

Abstract
Atrial fibrillation (AF) is the most common form of sustained cardiac arrhythmia in humans and is responsible for substantial morbidity and mortality worldwide. Emerging evidence indicates that abnormal cardiovascular development is involved in the pathogenesis of AF. In this study, the coding exons and splice sites of the NKX2-5 gene, which encodes a homeodomain-containing transcription factor essential for cardiovascular genesis, were sequenced in 146 unrelated patients with lone AF as well as the available relatives of the mutation carriers. A total of 700 unrelated ethnically matched healthy individuals used as controls were genotyped. The disease-causing potential of the identified NKX2-5 variations was predicted by MutationTaster and PolyPhen-2. The functional characteristics of the mutant NKX2-5 proteins were analyzed using a dual-luciferase reporter assay system. As a result, two heterozygous NKX2-5 mutations, including a previously reported p.E21Q and a novel p.T180A mutation, were identified in two families with AF transmitted in an autosomal dominant pattern. The mutations co-segregated with AF in the families with complete penetrance. The detected substitutions, which altered the amino acids highly conserved evolutionarily across species, were absent in 700 control individuals and were both predicted to be causative. Functional analyses demonstrated that the NKX2-5 mutants were associated with significantly decreased transcriptional activity compared with their wild-type counterpart. The findings expand the spectrum of NKX2-5 mutations linked to AF and provide additional evidence that dysfunctional NKX2-5 may confer vulnerability to AF, suggesting the potential benefit for the early prophylaxis and personalized treatment of AF.
Keywords: Atrial fibrillation, Transcription factor, NKX2-5, Molecular genetics, Mutation.
Introduction
Atrial fibrillation (AF), characterized by rapid and chaotic electrical activity in the atria with consequent deterioration of atrial mechanical function, is the most common form of cardiac arrhythmia seen in clinical practice, accounting for about one-third of hospitalizations for cardiac rhythm disturbances 1. The prevalence of AF is estimated to be approximately 1% in the general population, and increases markedly with advancing age, rising from less than 1% in individuals under 60 years of age to nearly 10% in subjects aged 80 years and above 1. According to a report from the Framingham Heart Study, the lifetime risk of developing AF is about 25% in males and females aged over 40 years 2. This supraventricular tachycardia, often resulting in an irregular ventricular response, is a major cause of substantial cardiovascular morbidity and mortality. AF is associated with a two-fold increase in the risk of death 3, and confers a five-fold increased risk of thromboembolic stroke 4. The annual incidence of stroke attributable to AF also increases with age, ranging from 1.5% of people in their fifties to 23.5% in octogenarians 4,5. In addition, AF may contribute to degraded quality of life, reduced exercise tolerance, impaired cognitive function, tachycardia-induced cardiomyopathy, and left ventricular dysfunction or even congestive heart failure 1,6. AF commonly occurs in association with various cardiac and systemic conditions, such as coronary artery disease, congenital heart disease, rheumatic heart disease, cardiac surgery, cardiomyopathy, myocarditis, pulmonary heart disease, hypertension, hyperthyroidism, and even electrolyte imbalance 1,7-9. However, in 2% to 45% of people with AF, there are no obvious pathologic substrates or risk factors, and such AF is termed lone AF 1, 10. Among patients with lone AF, at least 15% present with a positive family history, so diagnosed as familial AF 11. Increasing genetic epidemiological studies from around the world have consistently demonstrated the substantial familial aggregation of AF and a significantly increased risk for AF in the close relatives of patients with AF, indicating a strong genetic basis for AF 12-17. Genome-wide genetic linkage analysis with highly polymorphic microsatellite markers mapped AF-susceptibility loci on human chromosomes 10q22, 6q14-16, 11p15.5, 5p15, 10p11-q21, and 5p13, of which AF-causing mutations in two genes, including KCNQ1 on chromosome 11p15.5 and NUP155 on chromosome 5p13, were identified and functionally characterized 18. Analyses of candidate genes unveiled a long list of AF-associated genes, including KCNQ1, KCNE1, KCNE2, KCNA5, KCND3, KCNJ2, KCNH2, SCN5A, GJA1, GJA5, NPPA, and SCN1-4B 18-21. However, these established AF-related genes seem to be relatively rare causes of AF, and in an overwhelming majority of patients, the genetic defects predisposing to AF remain to be unknown.
Emerging evidence suggests that anomalous embryological development of the cardiovascular system, especially the pulmonary venous myocardium, is a common and key anatomic substrate for AF 22. Research in human developmental biology reveals the essential role of several transcription factors, including NKX2-5, GATA4, GATA5 and GATA6, in the cardiovascular genesis 23, and multiple mutations in GATA4, GATA5 and GATA6 have been causally implicated in the pathogenesis of AF 24-33. The expression and function of NKX2-5 overlap with those of the GATA family during cardiovascular development, especially in regulation of target gene expression synergistically with GATA4, which makes NKX2-5 a logical candidate gene for AF 34.
Materials and Methods
Ethics
This study was carried out in compliance with the ethical principles of the revised Declaration of Helsinki (Somerset West, Republic of South Africa, 1996). The research protocol was reviewed and approved by the local institutional ethics committee (the ethical approval number for cases and controls: KS1101; the date for the approval: April 12, 2011) and written informed consent was obtained from all participants prior to the study.
Study subjects
A cohort of 146 unrelated patients with lone AF was prospectively recruited from the Chinese Han population for the present study. In addition, if an NKX2-5 mutation was identified in a patient, the mutation carrier's available family members were also included. A total of 700 ethnically matched unrelated healthy individuals were enlisted as controls. All the subjects were assessed with regard to clinical and family history. Comprehensive physical examination, routine biological test, standard 12-lead electrocardiogram and transthoracic echocardiogram were performed. The clinical data including medical records, electrocardiogram and echocardiography reports were collected and reviewed. The study subjects were clinically classified as having AF using a consistently applied set of definitions 11.
Genetic screening
Peripheral venous blood specimens from all participants were prepared. Genomic DNA was isolated from blood lymphocytes of participants using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. The referential genomic DNA sequence of NKX2-5 was derived from GenBank (GenBank ID: NG_013340.1), a gene sequence database at the National Center for Biotechnical Information (NCBI; http://www.ncbi.nlm.nih.gov/). With the aid of the online Primer 3 program (http://frodo.wi.mit.edu/), the primer pairs used to amplify the coding exons (exon 1-2) and intron-exon boundaries of NKX2-5 by polymerase chain reaction (PCR) were designed as previously described 35. The NKX2-5 gene was screened for potential sequence variations by PCR-sequencing in 146 unrelated patients with lone AF. The available family members of the mutation carriers and the 700 unrelated healthy control individuals were also genotyped for NKX2-5. The PCR was performed using HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster, CA, USA) with standard conditions and concentrations of reagents. Amplified products were purified with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Amplicons were bi-directionally sequenced on an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems, Foster, CA, USA) with BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems, Foster, CA, USA). The sequencing primers were the same as designed previously for specific region amplifications. DNA sequences were viewed and analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems, Foster, CA, USA). Each variant was corroborated by bi-directional re-sequencing of an independent PCR-generated amplicon from the same subject and met the quality control threshold with a call rate exceeding 99%. For identified sequence variants, several standard databases including the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/) and the NCBI′s Single Nucleotide Polymorphism (SNP; http://www.ncbi.nlm.nih.gov/snp) and PubMed Databases (http://www.ncbi.nlm.nih.gov/pubmed) were queried to confirm their novelty.
Alignment of multiple NKX2-5 protein sequences
The conservation of the amino acids of NKX2-5 was evaluated by aligning human NKX2-5 to chimpanzee, monkey, dog, cow, mouse, rat, chicken, and zebrafish NKX2-5 using the HomoloGene and Show Multiple Alignment links on the NCBI's web site (http://www.ncbi.nlm.nih.gov/homologene).
Prediction of the disease-causing potential of NKX2-5 sequence variations
The disease-causing potential of NKX2-5 sequence variations was predicted by MutationTaster (an online program at website http://www.mutationtaster.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), automatically giving a probability score for each alteration to be either pathogenic or benign.
Expression plasmids and site-directed mutagenesis
The recombinant expression plasmids NKX2-5-pEFSA and pSSRa-GATA4, and the atrial natriuretic factor (ANF)-luciferase reporter plasmid, which contains the 2600-bp 5'-flanking region of the ANF gene, namely ANF(-2600)-Luc, were kindly provided by Dr. Ichiro Shiojima, from the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chuo-ku, Chiba, Japan. Each of the identified mutations was introduced into the wild-type NKX2-5 using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with a complementary pair of primers. The mutants were sequenced to confirm the desired mutations and to exclude any other sequence variations.
Luciferase reporter gene assays
COS-7 cells were cultured in DMEM supplemented with 10% fetal calf serum. The ANF(-2600)-Luc reporter construct and an internal control reporter plasmid pGL4.75 (hRluc/CMV, Promega, Madison, WI, USA) were used in transient transfection assays to examine the transcriptional activation function of the NKX2-5 mutants. COS-7 cells were transfected with 0.4 μg of wild-type or mutant NKX2-5-pEFSA expression vector, 1.0 μg of ANF(-2600)-Luc reporter construct, and 0.04 μg of pGL4.75 control reporter vector using PolyFect Transfection Reagent (Qiagen, Hilden, Germany). For co-transfection experiments, 0.2 μg of wild-type NKX2-5-pEFSA, 0.2 μg of mutant NKX2-5-pEFSA or empty vector pEFSA, 1.0 μg of ANF(-2600)-Luc, and 0.04 μg of pGL4.75 were used. Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega, Madison, WI, USA) 48h after transfection. The activity of the ANF promoter was presented as fold activation of Firefly luciferase relative to Renilla luciferase. Three independent experiments were performed at minimum for wild-type and mutant NKX2-5.
Given that NKX2-5 and GATA4 activate several cardiac gene promoters including the ANF promoter in a synergistic manner 36, similar experiments were performed in the presence of GATA4 to explore the synergistic effects of NKX2-5 and GATA4 on the ANF promoter. By using PolyFect Transfection Reagent (Qiagen, Hilden, Germany), COS-7 cells were transfected with 0.2 μg of wild-type NKX2-5-pEFSA, or 0.2μg of pSSRa-GATA4, or 0.2 μg of wild-type NKX2-5-pEFSA in combination with 0.2μg of pSSRa-GATA4, in the presence of 1.0 μg of ANF(-2600)-Luc and 0.02 μg of pGL4.75. For co-transfection assay, 0.1 μg of wild-type NKX2-5-pEFSA and 0.1 μg of each mutant NKX2-5-pEFSA were used, in the presence of 0.2μg of pSSRa-GATA4, 1.0 μg of ANF(-2600)-Luc, and 0.02 μg of pGL4.75.
Statistics
Data are presented as means ± SD. Continuous variables were tested for normal distribution and Student's unpaired t test was used to make comparison of numeric variables between two groups. Comparison of the categorical variables between two groups was performed using Pearson's χ2 test or Fisher's exact test when appropriate. A two-tailed p value < 0.05 was considered statistically significant.
Results
Clinical characteristics of the study population
A cohort of 146 unrelated patients with lone AF was enrolled and clinically investigated. A total of 700 ethnically matched unrelated healthy individuals were also clinically assessed. None of them had overt traditional risk factors for AF. There was no significant difference between patient and control groups in baseline characteristics including age, gender, body mass index, serum lipid, blood pressure, fasting blood glucose, left atrial dimension, left ventricular ejection fraction, heart rate at rest, as well as life style (data not shown). During five years of follow-up, seven patients were also diagnosed with hypertension in accordance to the criterion that the average systolic or diastolic blood pressure (two readings made after five minutes of rest in sitting position) was ≥140 mm Hg or ≥90 mm Hg, respectively, but at the time of initial diagnosis of AF, their blood pressures were normal. Besides, other traditional risk factors for AF such as structural heart disease, diabetes mellitus and hyperthyroidism were also absent at the time of first diagnosis of AF. The baseline clinical characteristics of the 146 patients with lone AF are summarized in Table 1.
Table 1.
Baseline demographics and clinical characteristics of the study population.
| Parameters | Statistics |
|---|---|
| Baseline demographics | |
| Age at first diagnosis of atrial fibrillation (years) | 44 ± 8 |
| Male (n, %) | 89 (61) |
| Body mass index (kg/m2) | 23 ± 1 |
| Left ventricular ejection fraction (%) | 62 ± 5 |
| Left atrial diameter (mm) | 36 ± 3 |
| Personal history of atrial fibrillation (n, %) | |
| Type of atrial fibrillation at presentation | |
| Paroxysmal | 97 (66) |
| Persistent | 32 (22) |
| Permanent | 17 (12) |
| History of cardioversion | 110 (75) |
| Positive family history of atrial fibrillation | 68 (47) |
| Medical history (n, %) | |
| History of syncope | 14 (10) |
| History of pacemaker | 10 (7) |
| Thromboembolic complication | 9 (6) |
| Hyperlipidemia | 8 (5) |
| Arterial hypertension | 7 (5) |
| Diabetes | 5 (3) |
| Medications (n, %) | |
| Amiodarone | 105 (72) |
| Warfarin | 84 (58) |
| Digitalis | 23 (16) |
| Aspirin | 18 (12) |
| Beta-blocker | 12 (8) |
| Calcium channel blocker | 6 (4) |
Continuous variables are expressed as means and standard deviation.
NKX2-5 mutations
Two heterozygous mutations in NKX2-5 were identified in 2 of 146 unrelated patients with lone AF, with a mutational prevalence of roughly 1.37%. Specifically, a substitution of cytosine (C) for guanine (G) in the first nucleotide of codon 21 (c.61G>C), predicting the replacement of glutamic acid (E) with glutamine (Q) at amino acid position 21 (p.E21Q), was identified in a patient from family 1. A substitution of G for adenine (A) at coding nucleotide 538 (c.538A>G), corresponding to the replacement of threonine (T) with alanine (A) at amino acid position 180 (p.T180A), was detected in a patient from family 2. The sequence chromatograms showing the detected heterozygous NKX2-5 mutations of c.61G>C and c.538A>G compared with their control sequences are shown in Figure 1. A schematic diagram of NKX2-5 showing the structural domains and the locations of the identified mutations is presented in Figure 2 37. The identified mutations were absent in the control individuals. The p.T180A mutation was not found in standard databases like HGMD, SNP, and PubMed, indicating a novel mutation. Although the p.E21Q mutation was previously identified in patients with congenital atrial septal defect or tetralogy of Fallot 38-40, it has not been associated with AF before.
Figure 1.

Sequence electropherograms showing the NKX2-5 mutations in contrast to their corresponding controls. The arrow indicates the heterozygous nucleotides of G/C and A/G in the probands from families 1 (A) and 2 (B), respectively (mutant) or the homozygous nucleotides of G/G (A) and A/A (B) in the corresponding control individuals (wild-type). The rectangle denotes the nucleotides comprising a codon of NKX2-5.
Figure 2.
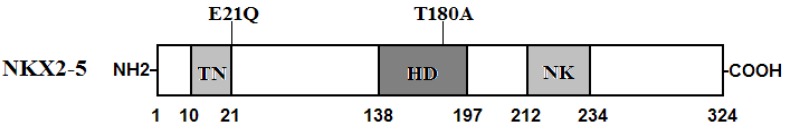
Schematic representation of NKX2-5 protein structure with the atrial fibrillation (AF)-related mutations indicated. The mutations identified in patients with lone atrial fibrillation are shown above the structural domains. NH2 means amino-terminus; TN, transcriptional activation domain; HD, homeodomain; NK, NK2-specific domain; and COOH, carboxyl-terminus.
Genetic scan of the relatives of each mutation carrier demonstrated that the mutation was present in all affected family members alive, but absent in unaffected family members examined. Analysis of the pedigrees showed that in each family the mutation co-segregated with AF transmitted in an autosomal dominant pattern with complete penetrance. The pedigree structures of the families are illustrated in Figure 3. The phenotypic characteristics and results of genetic screening of the affected family members available are listed in Table 2.
Figure 3.

Pedigree structures of the families with lone atrial fibrillation. Families are designated as family 1 and family 2, respectively. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, affected members; open symbols, unaffected members; arrows, probands; ''+'', carriers of the heterozygous mutations; and ''-'', non-carriers.
Table 2.
Phenotypic characteristics and status of NKX2-5 mutations of the affected pedigree members.
| Subject information | Phenotype | Electrocardiogram | Echocardiogram | Genotype | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Identity | Gender | Age at time of study (years) | Age at diagnosis of AF (years) | AF (Classification) |
Heart rate (beats/min) | QRS interval (ms) | QTc | LAD (mm) | LVEF (%) | NKX2-5 mutation |
| Family 1 | E21Q | |||||||||
| I-1 | M | 71 | 52 | Permanent | 69 | 96 | 454 | 39 | 62 | +/- |
| II-2 | F | 46 | 34 | Persistent | 72 | 98 | 416 | 37 | 66 | +/- |
| II-3 | M | 43 | 40 | Paroxysmal | 70 | 90 | 449 | 35 | 64 | +/- |
| Family 2 | T180A | |||||||||
| I-1 | M | 65 | 26 | Permanent | 66 | 110 | 458 | 42 | 60 | +/- |
| II-4 | F | 40 | 32 | Paroxysmal | 70 | 98 | 393 | 33 | 62 | +/- |
AF: atrial fibrillation, F: female, M: male, QTc: corrected QT interval, LAD: left atrial dimension, LVEF: left ventricular ejection fraction, +: present, -: absent.
Interestingly, congenital atrial septal defect was confirmed by the early echocardiogram and medical records of previous catheter-based closure in patient I-1 from family 2. In addition, two NKX2-5 polymorphisms, c.63A>G and c.606C>G, were observed in both AF patients and control individuals. However, there was no significant difference in either of the two allele frequencies between the AF patient and healthy control groups. All the sequence variants and their allele frequencies are listed in Table 3.
Table 3.
NKX2-5 sequence variations identified in this study.
| Location | Nucleotide | Amino acid | Allele frequency | |
|---|---|---|---|---|
| Patients | Controls | |||
| Exon 1 | c.61G>C | p.E21Q | (0.003) 1/292 | (0.000) 0/1400 |
| Exon 1 | c.63A>G | p.E21E | (0.223) 65/292 | (0.259) 362/1400 |
| Exon 2 | c.538A>G | p.T180A | (0.003) 1/292 | (0.000) 0/1400 |
| Exon 2 | c. 606C>G | p.L202L | (0.021) 6/292 | (0.016) 23/1400 |
Multiple NKX2-5 sequence alignments
Alignment of multiple NKX2-5 proteins across species showed that the altered amino acids were completely conserved evolutionarily, underscoring their functional importance, as shown in Figure 4.
Figure 4.

Alignment of multiple NKX2-5 protein sequences across species. The altered amino acids of p.E21 and p.T180 are completely conservative evolutionarily among various species.
Disease-causing potential of NKX2-5 sequence variations
The NKX2-5 mutations were both predicted to be disease-causing by MutationTaster, with p values of 0.98 and 1.00 for c.61G>C and c.538A>G, respectively, and they were predicted to be possibly damaging by PolyPhen-2, with a score of 0.95 (sensitivity: 0.79; specificity: 0.95) for c.61G>C and 1.00 (sensitivity: 0.14; specificity: 0.99) for c.538A>G, implying that mutated NKX2-5 might contribute to the development of AF in these families. No SNPs in the altered regions were found in the MutationTaster database.
Transcriptional activity of the mutant NKX2-5
The transcriptional activation characteristics of the mutated NKX2-5 in COS-7 cells were assayed using the promoter of ANF, one of the target genes regulated by NKX2-5, as a driver of luciferase reporter. The activity of the ANF promoter was expressed as fold activation of Firefly luciferase relative to Renilla luciferase. The same amounts of wild-type (0.4 μg), E21Q-mutant (0.4 μg), and T180A-mutant NKX2-5 (0.4 μg) activated the ANF promoter by ~13-fold, ~6-fold, and ~3-fold, respectively, with p values of 0.002 for E21Q-mutant and 0.001 for T180A-mutant when compared with wild-type NKX2-5. When the same amount of wild-type NKX2-5 (0.2 μg) was cotransfected with E21Q-mutant (0.2 μg) or T180A -mutant (0.2 μg), the induced activation of the ANF promoter was ~7-fold or ~5-fold, with p values of 0.003 for E21Q-mutant and 0.002 for T180A-mutant when compared with wild-type NKX2-5 (0.4 μg). These results suggest that the NKX2-5 mutations are associated with significantly reduced transactivational activity compared with their wild-type counterpart (Figure 5).
Figure 5.

Functional defects caused by NKX2-5 mutations. Activation of atrial natriuretic factor promoter driven luciferase reporter in COS-7 cells by NKX2-5 wild-type (WT), E21Q-mutant, or T180A-mutant, alone or in combination, demonstrated significantly reduced transactivational activity by mutant proteins. Experiments were performed in triplicate, and mean and standard deviations are shown. ** and * represent p < 0.0005 and p < 0.001, respectively, when compared with wild-type NKX2-5.
In addition, as shown in Figure 6, biological analysis demonstrated that NKX2-5 and GATA4 activated the ANF promoter in a synergistic manner. Cotransfection with wild-type NKX2-5 (0.2 μg) and GATA4 (0.2 μg) showed much stronger activation (~25-fold) of the ANF promoter than single transfection with 0.2 μg of wild-type NKX2-5 (~10-fold; p = 0.0004) or 0.2 μg of GATA4 (~3-fold; p = 0.0001). The synergistic activation (~18-fold) by E21Q-mutant NKX2-5 and GATA4 was significantly reduced by ~28% compared with that by wild-type NKX2-5 and GATA4 (p = 0.0094). The T180A-mutant had no synergistic effect on GATA4-induced activation of the ANF promoter (~3-fold), with a p value of 0.0001 when compared with wild-type NKX2-5 and GATA4.
Figure 6.

Reduced synergistic transcriptional activation by mutant NKX2-5 and GATA4. The synergistic activation by E21Q-mutant NKX2-5 and GATA4 was significantly reduced compared with that by wild-type NKX2-5 and GATA4. The E21Q-mutant NKX2-5 had significantly reduced synergistic effect while the T180A-mutant had no synergistic effect on GATA4-induced activation of the atrial natriuretic factor promoter. Experiments were performed in triplicate, and mean and standard deviations are shown. * represents p < 0.01 and ** represents p < 0.001, when compared with wild-type NKX2-5 and GATA4.
Discussion
In the present study, two heterozygous NKX2-5 mutations, p.E21Q and p.T180A, were identified in two families with lone AF, respectively. In each family the mutation co-segregated with AF and was absent in the 700 control individuals. A cross-species alignment of multiple NKX2-5 protein sequences displayed that the altered amino acids were completely conserved evolutionarily. Functional analyses revealed that both mutations were associated with a significantly decreased transcriptional activity. Therefore, it is likely that these two NKX2-5 variants predispose carriers to AF.
NKX2-5 has been identified in various vertebrate species from Xenopus to humans. It is highly expressed in the early cardiac progenitor cells in both primary and secondary heart fields during embryogenesis and continues to be expressed at a high level in the heart through adulthood, playing a pivotal role in normal cardiovascular development and maturation 41. Human NKX2-5 gene maps to chromosome 5q34 and consists of two exons coding for a protein of 324 amino acids 42. As shown in Figure 2, the NKX2-5 protein has a highly conserved structure composed of the N-terminal TN domain, the homeodomain (HD), and the C-terminal NK2-specific domain (NK). The HD is centrally located at amino acid positions 138-197 and contains a helix-turn-helix motif that recognizes and binds to the specific consensus DNA sequence 5'-T(C/T)AAGTG-3' 43. The TN and NK domains function to regulate the transcriptional activity, and the HD is involved in nuclear translocation, DNA binding, and interaction with other transcription factors as well 44. The NKX2-5 mutations p.E21Q and p.T180A identified in this study are located in TN and HD, respectively, and may influence the transcriptional activity of NKX2-5 by direct transcriptional inhibition or by interfering with its nuclear distribution or DNA-binding ability.
Previous studies have substantiated that NKX2-5 is an upstream regulator of several target genes expressed in the heart during embryogenesis including the genes that encode ANF, brain natriuretic peptide and α-cardiac actin 45. Therefore, the functional characteristics of an NKX2-5 mutation can be explored by analysis of the transcriptional activity of the ANF promoter in cells transfected with NKX2-5 mutant in contrast to its wild-type counterpart. In this study, the functional roles of the two NKX2-5 mutations identified in lone AF patients were analyzed by transcriptional activity assays and the results showed a significantly decreased transcriptional activity on a downstream gene. These findings support that dysfunctional NKX2-5 resulted from mutations is potentially an alternative pathogenic mechanism underlying AF.
The findings that functionally impaired NKX2-5 predisposes to AF may be partially ascribed to the abnormally developed pulmonary vein myocardium. The pulmonary venous vessels are ensheathed by a layer of myocardium referred to as pulmonary myocardial sleeve, which has been implicated in the initiation and perpetuation of AF by several projected arrhythmogenic mechanisms including enhanced intrinsic pacemaker activity and liability to reentrance 46-48. Genetic-labeling lineage tracing studies have shown that NKX2-5 is expressed in the atria as well as pulmonary myocardium and is indispensible for the localized formation of the sinoatrial node during embryogenesis. As a suppressor of the sinoatrial node lineage gene program, NKX2-5 may confine pacemaker activity to the sinoatrial and atrioventricular nodes. In a hypomorphic model with reduced NKX2-5 protein, the pulmonary cardiomyocytes shifted to connexin40-negative, HCN4-positive cells, resembling a nodal phenotype with pacemaker activity 47. In NKX2-5 knockout mouse embryos, HCN4 was activated along the entire embryonic heart tube; whereas connexin40 (Cx40) expression was inhibited, and ectopic pacemaker cells were present throughout the heart tube 48. Hence, NKX2-5 loss-of-function mutation presumably contributes to formation of the pulmonary myocardium sleeve and switch of the pulmonary myocardium to a sinoatrial node-like phenotype, yielding an atrial arrhythmogenic substrate that facilitates AF.
It has been verified that some downstream genes are regulated by NKX2-5, and mutations in multiple target genes have been linked to AF, including the genes encoding ANF 49, cardiac voltage-gated sodium channel pore-forming alpha subunit (SCN5A) 50-53, gap junction protein Cx40 54, and connexin43 (Cx43) 55. Therefore, mutated NKX2-5 may confer susceptibility to AF by reducing the expression of some target genes.
Association of NKX2-5 mutation with AF in humans was investigated previously. Ritchie and colleagues screened NKX2-5 in 160 unrelated index patients with familial AF and identified one novel heterozygous mutant (p.F145S) in a proband, with a mutational prevalence of around 0.63%. In this kindred the proband presented with early-onset paroxysmal AF. The proband's father also presented with lone AF in his 40s, and his brother, who carries the variant, has not been diagnosed with AF at age 52 years 56. The same mutation was also detected in 1 of 110 unrelated index patients with familial AF by Huang et al., with a mutational prevalence of about 0.91%, which co-segregated with AF in the family with complete penetrance. Functional analysis demonstrated that the F145S-mutant NKX2.5 was associated with significantly decreased transcriptional activity 57. Xie and co-workers scanned NKX2-5 in 136 unrelated patients with idiopathic AF and discovered two novel heterozygous NKX2-5 mutations (p.N19D and p.F186S) in two patients, respectively, with a mutational prevalence of approximately 1.47%. Functional assays revealed that the mutant NKX2-5 proteins were associated with significantly reduced transcriptional activity 58. Boldt et al. also sequenced NKX2-5 in 96 unrelated patients with idiopathic AF but did not find any mutation 59. These studies suggest that NKX2-5 mutations are a relatively rare cause of AF.
It was interesting that congenital atrial septal defect was documented in an AF patient carrying p.T180A mutation of NKX2-5. Similarly, congenital cardiovascular malformations were previously confirmed in AF patients carrying NKX2-5 mutations. Gutierrez-Roelens et al. sequenced NKX2-5 in seven unrelated patients with atrial septal defect and/or atrioventricular conduction block, and discovered one novel heterozygous mutation (p.Y256X) in an index patient. In the proband's family, all the five mutation carriers had atrioventricular block, of which three mutation carriers had also AF and cardiac structural anomalies 60. Watanabe et al. performed genetic analysis of NKX2-5 in two unrelated families with atrial septal defect and atrioventricular block, and found two novel deletion frameshift mutations (p.E72Afs and p.P77Ffs). In the family with p.E72Afs mutation, there were five family members who harbored the mutation, of which one had lone AF as the only phenotype, another had atrial septal defect and atrioventricular block, and the other three had atrial septal defect, atrioventricular block, and AF 61. Moreover, mutations in functionally synergistic partners of NKX2-5, including GATA4, GATA5 and GATA6, were also implicated in various congenital cardiovascular deformities with or without the presence of AF 24,28,31,62,63. Considering some congenital cardiac structural defects may close spontaneously, we cannot rule out the possibility that some mutation carriers with lone AF had minor cardiac septal defects that closed shortly after birth on their own. These observations imply that a subset of “lone” AF may share a common genetic origin with congenital heart disease.
Notably, a long list of NKX2-5 mutations were previously linked to a wide variety of congenital cardiovascular malformations, including atrial septal defect, ventricular septal defect, tetralogy of Fallot, double outlet right ventricle, L-transposition of the great artery, and hypoplastic left heart syndrome without AF, of which several mutations, including the p.E21Q mutation identified in present study, were also observed in unaffected family members 37-40,64,65. The pronounced variability in clinical presentation or penetrance of the genotypes may be explained by the following reasons. Firstly, AF occurs as rarely as a few times in a lifetime for some AF patients 66, suggesting that a longer duration of electrocardiographic monitoring is needed to record paroxysmal AF in these patients. Next, AF occurs more commonly in older patients, and these carriers may not be old enough to develop the disease. Thirdly, genomic imprinting results in preferential expression of the paternal or maternal allele of a certain gene, highlighting the effect of proband 67. Fourthly, the nature of a mutation (loss-of-function, dominant-negative, or gain-of-function effect) and its temporal and spatial effect during cardiac development (germline or somatic) are a potential explanation for this phenomenon 65, 68. Fifthly, different genetic backgrounds, including possibly common SNPs altering AF susceptibility, contribute to the variable penetrance of the AF-linked genotype 56. Finally, mutations like p.E21Q may be a genetic risk factor predisposing to AF, rather than a direct cause of AF, and environmental risk factors may be required for the onset of AF.
In conclusion, the current study expands the spectrum of NKX2-5 mutations linked to AF and provides additional evidence that functionally compromised NKX2-5 may be involved in the pathogenesis of AF, implying the potential application to the early prophylaxis and personalized therapy of this common arrhythmia.
Acknowledgments
We are really grateful to the participants for their devotion to the study. This work was supported in part by grants from the Shanghai Pujiang Program (09PJ1410400), the Natural Science Fund of Shanghai, China (13ZR1438400), The Fundamental Research Fund for Central University of Ministry of Education, China (15072), the Scientific Research Fund of Health Bureau, Shanghai, China (2010119), The Excellent Discipline Leader Training Foundation of Pudong New Strict, Shanghai, China (PWRd2012-05), and the National Natural Science Fund of China (81270161, 81270314, 81271927 and 81370301).
References
- 1.Fuster V, Rydén LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS, Smith SC Jr, Priori SG, Estes NA 3rd, Ezekowitz MD, Jackman WM, January CT, Lowe JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK, Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton RA, Halperin JL, Hochman JS, Kushner FG, Ohman EM, Stevenson WG, Tarkington LG, Yancy CW; American College of Cardiology Foundation/American Heart Association Task Force. 2011 ACCF/AHA/HRS Focused Updates Incorporated Into the ACC/AHA/ESC 2006 Guidelines for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;123:e269–367. doi: 10.1161/CIR.0b013e318214876d. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D'Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110:1042–6. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 3.Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–52. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 4.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–8. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 5.Dogan U, Dogan EA, Tekinalp M, Tokgoz OS, Aribas A, Akilli H, Ozdemir K, Gok H, Yuruten B. P-wave dispersion for predicting paroxysmal atrial fibrillation in acute ischemic stroke. Int J Med Sci. 2012;9:108–14. doi: 10.7150/ijms.9.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naji F, Pagliaruzzi M, Penko M, Kanic V, Vokac D. Changes in left ventricular filling in patients with persistent atrial fibrillation. Int J Med Sci. 2013;10:1876–9. doi: 10.7150/ijms.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu CY, Lin TH, Hsu PC, Lee WH, Lee HH, Chiu CA, Su HM, Lee CS, Yen HW, Voon WC, Lai WT, Sheu SH. Heart rate significantly influences the relationship between atrial fibrillation and arterial stiffness. Int J Med Sci. 2013;10:1295–300. doi: 10.7150/ijms.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai WC, Chen CY, Kuo HF, Wu MT, Tang WH, Chu CS, Lin TH, Su HM, Hsu PC, Jhuo SJ, Lin MY, Lee KT, Sheu SH, Lai WT. Areca nut chewing and risk of atrial fibrillation in Taiwanese men: a nationwide ecological study. Int J Med Sci. 2013;10:804–11. doi: 10.7150/ijms.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Du JL, Yuan J, Chen YQ. Statins therapy can reduce the risk of atrial fibrillation in patients with acute coronary syndrome: a meta-analysis. Int J Med Sci. 2013;10:198–205. doi: 10.7150/ijms.5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jahangir A, Lee V, Friedman PA, Trusty JM, Hodge DO, Kopecky SL, Packer DL, Hammill SC, Shen WK, Gersh BJ. Long-term progression and outcomes with aging in patients with lone atrial fibrillation: a 30-year follow-up study. Circulation. 2007;115:3050–6. doi: 10.1161/CIRCULATIONAHA.106.644484. [DOI] [PubMed] [Google Scholar]
- 11.Darbar D, Herron KJ, Ballew JD, Jahangir A, Gersh BJ, Shen WK, Hammill SC, Packer DL, Olson TM. Familial atrial fibrillation is a genetically heterogeneous disorder. J Am Coll Cardiol. 2003;41:2185–92. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 12.Fox CS, Parise H, D'Agostino RB Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–5. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 13.Ellinor PT, Yoerger DM, Ruskin JN, MacRae CA. Familial aggregation in lone atrial fibrillation. Hum Genet. 2005;118:179–84. doi: 10.1007/s00439-005-0034-8. [DOI] [PubMed] [Google Scholar]
- 14.Arnar DO, Thorvaldsson S, Manolio TA, Thorgeirsson G, Kristjansson K, Hakonarson H, Stefansson K. Familial aggregation of atrial fibrillation in Iceland. Eur Heart J. 2006;27:708–12. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 15.Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, Christensen K. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–83. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang YQ, Zhang XL, Wang XH, Tan HW, Shi HF, Fang WY, Liu X. Familial aggregation of lone atrial fibrillation in Chinese population. Intern Med. 2010;49:2385–91. doi: 10.2169/internalmedicine.49.4130. [DOI] [PubMed] [Google Scholar]
- 17.Lubitz SA, Yin X, Fontes JD, Magnani JW, Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D, Larson MG, Ellinor PT, Benjamin EJ. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. 2010;304:2263–9. doi: 10.1001/jama.2010.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahida S, Lubitz SA, Rienstra M, Milan DJ, Ellinor PT. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc Res. 2011;89:692–700. doi: 10.1093/cvr/cvq381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, Fang WY, Yang YQ. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med. 2013;32:144–50. doi: 10.3892/ijmm.2013.1355. [DOI] [PubMed] [Google Scholar]
- 20.Olesen MS, Nielsen MW, Haunsø S, Svendsen JH. Atrial fibrillation: the role of common and rare genetic variants. Eur J Hum Genet. 2014;22:297–306. doi: 10.1038/ejhg.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olesen MS, Andreasen L, Jabbari J, Refsgaard L, Haunsø S, Olesen SP, Nielsen JB, Schmitt N, Svendsen JH. Very early-onset lone atrial fibrillation patients have a high prevalence of rare variants in genes previously associated with atrial fibrillation. Heart Rhythm. 2014;11:246–51. doi: 10.1016/j.hrthm.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 22.Mommersteeg MT, Christoffels VM, Anderson RH, Moorman AF. Atrial fibrillation: a developmental point of view. Heart Rhythm. 2009;6:1818–24. doi: 10.1016/j.hrthm.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 23.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–77. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posch MG, Boldt LH, Polotzki M, Richter S, Rolf S, Perrot A, Dietz R, Ozcelik C, Haverkamp W. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur J Med Genet. 2010;53:201–3. doi: 10.1016/j.ejmg.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Yang YQ, Wang MY, Zhang XL, Tan HW, Shi HF, Jiang WF, Wang XH, Fang WY, Liu X. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin Chim Acta. 2011;412:1825–30. doi: 10.1016/j.cca.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 26.Jiang JQ, Shen FF, Fang WY, Liu X, Yang YQ. Novel GATA4 mutations in lone atrial fibrillation. Int J Mol Med. 2011;28:1025–32. doi: 10.3892/ijmm.2011.783. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Sun YM, Yang YQ. Mutation spectrum of the GATA4 gene in patients with idiopathic atrial fibrillation. Mol Biol Rep. 2012;39:8127–35. doi: 10.1007/s11033-012-1660-6. [DOI] [PubMed] [Google Scholar]
- 28.Yang YQ, Wang J, Wang XH, Wang Q, Tan HW, Zhang M, Shen FF, Jiang JQ, Fang WY, Liu X. Mutational spectrum of the GATA5 gene associated with familial atrial fibrillation. Int J Cardiol. 2012;157:305–7. doi: 10.1016/j.ijcard.2012.03.132. [DOI] [PubMed] [Google Scholar]
- 29.Gu JY, Xu JH, Yu H, Yang YQ. Novel GATA5 loss-of-function mutations underlie familial atrial fibrillation. Clinics (Sao Paulo) 2012;67:1393–9. doi: 10.6061/clinics/2012(12)08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang XH, Huang CX, Wang Q, Li RG, Xu YJ, Liu X, Fang WY, Yang YQ. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int J Mol Med. 2013;31:43–50. doi: 10.3892/ijmm.2012.1189. [DOI] [PubMed] [Google Scholar]
- 31.Yang YQ, Wang XH, Tan HW, Jiang WF, Fang WY, Liu X. Prevalence and spectrum of GATA6 mutations associated with familial atrial fibrillation. Int J Cardiol. 2012;155:494–6. doi: 10.1016/j.ijcard.2011.12.091. [DOI] [PubMed] [Google Scholar]
- 32.Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu YJ, Tan HW, Wang XH, Jiang JQ, Fang WY, Liu X. GATA6 loss-of-function mutation in atrial fibrillation. Eur J Med Genet. 2012;55:520–6. doi: 10.1016/j.ejmg.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Liu WD, Yang ZL, Yang YQ. Novel GATA6 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2012;30:783–90. doi: 10.3892/ijmm.2012.1068. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Rath N, Hannenhalli S, Wang Z, Cappola T, Kimura S, Atochina-Vasserman E, Lu MM, Beers MF, Morrisey EE. GATA and Nkx factors synergistically regulate tissue-specific gene expression and development in vivo. Development. 2007;134:189–98. doi: 10.1242/dev.02720. [DOI] [PubMed] [Google Scholar]
- 35.Liu XY, Wang J, Yang YQ, Zhang YY, Chen XZ, Zhang W, Wang XZ, Zheng JH, Chen YH. Novel NKX2-5 mutations in patients with familial atrial septal defects. Pediatr Cardiol. 2011;32:193–201. doi: 10.1007/s00246-010-9859-6. [DOI] [PubMed] [Google Scholar]
- 36.Zhu W, Shiojima I, Hiroi Y, Zou Y, Akazawa H, Mizukami M, Toko H, Yazaki Y, Nagai R, Komuro I. Functional analyses of three Csx/Nkx-2.5 mutations that cause human congenital heart disease. J Biol Chem. 2000;275:35291–6. doi: 10.1074/jbc.M000525200. [DOI] [PubMed] [Google Scholar]
- 37.Stallmeyer B, Fenge H, Nowak-Göttl U, Schulze-Bahr E. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet. 2010;78:533–40. doi: 10.1111/j.1399-0004.2010.01422.x. [DOI] [PubMed] [Google Scholar]
- 38.Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–8. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 39.Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, Feneley M, Harvey RP. Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. 2003;41:2072–6. doi: 10.1016/s0735-1097(03)00420-0. [DOI] [PubMed] [Google Scholar]
- 40.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–5. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Akazawa H, Komuro I. Cardiac transcription factor Csx/Nkx2-5: Its role in cardiac development and diseases. Pharmacol Ther. 2005;107:252–68. doi: 10.1016/j.pharmthera.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Shiojima I, Komuro I, Inazawa J, Nakahori Y, Matsushita I, Abe T, Nagai R, Yazaki Y. Assignment of cardiac homeobox gene CSX to human chromosome 5q34. Genomics. 1995;27:204–6. doi: 10.1006/geno.1995.1027. [DOI] [PubMed] [Google Scholar]
- 43.Chen CY, Schwartz R J. Identification of novel DNA binding targets and regulatory domains of a murine tinman homeodomain factor, nkx-2.5. J Biol Chem. 1995;270:15628–33. doi: 10.1074/jbc.270.26.15628. [DOI] [PubMed] [Google Scholar]
- 44.Pradhan L, Genis C, Scone P, Weinberg EO, Kasahara H, Nam HJ. Crystal structure of the human NKX2.5 homeodomain in complex with DNA target. Biochemistry. 2012;51:6312–9. doi: 10.1021/bi300849c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–80. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 46.Haïssaguerre M, Jaïs P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P, Clémenty J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–66. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 47.Mommersteeg MT, Brown NA, Prall OW, de Gier-de Vries C, Harvey RP, Moorman AF, Christoffels VM. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ Res. 2007;101:902–9. doi: 10.1161/CIRCRESAHA.107.161182. [DOI] [PubMed] [Google Scholar]
- 48.Mommersteeg MT, Hoogaars WM, Prall OW, de Gier-de Vries C, Wiese C, Clout DE, Papaioannou VE, Brown NA, Harvey RP, Moorman AF, Christoffels VM. Molecular pathway for the localized formation of the sinoatrial node. Circ Res. 2007;100:354–62. doi: 10.1161/01.RES.0000258019.74591.b3. [DOI] [PubMed] [Google Scholar]
- 49.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC Jr, Olson TM. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briggs LE, Takeda M, Cuadra AE, Wakimoto H, Marks MH, Walker AJ, Seki T, Oh SP, Lu JT, Sumners C, Raizada MK, Horikoshi N, Weinberg EO, Yasui K, Ikeda Y, Chien KR, Kasahara H. Perinatal loss of Nkx2-5 results in rapid conduction and contraction defects. Circ Res. 2008;103:580–90. doi: 10.1161/CIRCRESAHA.108.171835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terada R, Warren S, Lu JT, Chien KR, Wessels A, Kasahara H. Ablation of Nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc Res. 2011;91:289–99. doi: 10.1093/cvr/cvr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–35. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, Hedley PL, Christiansen M, Olesen SP, Haunsø S, Schmitt N, Jespersen T, Svendsen JH. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–9. doi: 10.1161/CIRCGENETICS.111.962597. [DOI] [PubMed] [Google Scholar]
- 54.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–88. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 55.Thibodeau IL, Xu J, Li Q, Liu G, Lam K, Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ, Gollob MH. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation. 2010;122:236–44. doi: 10.1161/CIRCULATIONAHA.110.961227. [DOI] [PubMed] [Google Scholar]
- 56.Ritchie MD, Rowan S, Kucera G, Stubblefield T, Blair M, Carter S, Roden DM, Darbar D. Chromosome 4q25 variants are genetic modifiers of rare ion channel mutations associated with familial atrial fibrillation. J Am Coll Cardiol. 2012;60:1173–81. doi: 10.1016/j.jacc.2012.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2013;31:1119–26. doi: 10.3892/ijmm.2013.1316. [DOI] [PubMed] [Google Scholar]
- 58.Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang WY, Liu X, Yang YQ. Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics (Sao Paulo) 2013;68:777–84. doi: 10.6061/clinics/2013(06)09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boldt LH, Posch MG, Perrot A, Polotzki M, Rolf S, Parwani AS, Huemer M, Wutzler A, Ozcelik C, Haverkamp W. Mutational analysis of the PITX2 and NKX2-5 genes in patients with idiopathic atrial fibrillation. Int J Cardiol. 2010;145:316–7. doi: 10.1016/j.ijcard.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 60.Gutierrez-Roelens I, De Roy L, Ovaert C, Sluysmans T, Devriendt K, Brunner HG, Vikkula M. A novel CSX/NKX2-5 mutation causes autosomal-dominant AV block: are atrial fibrillation and syncopes part of the phenotype? Eur J Hum Genet. 2006;14:1313–6. doi: 10.1038/sj.ejhg.5201702. [DOI] [PubMed] [Google Scholar]
- 61.Watanabe Y, Benson DW, Yano S, Akagi T, Yoshino M, Murray JC. Two novel frameshift mutations in NKX2.5 result in novel features including visceral inversus and sinus venosus type ASD. J Med Genet. 2002;39:807–11. doi: 10.1136/jmg.39.11.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–7. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM, Wang Q, Li RG, Fang WY, Wang XZ, Yang YQ. Novel GATA6 mutations associated with congenital ventricular septal defect or tetralogy of Fallot. DNA Cell Biol. 2012;31:1610–7. doi: 10.1089/dna.2012.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–11. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 65.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–73. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chugh SS, Blackshear JL, Shen WK, Hammill SC, Gersh BJ. Epidemiology and natural history of atrial fibrillation: clinical implications. J Am Coll Cardiol. 2001;37:371–8. doi: 10.1016/s0735-1097(00)01107-4. [DOI] [PubMed] [Google Scholar]
- 67.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 68.Kasahara H, Lee B, Schott JJ, Benson DW, Seidman JG, Seidman CE, Izumo S. Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest. 2000;106:299–308. doi: 10.1172/JCI9860. [DOI] [PMC free article] [PubMed] [Google Scholar]