, Gibson GE, Kark RA, Carrel RE. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics. 1976;58:713–721. [PubMed] [Google Scholar]
- 24.Robinson B, Taylor J, Sherwood W. Deficiency of dihydrolipoyl dehydrogenase (a component of the pyruvate and alpha ketoglutarate dehydrogenase complex): a cause of congenital chronic lactic acidosis in infancy. Pediatrics. 1977;11:1198–1202. doi: 10.1203/00006450-197712000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Wick H, Schweizer K, Baumgartner R. Thiamine dependency in a patient with congenital lactic acidaemia due to pyruvate dehydrogenase deficiency. Agents Actions. 1977;7:405–410. doi: 10.1007/BF01969575. [DOI] [PubMed] [Google Scholar]
- 26.DeVivo DC, Haymond MW, Obert KA, Nelson JS, Pagliara AS. Defective activation of the pyruvate dehydrogenase complex in subacute necrotizing encephalomyelopathy. Ann Neurol. 1979;6:483–494. doi: 10.1002/ana.410060605. [DOI] [PubMed] [Google Scholar]
- 27.Robinson BH, Taylor J, Kahler SG, Kirkman HN. Lactic acidemia, neurologic deterioration and carbohydrate dependence in a girl with dihydrolipoyl dehydrogenase deficiency. Eur J Pediatr. 1981;136:35–39. doi: 10.1007/BF00441708. [DOI] [PubMed] [Google Scholar]
- 28.Prick M, Gabreels F, Renier W, Trijbels F, Jaspar H, Lamers K, Kok J. Pyruvate dehydrogenase deficiency restricted to brain. Neurology. 1981;31:398–404. doi: 10.1212/wnl.31.4.398. [DOI] [PubMed] [Google Scholar]
- 29.Uziel G, Bottacchi E, Moschen G, Giovanardi-Rossi P, Cardace G, Di Donato S. Pyruvate–dehydrogenase complex in ataxic patients: enzyme deficiency in ataxic encephalopathy plus lactic acidosis and normal activity in Friedreich ataxia. Ital J Neurol Sci. 1982;3:317–321. doi: 10.1007/BF02043580. [DOI] [PubMed] [Google Scholar]
- 30.Ohtake M, Takada G, Miyabayashi S, Arai N, Tada K, Morinaga S. Pyruvate decarboxylase deficiency in a patient with Leigh’s encephalomyelopathy. Tohoku J Exp Med. 1982;137:379–386. doi: 10.1620/tjem.137.379. [DOI] [PubMed] [Google Scholar]
- 31.Hansen TL, Christensen E, Brandt NJ. Studies on pyruvate carboxylase, pyruvate decarboxylase and lipoamide dehydrogenase in subacute necrotizing encephalomyelopathy. Acta Paediatr Scand. 1982;71:263–267. doi: 10.1111/j.1651-2227.1982.tb09412.x. [DOI] [PubMed] [Google Scholar]
- 32.Kodama S, Yagi R, Ninomiya M, Goji K, Takahashi T, Morishita Y, Matsuo T. The effect of a high fat diet on pyruvate decarboxylase deficiency without central nervous system involvement. Brain Dev. 1983;5:381–389. doi: 10.1016/s0387-7604(83)80043-6. [DOI] [PubMed] [Google Scholar]
- 33.Dierdorf SF, McNiece WL. Anaesthesia and pyruvate dehydrogenase deficiency. Can Anaesth Soc J. 1983;30:413–416. doi: 10.1007/BF03007865. [DOI] [PubMed] [Google Scholar]
- 34.Kuhara T, Shinka T, Inoue Y, Matsumoto M, Yoshino M, Sakaguchi Y, Matsumoto I. Studies of urinary organic acid profiles of a patient with dihydrolipoyl dehydrogenase deficiency. Clin Chim Acta. 1983;133:133–140. doi: 10.1016/0009-8981(83)90398-4. [DOI] [PubMed] [Google Scholar]
- 35.Evans OB. Episodic weakness in pyruvate decarboxylase deficiency. J Pediatr. 1984;105:961–963. doi: 10.1016/s0022-3476(84)80090-6. [DOI] [PubMed] [Google Scholar]
- 36.Matuda S, Kitano A, Sakaguchi Y, Yoshino M, Saheki T. Pyruvate dehydrogenase subcomplex with lipoamide dehydrogenase deficiency in a patient with lactic acidosis and branched chain ketoaciduria. Clin Chim Acta. 1984;140:59–64. doi: 10.1016/0009-8981(84)90151-7. [DOI] [PubMed] [Google Scholar]
- 37.Johnston K, Newth CJ, Sheu KF, Patel MS, Heldt GP, Schmidt KA, Packman S. Central hypoventilation syndrome in pyruvate dehydrogenase complex deficiency. Pediatrics. 1984;74:1034–1040. [PubMed] [Google Scholar]
- 38.Matalon R, Stumpf DA, Michals K, Hart RD, Parks JK, Goodman SI. Lipoamide dehydrogenase deficiency with primary lactic acidosis: favorable response to treatment with oral lipoic acid. J Pediatr. 1984;104:65–69. doi: 10.1016/s0022-3476(84)80591-0. [DOI] [PubMed] [Google Scholar]
- 39.Matsuo M, Ookita K, Takemine H, Koike K, Koike M. Fatal case of pyruvate dehydrogenase deficiency. Acta Paediatr Scand. 1985;74:140–142. doi: 10.1111/j.1651-2227.1985.tb10937.x. [DOI] [PubMed] [Google Scholar]
- 40.McCormick K, Viscardi RM, Robinson B, Heininger J. Partial pyruvate decarboxylase deficiency with profound lactic acidosis and hyperammonemia: responses to dichloroacetate and benzoate. Am J Med Genet. 1985;22:291–299. doi: 10.1002/ajmg.1320220211. [DOI] [PubMed] [Google Scholar]
- 41.Ho L, Hu CW, Packman S, Patel MS. Deficiency of the pyruvate dehydrogenase component in pyruvate dehydrogenase complex-deficient human fibroblasts. J Clin Invest. 1986;78:844–847. doi: 10.1172/JCI112651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKay N, Petrova-Benedict R, Thoene J, Bergen B, Wilson W, Robinson B. Lactic acidaemia due to pyruvate dehydrogenase deficiency, with evidence of protein polymorphism in the α-subunit of the enzyme. Eur J Pediatr. 1986;144:445–450. doi: 10.1007/BF00441736. [DOI] [PubMed] [Google Scholar]
- 43.Wicking C, Scholem R, Hunt S, Brown G. Immunochemical analysis of new and mutant forms of human pyruvate dehydrogenase. J Biochem. 1986;239:89–96. doi: 10.1042/bj2390089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung SJ, Asoh S, Yamanaka T, Okamura-Oho Y, Toshima K, Woo M, Nonaka I. Muscle involvement in pyruvate dehydrogenase complex (PDHC) deficiency. Brain Dev. 1987;9:9–15. doi: 10.1016/s0387-7604(87)80003-7. [DOI] [PubMed] [Google Scholar]
- 45.Brown GK, Scholem RD, Hunt SM, Harrison JR, Pollard AC. Hyperammonaemia and lactic acidosis in a patient with pyruvate dehydrogenase deficiency. J Inherit Metab Dis. 1987;10:359–366. doi: 10.1007/BF01799978. [DOI] [PubMed] [Google Scholar]
- 46.Kretzschmar HA, Dearmond SJ, Koch TK, Patel MS, Newth CJ, Schmidt KA, Packman S. Pyruvate dehydrogenase complex deficiency as a cause of subacute necrotizing encephalopathy (Leigh disease) Pediatrics. 1987;79:370–373. [PubMed] [Google Scholar]
- 47.Robinson BH, MacMillan H, Petrova-Benedict R, Sherwood WG. Variable clinical presentation in patients with defective E1 component of pyruvate dehydrogenase complex. J Pediatr. 1987;111:525–533. doi: 10.1016/s0022-3476(87)80112-9. [DOI] [PubMed] [Google Scholar]
- 48.Brown GK, Haan EA, Kirby DM, Scholem RD, Wraith JE, Rogers JG, Danks DM. “Cerebral” lactic acidosis: defects in pyruvate metabolism with profound brain damage and minimal systemic acidosis. Eur J Pediatr. 1988;147:10–14. doi: 10.1007/BF00442603. [DOI] [PubMed] [Google Scholar]
- 49.Birch-Machin MA, Shepherd IM, Solomon M, Yeaman SJ, Gardner-Medwin D, Sherratt HS, Lindsay JG, Aynsley-Green A, Turnbull DM. Fatal lactic acidosis due to deficiency of E1 component of the pyruvate dehydrogenase complex. J Inherit Metab Dis. 1988;11:207–217. doi: 10.1007/BF01799876. [DOI] [PubMed] [Google Scholar]
- 50.Kerr DS, Berry SA, Lusk MM, Ho L, Patel M. A deficiency of both subunits of pyruvate dehydrogenase which is not expressed in fibroblasts. Pediatr Res. 1988;24:95–100. doi: 10.1203/00006450-198807000-00022. [DOI] [PubMed] [Google Scholar]
- 51.Byrd DJ, Krohn HP, Winkler L, Steinborn C, Hadam M, Brodehl J, Hunneman DH. Neonatal pyruvate dehydrogenase deficiency with lipoate responsive lactic acidemia and hyperammonemia. Eur J Pediatr. 1989;148:543–547. doi: 10.1007/BF00441554. [DOI] [PubMed] [Google Scholar]
- 52.Kitano A, Endo F, Kuroda Y, Aso S, Kawasaki T, Matsuda I. Biochemical nature of pyruvate dehydrogenase complex in the patient with primary lactic acidaemia. J Inherit Metab Dis. 1989;12:379–385. doi: 10.1007/BF01802031. [DOI] [PubMed] [Google Scholar]
- 53.Kitano A, Endo F, Matsuda I, Miyabayashi S, Dahl HH. Mutation of the E1 alpha subunit of the pyruvate dehydrogenase complex, in relation to heterogeneity. J Inherit Metab Dis. 1989;12:97–107. doi: 10.1007/BF01800710. [DOI] [PubMed] [Google Scholar]
- 54.Old SE, DeVivo DC. Pyruvate dehydrogenase complex deficiency: biochemical and immunoblot analysis of cultured skin fibroblasts. Ann Neurol. 1989;26:746–751. doi: 10.1002/ana.410260610. [DOI] [PubMed] [Google Scholar]
- 55.Endo H, Hasegawa K, Narisawa K, Tada K, Kagawa Y, Ohta S. Defective gene in lactic acidosis: abnormal pyruvate dehydrogenase E1α-subunit caused by a frame shift. Am J Hum Genet. 1989;44:358–364. [PMC free article] [PubMed] [Google Scholar]
- 56.Kitano A, Endo F, Matsuda I. Immunochemical analysis of pyruvate dehydrogenase complex in 2 boys with primary lactic acidemia. Neurology. 1990;40:1312–1314. doi: 10.1212/wnl.40.8.1312. [DOI] [PubMed] [Google Scholar]
- 57.Sperl W, Ruitenbeek W, Kerkhof CMC, Sengers RCA, Trijbels JMF, Guggenbichler JP, Janssen AJM, Bakkeren JAJM. Eur J Pediatr. 1990;149:487–492. doi: 10.1007/BF01959401. [DOI] [PubMed] [Google Scholar]
- 58.Robinson BH, MacKay N, Petrova-Benedict R, Ozalp I, Coskun T, Stacpoole PW. Defects in the E2 lipoyl transacetylase and the X-lipoyl containing component of the pyruvate dehydrogenase complex in patients with lactic acidemia. J Clin Invest. 1990;85:1821–1824. doi: 10.1172/JCI114641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Federico A, Dotti MT, Fabrizi GM, Palmeri S, Massimo L, Robinson BH, Malandrini A, Guazzi GC. Congenital lactic acidosis due to a deficiency of pyruvate dehydrogenase complex (E1) Eur Neurol. 1990;30:123–127. doi: 10.1159/000117327. [DOI] [PubMed] [Google Scholar]
- 60.Dahl HHM, Maragos C, Brown RM, Hansen LL, Brown GK. Pyruvate dehydrogenase deficiency caused by deletion of a 7-bp repeat sequence in the E1 gene. Am J Hum Genet. 1990;47:286–293. [PMC free article] [PubMed] [Google Scholar]
- 61.de Meirleir LJ, Lissens W, Vamos E, Liebaers I. Pyruvate dehydrogenase deficiency due to a mutation of the E1 alpha subunit. J Inherit Metab Dis. 1991;14:301–304. doi: 10.1007/BF01811687. [DOI] [PubMed] [Google Scholar]
- 62.Mahbubul Huq AHM, Ito M, Naito E, Saijo T, Takeda E, Kuroda Y. Demonstration of an unstable variant of pyruvate dehydrogenase protein (E1) in cultured fibroblasts from a patient with congenital lactic acidosis. Pediatr Res. 1991;30:11–14. doi: 10.1203/00006450-199107000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Endo H, Miyabayashi S, Tada K, Narisawa K. A four nucleotide insertion at the E1α gene in a patient with pyruvate dehydrogenase deficiency. J Inherit Metab Dis. 1991;14:793–799. doi: 10.1007/BF01799952. [DOI] [PubMed] [Google Scholar]
- 64.Chun K, MacKay N, Petrova-Benedict R, Robinson BH. Pyruvate dehydrogenase deficiency due to a 20-bp deletion in exon II of the pyruvate dehydrogenase (PDH) E1 alpha gene. Am J Hum Genet. 1991;49:414–420. [PMC free article] [PubMed] [Google Scholar]
- 65.Hansen L, Brown GK, Kirby DM, Dahl HHM. Characterization of the mutation of three patients with pyruvate dehydrogenase E1α deficiency. J Inherit Metab Dis. 1991;14:140–151. doi: 10.1007/BF01800586. [DOI] [PubMed] [Google Scholar]
- 66.Narisawa K, Endo H, Miyabayashi S, Tada K. Thiamine responsive pyruvate dehydrogenase deficiency. J Nutr Sci Vitaminol (Tokyo) 1992;(Spec No):585–8. doi: 10.3177/jnsv.38.special_585. [DOI] [PubMed] [Google Scholar]
- 67.De Meirleir L, Lissens W, Vamos E, Liebaers I. Pyruvate dehydrogenase (PDH) deficiency caused by a 21-base pair insertion mutation in the E1α subunit. Hum Genet. 1992;88:649–652. doi: 10.1007/BF02265291. [DOI] [PubMed] [Google Scholar]
- 68.Wijburg FA, Barth PG, Bindoff LA, Birch-Machin MA, van der Blij JF, Ruitenbeek W, Turnbull DM, Schutgens RB. Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: results of treatment with a ketogenic diet. Neuropediatrics. 1992;23:147–152. doi: 10.1055/s-2008-1071331. [DOI] [PubMed] [Google Scholar]
- 69.Dahl HHM, Hansen LL, Brown RM, Danks DM, Rogers JG, Brown GK. X-linked pyruvate dehydrogenase subunit deficiency in heterozygous females: variable manifestation of the same mutation. J Inherit Metab Dis. 1992;15:835–847. doi: 10.1007/BF01800219. [DOI] [PubMed] [Google Scholar]
- 70.Ito M, Huq AH, Naito E, Saijo T, Takeda E, Kuroda Y. Mutation of the E1α gene in a female patient with pyruvate dehydrogenase deficiency due to rapid degradation of E1 protein. J Inherit Metab Dis. 1992;15:848–856. doi: 10.1007/BF01800220. [DOI] [PubMed] [Google Scholar]
- 71.Hansen LL, Brown GK, Brown RM, Dahl HH. Pyruvate dehydrogenase deficiency caused by a 5 base pair duplication in the E1 alpha subunit. Hum Mol Genet. 1993;2:805–807. doi: 10.1093/hmg/2.6.805. [DOI] [PubMed] [Google Scholar]
- 72.Hansikova H, Zeman J, et al. Deficiency of pyruvate dehydrogenase complex in tissues of an eight month old infant. Biochem Mol Biol Int. 1993;31:1157–1166. [PubMed] [Google Scholar]
- 73.Michotte A, De Meirleir L, Lissens W, Denis R, Wayenberg JL, Liebaers I, Brucher JM. Neuropathological findings of a patient with pyruvate dehydrogenase E1α deficiency presenting as a cerebral lactic acidosis. Acta Neuropathol. 1993;85:674–678. doi: 10.1007/BF00334680. [DOI] [PubMed] [Google Scholar]
- 74.DeMeirleir L, Lissens W, Denis R, Wayenberg JL, Michotte A, Brucher JM, Vamos E, Gerlo E, Liebaers I. Pyruvate dehydrogenase deficiency: clinical and biochemical diagnosis. Pediatr Neurol. 1993;9:216–220. doi: 10.1016/0887-8994(93)90088-t. [DOI] [PubMed] [Google Scholar]
- 75.Chun K, MacKay N, Petrova-Benedict R, Robinson BH. Mutations in the X-linked E1 alpha subunit of pyruvate dehydrogenase leading to deficiency of the pyruvate dehydrogenase complex. Hum Mol Genet. 1993;2:449–454. doi: 10.1093/hmg/2.4.449. [DOI] [PubMed] [Google Scholar]
- 76.Iso A, Murakami N, Yoneyama H, Hanaoka S, Kurokawa T, Nonaka I. Idiopathic lactic acidemia with developmental delay and type I muscle fiber atrophy: report of two patients. Brain Dev. 1993;15:384–386. doi: 10.1016/0387-7604(93)90127-t. [DOI] [PubMed] [Google Scholar]
- 77.Takakubo F, Thorburn DR, Dahl HHM. A four-nucleotide insertion hotspot in the X chromosome located pyruvate dehydrogenase E1α gene (PDHA1) Hum Mol Genet. 1993;2:473–474. doi: 10.1093/hmg/2.4.473. [DOI] [PubMed] [Google Scholar]
- 78.Marsac C, Stansbie D, Bonne G, Cousin J, Jehenson P, Benelli C, Leroux JP, Lindsay G. Defect in the lipoyl bearing protein X subunit of the pyruvate dehydrogenase complex in two patients with encephalomyelopathy. J Pediatr. 1993;123:915–920. doi: 10.1016/s0022-3476(05)80387-7. [DOI] [PubMed] [Google Scholar]
- 79.Tóth PP, el-Shanti H, Eivins S, Rhead WJ, Klein JM. Transient improvement of congenital lactic acidosis in a male infant with pyruvate decarboxylate deficiency treated with dichloroacetate. J Pediatr. 1993;123:427–430. doi: 10.1016/s0022-3476(05)81751-2. [DOI] [PubMed] [Google Scholar]
- 80.Bonne G, Benelli C, De Meirleir L, Lissens W, Chaussain M, Diry M, Clot JP, Ponsot G, Geoffroy V, Leroux JP, et al. E1 pyruvate dehydrogenase deficiency in a child with motor neuropathy. Pediatr Res. 1993;33:284–288. doi: 10.1203/00006450-199303000-00016. [DOI] [PubMed] [Google Scholar]
- 81.Dahl HH, Brown GK. Pyruvate dehydrogenase deficiency in a male caused by a point mutation (F205L) in the E1 alpha subunit. Hum Mutat. 1994;3:152–155. doi: 10.1002/humu.1380030210. [DOI] [PubMed] [Google Scholar]
- 82.Cross JH, Connelly A, Gadian DG, Kendall BE, Brown GK, Brown RM, Leonard JV. Clinical diversity of pyruvate dehydrogenase deficiency. Pediatr Neurol. 1994;10:276–283. doi: 10.1016/0887-8994(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 83.Naito E, Ito M, Yokota I, Matsuda J, Yara A, Kuroda Y. Pyruvate dehydrogenase deficiency caused by a four-nucleotide insertion in the E1α subunit gene. Hum Mol Genet. 1994;3:1193–1194. doi: 10.1093/hmg/3.7.1193. [DOI] [PubMed] [Google Scholar]
- 84.Matthews PM, Brown RM, Otero LJ, Marchington DR, LeGris M, Howes R, Meadows LS, Shevell M, Scriver CR, Brown GK. Pyruvate dehydrogenase deficiency. Clinical presentation and molecular genetic characterization of five new patients. Brain. 1994;117:435–443. doi: 10.1093/brain/117.3.435. [DOI] [PubMed] [Google Scholar]
- 85.Naito E, Ito M, Takeda E, Yokota I, Yoshijima S, Kuroda Y. Molecular analysis of abnormal pyruvate dehydrogenase in a patient with thiamine-responsive lactic acidemia. Pediatr Res. 1994;36:340–346. doi: 10.1203/00006450-199409000-00013. [DOI] [PubMed] [Google Scholar]
- 86.Awata H, Endo F, Tanoue A, Kitano A, Matsuda I. Characterization of a point mutation in the pyruvate dehydrogenase E1 alpha gene from two boys with primary lactic acidaemia. J Inherit Metab Dis. 1994;17:189–195. doi: 10.1007/BF00711616. [DOI] [PubMed] [Google Scholar]
- 87.Chabrol B, Mancini J, Benelli C, Gire C, Munnich A. Leigh syndrome: pyruvate dehydrogenase defect. A case with peripheral neuropathy. J Child Neurol. 1994;9:52–55. doi: 10.1177/088307389400900113. [DOI] [PubMed] [Google Scholar]
- 88.Takakubo F, Thorburn DR, Brown RM, Brown GK, Dahl HH. A novel mutation (P316L) in a female with pyruvate dehydrogenase E1α deficiency. Hum Mutat. 1995;6:274–275. doi: 10.1002/humu.1380060317. [DOI] [PubMed] [Google Scholar]
- 89.Lissens W, Desguerre I, Benelli C, Marsac C, Fouque F, Haenggeli C, Ponsot G, Seneca S, Liebaers I, De Meirleir L. Pyruvate dehydrogenase deficiency in a female due to a 4 base pair deletion in exon 10 of the E1α gene. Hum Mol Genet. 1995;4:307–308. doi: 10.1093/hmg/4.2.307. [DOI] [PubMed] [Google Scholar]
- 90.Otero LJ, Brown GK, Silver K, Arnold DL, Mathews PM. Association of cerebral dysgenesis and lactic acidemia with X-linked PDH E1α subunit mutations in females. Pediatr Neurol. 1995;13:327–332. doi: 10.1016/0887-8994(95)00222-7. [DOI] [PubMed] [Google Scholar]
- 91.Chun K, MacKay N, Petrova-Benedict R, Federico A, Fois A, Cole DE, Robertson E, Robinson BH. Mutations in the X-linked E1 alpha subunit of pyruvate dehydrogenase: exon skipping, insertion of duplicate sequence, and missense mutations leading to the deficiency of the pyruvate dehydrogenase complex. Am J Hum Genet. 1995;56:558–569. [PMC free article] [PubMed] [Google Scholar]
- 92.Hemalatha SG, Kerr DS, Wexler ID, Lusk MM, Kaung M, Du Y, Kolli M, Schelper RL, Patel MS. Pyruvate dehydrogenase complex deficiency due to a point mutation (P188L) within the thiamine pyrophosphate binding loop of the E1 alpha subunit. Hum Mol Genet. 1995;4:315–318. doi: 10.1093/hmg/4.2.315. [DOI] [PubMed] [Google Scholar]
- 93.Elpeleg ON, Ruitenbeek W, Jakobs C, Barash V, De Vivo DC, Amir N. Congenital lactic acidemia caused by lipoamide dehydrogenase deficiency with favorable outcome. J Pediatr. 1995;126:72–74. doi: 10.1016/s0022-3476(95)70506-6. [DOI] [PubMed] [Google Scholar]
- 94.Matsuda J, Ito M, Naito E, Yokota I, Kuroda Y. DNA diagnosis of pyruvate dehydrogenase deficiency in female patients with congenital lactic acidaemia. J Inherit Metab Dis. 1995;18:534–546. doi: 10.1007/BF02435998. [DOI] [PubMed] [Google Scholar]
- 95.Takakubo F, Cartwright P, Hoogenraad N, Thorburn DR, Collins F, Lithgow T, Dahl HHM. An amino acid substitution in the pyruvate dehydrogenase E1 gene, affecting mitochondrial import of the precursor protein. Am J Hum Genet. 1995;57:772–780. [PMC free article] [PubMed] [Google Scholar]
- 96.Briones P, Lopez MJ, DeMeirleir L, Ribes A, Rodés M, Martinez-Costa C, Peris M, Lissens W. Leigh syndrome due to pyruvate dehydrogenase E1α deficiency (point mutation R263G) in a Spanish boy. J Inherit Metab Dis. 1996;19:795–796. doi: 10.1007/BF01799177. [DOI] [PubMed] [Google Scholar]
- 97.Pastoris O, Savasta S, Foppa P, Catapano M, Dossena M. Pyruvate dehydrogenase deficiency in a child responsive to thiamine treatment. Acta Paediatr. 1996;85:625–628. doi: 10.1111/j.1651-2227.1996.tb14104.x. [DOI] [PubMed] [Google Scholar]
- 98.Lissens W, De Meirleir L, Seneca S, Benelli C, Marsac C, Poll-The BT, Briones P, Ruitenbeek W, van Diggelen O, Chaigne D, Ramaekers V, Liebaers I. Mutation analysis of the pyruvate dehydrogenase E1α gene in eight patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat. 1996;7:46–51. doi: 10.1002/(SICI)1098-1004(1996)7:1<46::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 99.Geoffroy V, Fouque F, Benelli C, Poggi F, Saudubray JM, Lissens W, Meirleir LD, Marsac C, Lindsay JG, Sanderson SJ. Defect in the X-lipoyl-containing component of the pyruvate dehydrogenase complex in a patient with a neonatal lactic acidemia. Pediatrics. 1996;97:267–272. [PubMed] [Google Scholar]
- 100.Hong YS, Kerr DS, Craigen WJ, Tan J, Pan Y, Lusk M, Patel MS. Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum Mol Genet. 1996;5:1925–1930. doi: 10.1093/hmg/5.12.1925. [DOI] [PubMed] [Google Scholar]
- 101.Craigen W. Leigh disease with deficiency of lipoamide dehydrogenase: treatment failure with dichloroacetate. Pediatr Neurol. 1996;24:69–71. doi: 10.1016/0887-8994(96)00005-7. [DOI] [PubMed] [Google Scholar]
- 102.Tripatara A, Kerr DS, Lusk MM, Kolli M, Tan J, Patel MS. Three new mutations of the pyruvate dehydrogenase alpha subunit: a point mutation (M181V), 3 bp deletion (-R282), and 16 bp insertion/frameshift (K358SVS>TVDQS) Hum Mutat. 1996;8:180–182. doi: 10.1002/(SICI)1098-1004(1996)8:2<180::AID-HUMU11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 103.Fujii T, Garcia-Alvarez MB, Sheu KF, Kranz-Eble PJ, De Vivo DC. Pyruvate dehydrogenase deficiency: the relation of the E1 alpha mutation to the E1 beta subunit deficiency. Pediatr Neurol. 1996;14:328–334. doi: 10.1016/0887-8994(96)00058-6. [DOI] [PubMed] [Google Scholar]
- 104.Kinoshita H, Sakuragawa N, Tada H, Naito E, Kuroda Y, Nonaka I. Recurrent muscle weakness and ataxia in thiamine-responsive pyruvate dehydrogenase complex deficiency. J Child Neurol. 1997;12:141–144. doi: 10.1177/088307389701200212. [DOI] [PubMed] [Google Scholar]
- 105.Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Osaka H, Kimura S, Kuroda Y. Biochemical and molecular analysis of an X-linked case of Leigh syndrome associated with thiamin-responsive pyruvate dehydrogenase deficiency. J Inherit Metab Dis. 1997;20:539–548. doi: 10.1023/a:1005305614374. [DOI] [PubMed] [Google Scholar]
- 106.Aral B, Benelli C, Ait-Ghezala G, Amessou M, Fouque F, Maunoury C, Creau N, Kamoun P, Marsac C. Mutations in PDX1, the human lipoyl-containing component X of the pyruvate dehydrogenase-complex gene on chromosome 11p1, in congenital lactic acidosis. Am J Hum Genet. 1997;61:1318–1326. doi: 10.1086/301653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aptowitzer I, Saada A, Faber J, Kleid D, Elpeleg ON. Liver disease in the Ashkenazi-Jewish lipoamide dehydrogenase deficiency. J Pediatr Gastroenterol Nutr. 1997;24:599–601. doi: 10.1097/00005176-199705000-00019. [DOI] [PubMed] [Google Scholar]
- 108.Elpeleg ON, Shaag A, Glustein JZ, Anikster Y, Joseph A, Saada A. Lipoamide dehydrogenase deficiency in Ashkenazi Jews: an insertion mutation in the mitochondrial leader sequence. Hum Mutat. 1997;10:256–257. doi: 10.1002/(SICI)1098-1004(1997)10:3<256::AID-HUMU16>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 109.Hong YS, Kerr DS, Liu TC, Lusk M, Powell BR, Patel MS. Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340 K and G101del). Analysis of a family and prenatal testing. Biochim Biophys Acta. 1997;1362:160–168. doi: 10.1016/s0925-4439(97)00073-2. [DOI] [PubMed] [Google Scholar]
- 110.Marsac C, Benelli C, Desguerre I, Diry M, Fouque F, De Meirleir L, Ponsot G, Seneca S, Poggi F, Saudubray JM, Zabot MT, Fontan D, Lissens W. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1α deficiency. Hum Genet. 1997;99:785–792. doi: 10.1007/s004390050449. [DOI] [PubMed] [Google Scholar]
- 111.Wexler ID, Hemalathu SG, McConnell J, Buist NR, Dahl HH, Berry SA, Cederbaum SD, Patel MS, Kerr DS. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: studies in patients with identical mutations. Neurology. 1997;49:1655–1661. doi: 10.1212/wnl.49.6.1655. [DOI] [PubMed] [Google Scholar]
- 112.De Meirleir L, Specola N, Seneca S, Lissens W. Pyruvate dehydrogenase E1α deficiency in a family: different clinical presentation in two siblings. J Inherit Metab Dis. 1998;21:224–226. doi: 10.1023/a:1005347501111. [DOI] [PubMed] [Google Scholar]
- 113.Ling M, McEachern G, Seyda A, MacKay N, Scherer SW, Bratinova S, Beatty B, Giovannucci-Uzielli ML, Robinson BH. Detection of a homozygous four base pair deletion in the protein X gene in a case of pyruvate dehydrogenase complex deficiency. Hum Mol Genet. 1998;7:501–505. doi: 10.1093/hmg/7.3.501. [DOI] [PubMed] [Google Scholar]
- 114.De Meirleir L, Lissens W, Benelli C, Marsac C, De Klerk J, Scholte J, van Diggelen O, Kleijer W, Seneca S, Liebaers I. Pyruvate dehydrogenase complex deficiency and absence of subunit X. J Inherit Metab Dis. 1998;21:9–16. doi: 10.1023/a:1005351012066. [DOI] [PubMed] [Google Scholar]
- 115.Otero LJ, Brown RM, Brown GK. Arginine 302 mutations in the pyruvate dehydrogenase E1 subunit gene: identification of further patients and in vitro demonstration of pathogenicity. Hum Mutat. 1998;12:114–121. doi: 10.1002/(SICI)1098-1004(1998)12:2<114::AID-HUMU6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 116.Rubio-Gozalbo ME, Heerschap A, Trijbels JMF, De Meirleir L, Thijssen HOM, Smeitink JAM. Proton MR spectroscopy in a child with pyruvate dehydrogenase complex deficiency. Magn Reson Imaging. 1999;17:939–944. doi: 10.1016/s0730-725x(99)00002-8. [DOI] [PubMed] [Google Scholar]
- 117.Shany E, Saada A, Landau D, Shaag A, Hershkovitz E, Elpeleg ON. Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem Biophys Res Commun. 1999;262:163–166. doi: 10.1006/bbrc.1999.1133. [DOI] [PubMed] [Google Scholar]
- 118.Naito E, Ito M, Yokota I, Saijo T, Chen S, Maehara M, Kuroda Y. Concomitant administration of sodium dichloroacetate and thiamine in west syndrome caused by thiamine-responsive pyruvate dehydrogenase complex deficiency. J Neurol Sci. 1999;171:56–59. doi: 10.1016/s0022-510x(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 119.Lissens W, Vreken P, Barth PG, Wijburg FA, Ruitenbeek W, Wanders RJA, Seneca S, Liebaers I, De Meirleir L. Cerebral palsy and pyruvate dehydrogenase deficiency: identification of two new mutations in the E1 gene. Eur J Pediatr. 1999;158:853–857. doi: 10.1007/s004310051222. [DOI] [PubMed] [Google Scholar]
- 120.Naito E, Ito M, Yokota I, Saijo T, Ogawa Y, Shinahara K, Kuroda Y. Gender-specific occurrence of West syndrome in patients with pyruvate dehydrogenase complex deficiency. Neuropediatrics. 2001;32:295–298. doi: 10.1055/s-2001-20404. [DOI] [PubMed] [Google Scholar]
- 121.Seyda A, Chun K, Packman S, Robinson BH. A case of PDH-E1 alpha mosaicism in a male patient with severe metabolic lactic acidosis. J Inherit Metab Dis. 2001;24:551–559. doi: 10.1023/a:1012463726810. [DOI] [PubMed] [Google Scholar]
- 122.Benelli C, Fouque F, Redonnet-Vernhet I, Malgat M, Fontan D, Marsac C, Dey R. A novel Y243S mutation in the pyruvate dehydrogenase E1 alpha gene subunit: correlation with thiamine pyrophosphate interaction. J Inherit Metab Dis. 2002;25:325–327. doi: 10.1023/a:1016570828778. [DOI] [PubMed] [Google Scholar]
- 123.Naito E, Ito M, Yokota I, Saijo T, Ogawa Y, Kuroda Y. Diagnosis and molecular analysis of three male patients with thiamine-responsive pyruvate dehydrogenase complex deficiency. J Neurol Sci. 2002;201:33–37. doi: 10.1016/s0022-510x(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 124.Dey R, Aral B, Abitbol M, Marsac C. Pyruvate dehydrogenase deficiency as a result of splice-site mutations in the PDX1 gene. Mol Genet Metab. 2002;76:344–347. doi: 10.1016/s1096-7192(02)00104-x. [DOI] [PubMed] [Google Scholar]
- 125.Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Ogawa Y, Kitamura S, Takada E, Horii Y, Kuroda Y. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim Biophys Acta. 2002;1588:79–84. doi: 10.1016/s0925-4439(02)00142-4. [DOI] [PubMed] [Google Scholar]
- 126.Hargreaves IP, Heales SJR, Briddon A, Lee PJ, Hanna MG, Land JM. Primary pyruvate dehydrogenase deficiency with mild hyperlactatemia and hyperalaninemia. J Inherit Metab Dis. 2003;26:505–506. doi: 10.1023/a:1025181512847. [DOI] [PubMed] [Google Scholar]
- 127.Zand DJ, Simon EM, Pulitzer SB, Wang DJ, Wang ZJ, Rorke LB, Palmieri M, Berry GT. In vivo pyruvate detected by MR spectroscopy in neonatal pyruvate dehydrogenase deficiency. AJNR Am J Neuroradiol. 2003;24:1471–1474. [PMC free article] [PubMed] [Google Scholar]
- 128.Dey R, Mine M, Desguerre I, Slama A, Van Den Berghe L, Brivet M, Aral B, Marsac C. A new case of pyruvate dehydrogenase deficiency due to a novel mutation in the PDX1 gene. Ann Neurol. 2003;53:273–277. doi: 10.1002/ana.10478. [DOI] [PubMed] [Google Scholar]
- 129.Grafakou O, Oexle K, Van den Heuvel L, Smeets R, Trijbels F, Goebel HH, Bosshard N, Superti-Furga A, Steinmann B, Smeitink J. Leigh syndrome due to compound heterozygosity of dihydrolipoamide dehydrogenase gene mutations: description of the first E3 splice site mutation. Eur J Pediatr. 2003;163:714–718. doi: 10.1007/s00431-003-1282-z. [DOI] [PubMed] [Google Scholar]
- 130.Brown RM, Head RA, Bourbriak II, Leonard JV, Brown GK. A pathogenic glutamate to aspartate substitution (D296E) in the pyruvate dehydrogenase E1 subunit gene PDHA1. Hum Mutat. 2003;22:496–497. doi: 10.1002/humu.9198. [DOI] [PubMed] [Google Scholar]
- 131.Mellick G, Price L, Boyle R. Late-onset presentation of pyruvate dehydrogenase deficiency. Mov Disord. 2004;19:727–729. doi: 10.1002/mds.20063. [DOI] [PubMed] [Google Scholar]
- 132.Brown RM, Head RA, Boubriak II, Leonard JV, Thomas NH, Brown GK. Mutations in the gene for the E1β subunit: a novel cause of pyruvate dehydrogenase deficiency. Hum Genet. 2004;115:123–127. doi: 10.1007/s00439-004-1124-8. [DOI] [PubMed] [Google Scholar]
- 133.Cameron JM, Levandovskiy V, Mackay N, Tein I, Robinson BH. Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1α subunit. Am J Med Genet A. 2004;131:59–66. doi: 10.1002/ajmg.a.30287. [DOI] [PubMed] [Google Scholar]
- 134.Head RA, de Goede CGEL, Newton RWN, Walter JH, McShane MA, Brown RM, Brown GK. Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev Med Child Neurol. 2004;46:710–712. doi: 10.1017/s0012162204001197. [DOI] [PubMed] [Google Scholar]
- 135.Wada N, Matsuishi T, Nonaka M, Naito E, Yoshino M. Pyruvate dehydrogenase E1α subunit deficiency in a female patient: evidence of antenatal origin of brain damage and possible etiology of infantile spasms. Brain Dev. 2004;26:57–60. doi: 10.1016/s0387-7604(03)00072-x. [DOI] [PubMed] [Google Scholar]
- 136.Head RA, Brown RM, Zolkipli Z, Shahdadpuri R, King MD, Clayton PT, Brown GK. Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann Neurol. 2005;58:234–241. doi: 10.1002/ana.20550. [DOI] [PubMed] [Google Scholar]
- 137.Tulinius M, Darin N, Wiklund LM, Holmberg E, Eriksson JE, Lissens W, De Meirleir L, Holme E. A family with pyruvate dehydrogenase complex deficiency due to a novel C>T substitution at nucleotide position 407 in exon 4 of the X linked E1α gene. Eur J Pediatr. 2005;164:99–103. doi: 10.1007/s00431-004-1570-2. [DOI] [PubMed] [Google Scholar]
- 138.Brivet M. First characterization of large deletion of the PDHA1 gene. Mol Genet Metab. 2005;89:456–461. doi: 10.1016/j.ymgme.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 139.Hansen LL, Horn N, Dahl HH, Kruse TA. Pyruvate dehydrogenase deficiency caused by a 33 base pair duplication in the PDH E1 alpha subunit. Hum Mol Genet. 1994;3:1021–1022. doi: 10.1093/hmg/3.6.1021. [DOI] [PubMed] [Google Scholar]
- 140.Brown RM, Head RA, Morris AAM, Raiman JAJ, Walter JH, Whitehouse WP, Brown GK. Pyruvate dehydrogenase E3 binding protein (protein X) deficiency. Dev Med Child Neurol. 2006;48:756–760. doi: 10.1017/S0012162206001617. [DOI] [PubMed] [Google Scholar]
- 141.Strassburg HM, Koch J, Mayr J, Sperl W, Boltshauser E. Acute flaccid paralysis as initial symptom in 4 patients with novel E1α mutations of the pyruvate dehydrogenase complex. Neuropediatrics. 2006;37:137–141. doi: 10.1055/s-2006-924555. [DOI] [PubMed] [Google Scholar]
- 142.Cameron JM, Levandovskiy V, Mackay N, Raiman J, Renaud DL, Clarke JTR, Feigenbaum A, Elpeleg O, Robinson BH. Novel mutations in dihydrolipoamide dehydrogenase deficiency in two cousins with borderline-normal complex activity. Am J Med Genet A. 2006;140:1542–1552. doi: 10.1002/ajmg.a.31313. [DOI] [PubMed] [Google Scholar]
- 143.Schiff M, Miné M, Brivet M, Marsac C, Elmaleh-Bergés M, Evrard P, Ogier de Baulny H. Leigh’s disease due to a new mutation in PDHX gene. Ann Neurol. 2006;59:709–714. doi: 10.1002/ana.20818. [DOI] [PubMed] [Google Scholar]
- 144.Okajima K, Warman ML, Byrne LC, Kerr DS. Somatic mosaicism in a male with an exon skipping mutation in PDHA1 of the pyruvate dehydrogenase complex results in a milder phenotype. Mol Genet Metab. 2006;87:162–168. doi: 10.1016/j.ymgme.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 145.Willemsen M, Rodenburg RJ, Teszas A, van den Heuvel L, Kosztolanyi G, Morava E. Females with PDHA1 gene mutations: a diagnostic challenge. Mitochondrion. 2006;6:155–159. doi: 10.1016/j.mito.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 146.Debray FG, Lambert M, Vanasse M, Decarie JC, Cameron J, Levandovskiy V, Robinson BH, Mitchell GA. Intermittent peripheral weakness as the presenting feature of pyruvate dehydrogenase deficiency. Eur J Pediatr. 2006;165:462–466. doi: 10.1007/s00431-006-0104-5. [DOI] [PubMed] [Google Scholar]
- 147.Soares-Fernandes JP, Teixeira-Gomes R, Cruz R, Ribeiro M, Magalhães Z, Rocha JF, Leijser LM. Neonatal pyruvate dehydrogenase deficiency due to a R302H mutation in the PDHA1 gene: MRI findings. Pediatr Radiol. 2008;38:559–562. doi: 10.1007/s00247-007-0721-9. [DOI] [PubMed] [Google Scholar]
- 148.Ridout CK, Brown RM, Walter JH, Brown GK. Somatic mosaicism for a PDHA1 mutation in a female with pyruvate dehydrogenase deficiency. Hum Genet. 2008;124:187–193. doi: 10.1007/s00439-008-0538-0. [DOI] [PubMed] [Google Scholar]
- 149.Sedel F, Challe G, Mayer JM, Boutron A, Fontaine B, Saudubray JM, Brivet M. Thiamine responsive pyruvate dehydrogenase deficiency in an adult with peripheral neuropathy and optic neuropathy. J Neurol Neurosurg Psychiatry. 2008;79:846–847. doi: 10.1136/jnnp.2007.136630. [DOI] [PubMed] [Google Scholar]
- 150.Han Z, Zhong L, Srivastava A, Stacpoole PW. Pyruvate dehydrogenase complex deficiency caused by ubiquitination and proteasome-mediated degradation of the E1β subunit. J Biol Chem. 2008;283:237–243. doi: 10.1074/jbc.M704748200. [DOI] [PubMed] [Google Scholar]
- 151.Okajima K, Korotchkina LG, Prasad C. Mutations of the E1β subunit gene (PDHB) in four families with pyruvate dehydrogenase deficiency. Mol Genet Metab. 2008;93:371–380. doi: 10.1016/j.ymgme.2007.10.135. [DOI] [PubMed] [Google Scholar]
- 152.Boichard A, Venet L, Naas T, Boutron A, Chevret L, de Baulny HO, De Lonlay P, Legrand A, Nordman P, Brivet M. Two silent substitutions in the PDHA1 gene cause exon 5 skipping by disruption of putative exonic splicing enhancer. Mol Genet Metab. 2008;93:323–330. doi: 10.1016/j.ymgme.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 153.Debray FG, Lambert M, Gagne R, Maranda B, Laframboise R, MacKay N, Robinson BH, Mitchell GA. Pyruvate dehydrogenase deficiency presenting as intermittent isolated acute ataxia. Neuropediatrics. 2008;39:20–23. doi: 10.1055/s-2008-1077084. [DOI] [PubMed] [Google Scholar]
- 154.Ostergaard E, Moller LB, Kalkanoglu-Sivri HS, Dursun A, Kibaek M, Thelle T, Christensen E, Duno M, Wibrand F. Four novel PDHA1 mutations in pyruvate dehydrogenase deficiency. J Inherit Metab. 2009;115:123–127. doi: 10.1007/s10545-009-1179-8. [DOI] [PubMed] [Google Scholar]
- 155.Small J, Gonzales G, Nagao K, Walton DS, Caruso PA. Optic neuropathy in a patient with pyruvate dehydrogenase deficiency. Pediatr Radiol. 2009;39:1114–1117. doi: 10.1007/s00247-009-1344-0. [DOI] [PubMed] [Google Scholar]
- 156.Bachmann-Gagescu R, Lawrence Merritt J, II, Hahn SH. A cognitively normal PDH-deficient 18-year old man carrying the R263G mutation in the PDHA1 gene. J Inherit Metab Dis. doi: 10.1007/s10545-009-1101-4. (in press) [DOI] [PubMed] [Google Scholar]
- 157.Quintana E, Mayr JA, García Silva MT, Font A, Tortoledo MA, Moliner S, Ozaez L, Lluch M, Cabello A, Ricoy JR, Koch J, Ribes A, Sperl W, Briones P. PDH E(1)beta deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis. doi: 10.1007/s10545-009-1343-1. (in press) [DOI] [PubMed] [Google Scholar]
- 158.João Silva M, Pinheiro A, Eusébio F, Gaspar A, Tavares de Almeida I, Rivera I. Pyruvate dehydrogenase deficiency: identification of a novel mutation in the PDHA1 gene which responds to amino acid supplementation. Eur J Pediatr. 2009;168:17–22. doi: 10.1007/s00431-008-0700-7. [DOI] [PubMed] [Google Scholar]
- 159.Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G, Marsac C, Brivet M, Desguerre I. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52:e1–e9. doi: 10.1111/j.1469-8749.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- 160.Coughlin CR, Krantz ID, Scmitt ES, Zhang S, Wong LJ, Kerr DS, Ganesh J. Somatic mosaicism for PDHA1 mutation in a male with pyruvate dehydrogenase complex deficiency. Mol Genet Metab. 2010;100:296–299. doi: 10.1016/j.ymgme.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 161.Sato S, Ioi H, Kashiwagi Y, Kawashima H, Miyajima T, Naito E, Hoshika A. Novel mutation (R263X) of the E1α subunit in pyruvate dehydrogenase complex deficiency. Pediatr Int. 2010;52:e181–e183. doi: 10.1111/j.1442-200X.2010.03112.x. [DOI] [PubMed] [Google Scholar]
- 162.McWilliam CA, Ridout CK, Brown RM, McWilliam RC, Tolmie J, Brown GK. Pyruvate dehydrogenase E2 deficiency: a potentially treatable cause of episodic dystonia. Eur J Paediatr Neurol. 2010;14:349–353. doi: 10.1016/j.ejpn.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 163.Quintana E, Gort L, Busquets C, Navarro-Sastre A, Lissens W, Moliner S, Lluch M, Vilaseca MA, De Meirleir L, Ribes A, Briones P PDH Working Group. Mutational study in the PDHA1 gene of 40 patients suspected of pyruvate dehydrogenase complex deficiency. Clin Genet. 2010;77:474–482. doi: 10.1111/j.1399-0004.2009.01313.x. [DOI] [PubMed] [Google Scholar]
- 164.Karppa M, Syrjala P, Tolonen U, Majamaa K. Peripheral neuropathy in patients with the 3243A>G mutation in mitochondrial DNA. J Neurol. 2003;250:216–221. doi: 10.1007/s00415-003-0981-8. [DOI] [PubMed] [Google Scholar]
- 165.Stickler D, Valenstein E, Neiberger RE, Perkins LA, Carney PR, Shuster JJ, Theriaque DW, Stacpoole PW. Peripheral neuropathy in genetic mitochondrial diseases. Pediatr Neurology. 2006;34:127–131. doi: 10.1016/j.pediatrneurol.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 166.Kaufmann P, Pascual JM, Anziska Y, Gooch CL, et al. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch Neurol. 2006;63:746–748. doi: 10.1001/archneur.63.5.746. [DOI] [PubMed] [Google Scholar]
- 167.Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- 168.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 169.Lib MY, Brown RM, Brown GK, Marusich MF, et al. Detection of pyruvate dehydrogenase E1α-subunit deficiencies in females by immunohistochemical demonstration of mosaicism in cultured fibroblasts. J Histochem Cytochem. 2002;50:877–884. doi: 10.1177/002215540205000701. [DOI] [PubMed] [Google Scholar]
- 170.Lissens W, De Meirleir L, Seneca S, Liebaers I, et al. Mutations in the X-linked pyruvate dehydrogenase (E1) alpha subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat. 2000;15:209–219. doi: 10.1002/(SICI)1098-1004(200003)15:3<209::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 171.Mine M, Brivet M, Schiff M, Ogier H, et al. A novel gross deletion caused by non-homologous recombination of the PDHX gene in a patient with pyruvate dehydrogenase deficiency. Mol Genet Metab. 2006;89:106–110. doi: 10.1016/j.ymgme.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 172.Ridout CK, Keighley P, Krywawych S, Brown RM, et al. A putative exonic splicing enhancer in exon 7 of the PDHA1 gene affects splicing of adjacent exons. Hum Mutat. 2008;29:451–463. doi: 10.1002/humu.9525. [DOI] [PubMed] [Google Scholar]
- 173.Kerr DS. Treatment of congenital lactic acidosis: a review. Int Pediatr. 1995;10:75–81. [Google Scholar]
- 174.Kossoff EH, Zupec-Kania BA, Amark PE, Ballaban-Gil KR, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50:304–317. doi: 10.1111/j.1528-1167.2008.01765.x. [DOI] [PubMed] [Google Scholar]
- 175.Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38:1124–1144. doi: 10.1016/0026-0495(89)90051-6. [DOI] [PubMed] [Google Scholar]
- 176.Han Z, Berendzen K, Zhong L, Surolia I, et al. A combined therapeutic approach for pyruvate dehydrogenase deficiency using self-complementary adeno-associated virus serotype-specific vectors and dichloroacetate. Mol Genet Metab. 2008;93:381–387. doi: 10.1016/j.ymgme.2007.10.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Duncan GE, Perkins LA, Theriaque DW, Neiberger RE, et al. Dichloroacetate therapy attenuates the blood lactate response to submaximal exercise in patients with defects in mitochondrial energy metabolism. J Clin Endocrinol Metab. 2004;89:1733–1738. doi: 10.1210/jc.2003-031684. [DOI] [PubMed] [Google Scholar]
- 178.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, et al. A controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 179.Kaufmann P, Engelstad K, Wei Y, Jhung S, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.