

Abstract
A simple procedure for simultaneously derivatizing non-hydroxy and hydroxy fatty acids prior to GC analysis [I. Ciucanu and F. Kerek, J. Chromatogr., 284 (1984) 179] has been evaluated for its usefulness in determining sphingolipid acyl composition. The method uses methyl iodide in polar aprotic solvents to generate methyl esters of carboxyl groups and methyl ethers of hydroxyl groups. Methylation efficiency is examined as a function of hydroxyl group presence and location in free fatty acids as well as a function of 2-hydroxy fatty acid chain length. Conditions are also reported for efficient saponification and derivatization of sphingolipid fatty acyl chains as is illustrated using bovine brain galactosylceramide.
INTRODUCTION
Compared to glycerol-based lipids, sphingolipids require special conditions for accurate determination of their fatty acyl composition, in part because relatively large amounts of 2-hydroxy fatty acyl residues often are present [1–9]. Accordingly, derivatization prior to gas chromatographic analysis is typically accomplished by converting the carboxyl groups to methyl esters and the hydroxyl groups to trimethylsilyl (TMS) ether derivatives, which often are water-labile [10–13]. An alternative one-step procedure utilizing methyl iodide in polar aprotic organic solvents has been described by Ciucanu and Kerek [14] for simultaneously generating methyl esters of carboxyl groups and methyl ethers of hydroxyl groups in non-hydroxy and hydroxy fatty acids. Advantages of the methyl iodide method include simplicity and stability of the resulting methyl ether derivatives to water. However, the applicability to sphingolipids was not addressed in this initial report. For example, of the three hydroxy fatty acid standards studied, only one was a 2-hydroxy fatty acid and that was 2-hydroxypalmitic acid. Also, although the saponification rate of ester-linked fatty acyl chains in phosphoglycerides, glycerides, cholesteryl esters and methyl esters was reported, no information was provided for amide-linked fatty acyl chains typically found in sphingolipids. To assess the utility of the methyl iodide method for characterizing sphingolipid fatty acyl composition, we have examined methylation efficiency as a function of chain length in 2-hydroxy fatty acids and as a function of hydroxyl group presence and location. We have also defined conditions for application to sphingolipids as demonstrated using bovine brain galactosylceramide.
EXPERIMENTAL
Materials
Non-hydroxy fatty acid standards were obtained from NuChek Prep (Elysian, MN, USA) and 2-hydroxy fatty acid standards (C18:0, 20:0, 22:0) were supplied by Analabs (South Norwalk, CT, USA). Bovine brain galactosylceramide (GalCer) was obtained from Avanti Polar Lipids (Birmingham, AL, USA). Bovine brain GalCer containing 99% 2-hydroxy fatty acids (Type I) or 99% non-hydroxy fatty acids (Type II) were supplied by Sigma (St. Louis, MO, USA). Dimethylacetamide and iodomethane were obtained from Sigma and Aldrich (Milwaukee, WI, USA), respectively. Other reagents were analytical grade.
Methods
Lipid purity was checked by thin-layer chromatography (TLC) using two solvent systems for GalCer [chloroform–methanol–water (85:15:1.5, v/v/v) or (50:21:3, v/v/v)] and one for free fatty acids [light petroleum (b.p. 00–00°C)–diethyl ether–acetic acid (80:20:1, v/v/v)]. Visualization of lipids was achieved by charring TLC plates after spraying with concentrated chromic–sulfuric acid (detection limit = 0.1 μg fatty acid), or by viewing under ultraviolet light after spraying with primulin (detection limit = 50 pmol diacyl lipid) or after spraying with orcinol (sugar specific; detection limit = 0.5 nmol). Scanning densitometry was performed with a Hoefer GS 300 instrument (San Francisco, CA, USA). All lipids were judged to be greater than 99% pure.
Free fatty acid derivatization
After dissolving free fatty acids in dimethylacetamide (1.0 ml), crushed NaOH (approx. 160 mg) was added and the sample was heated at 80°C for 1 h. Upon cooling, iodomethane (1.0 ml) was added and the sample was again heated for 1 hr at 80°C [14]. Fatty acid methy esters (FAMEs) were extracted using light petroleum (3×2 ml) after adding water (2 ml). The light petroleum phase was washed three times with water (2 ml) to remove residual NaOH. The resulting FAMEs are known to be stable for extended time periods in contrast to the limited stability of other derivatives (e.g., TMS) [10–13]. As pointed out by Ciucanu and Kerek [14], the extremely low acidity of the hydroxyl group necessitates the use of a very strong base and the heating step was necessary for samples containing cholesteryl esters. While such conditions may induce isomerization of polyunsaturated fatty acyl residues, sphingolipids lack these acyl chains.
GalCer acyl chain derivatization
After drying aliquots (0.5 mg) of bovine brain GalCer in PTFE-lined screw-capped glass tubes under nitrogen, samples were hydrolyzed for 1 h at 100°C under nitrogen using 0.5 M HCl in acetonitrile–water [1 ml (9:1, v/v)] [15] and then were taken to dryness under nitrogen. Subsequent derivatization steps were performed as described above for free fatty acids.
Fatty acid analysis
After derivatization, FAMEs were quantitated using a Packard 427 gas Chromatograph equipped with a 50 m × 0.25 mm free fatty acid phase (FFAP)–fused-silica capillary column (0.25 μm film thickness; Quadrex). The carrier gas was helium at a flow of 47.5 cm/s and the split ratio was set at 1:66. The column was programmed from 180–220°C at 2°C/min with an initial hold at 180°C for 13 min and a final hold at 220°C for 80 min. Peaks were integrated using a dedicated microprocessor.
In control experiments, the fatty acyl composition of bovine brain GalCer was determined either by simultaneously resolving non-hydroxy and 2-methoxy FAMEs by capillary gas chromatography (GC) or by separating non-hydroxy and 2-methoxy FAMEs by TLC prior to GC analysis. No significant differences were evident upon comparing results. In instances of partial peak overlap (e.g., C23:0(2-OH) and C25:0; C26:0 and C24:0(2-OH); C24:1(2-OH) and C26:1), integration of deconvoluted peaks yielded results similar to those obtained when non-hydroxy and 2-methoxy FAMEs were separated by TLC and analyzed independently by GC. Other workers have reported complete resolution of non-hydroxy and 2-methoxy FAMEs using capillary GC [16].
A DuPont DP-102 mass spectrometer equipped with an all-glass jet separator and a SP-1000 fused capillary GC column (0.25 mm × 30 m; 0.25 μm film thickness; Supelco) was used for positive identification of each FAME by electron impact mass spectrometric (MS) analysis. The column temperature was programmed from 120–220°C at 2°C/min with a final hold time of 120 min. Helium was used as the carrier gas (34 cm/s). Mass spectra were recorded at an ionization voltage of 70 eV. Ions from 43 to 500 were scanned at 300 amu/s.
Identification of derivatized fatty acids, when based solely on GC retention times, is not straight-forward because certain 2-hydroxy fatty acid derivatives elute similarly to non-hydroxy fatty acid derivatives (Fig. 1). However, the electron impact mass spectral fragmentation patterns of non-hydroxy and 2-methoxy FAMEs are distinctly different (Fig. 2). The intense m – 59 peak and the lack of the base peak at m/z 74 are diagnostic for 2-methoxy FAMEs and clearly distinguishes them from non-hydroxy FAMEs [11]. This information, along with the mass of the molecular ion, allows definitive identification of each FAME.
Fig. 1.
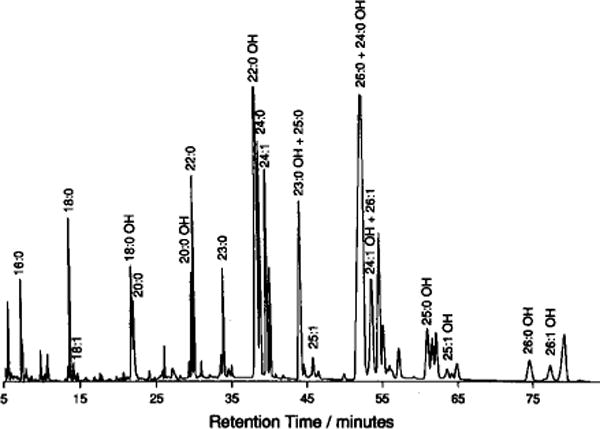
Capillary GC profile of derivatized FAMEs from bovine brain GalCer. A FFAP-fused silica capillary column (Quadrex) was used as described in the Methods section.
Fig. 2.
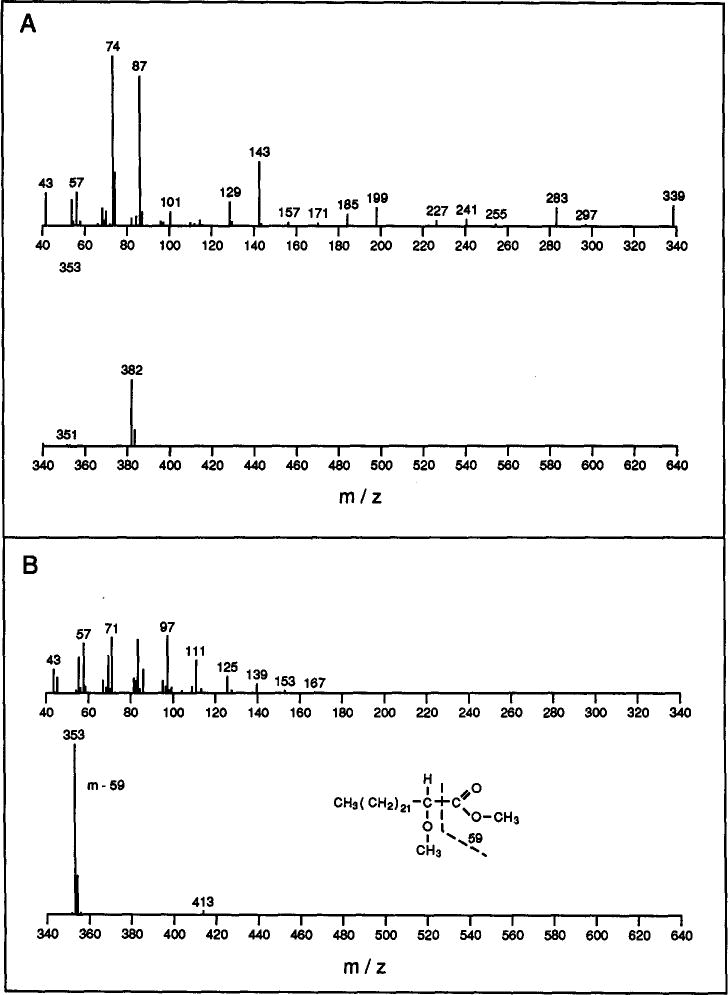
Typical fragmentation patterns of non-hydroxy and 2-methoxy fatty acid methyl esters obtained by mass spectral analysis. (A) Tetracosanoic (24:0) methyl ester (mol. wt. 382); (B) 2-methoxy-tetracosanoic methyl ester (mol. wt. 412).
RESULTS AND DISCUSSION
Free fatty acids studies
Initially, control experiments were performed to assess the feasibility of the methyl iodide method for determining sphingolipid fatty acyl composition. For example, because sphingolipids often contain long, saturated 2-hydroxyacyl chains, the effect of acyl chain length on derivatization efficiency was checked using representative 2-hydroxy fatty acids other than 2-hydroxypalmitic acid [14]. Following derivatization and TLC separation using light petroleum–diethyl ether–acetic acid (80:20:1, v/v/v), lipids were visualized by charring with concentrated chromic–sulfuric acid. Based on inspection and scanning densitometry, nearly quantitative methylation (>95%) was observed for 2-hydroxy fatty acids regardless of chain length (C18:0, C20:0, C22:0).
To determine whether the presence and position of the hydroxyl group affects the extent of methylation, stearic acid, 2-hydroxybehenic acid, and 11-hydroxystearic acid derivatization (0.5 mg each) were compared by TLC analysis as described above. Both the stearic acid and 2-hydroxybehenic acids were methylated almost quantitatively (>95%), but methylation of 11-hydroxystearic acid was only about 50%. Increasing the reaction temperature to 100°C did not improve the extent of methylation.
Galactosylceramide studies
Based on the results obtained above with the free fatty acids, we anticipated that the methyl iodide method would work well if the amide-linked fatty acyl residues were released efficiently. Relatively harsh conditions are needed to release amide-linked acyl chains compared to those used with ester-linked fatty acids [1–6]. One method that reportedly releases sphingomyelin’s acyl chains quantitatively involves heating in acidic aqueous acetonitrile [15]. To verify that this method is appropriate for galactosylceramides, experiments were run as described in the Methods section. Aliquots of the reaction mixture were analyzed by TLC using chloroform–methanol–water (85:15:1.5, v/v/v) to assess release of free fatty acid, and lipid products were visualized by charring with concentrated chromic–sulfuric acid. Based on inspection and scanning densitometry, over 90% release of fatty acids was achieved.
These findings were confirmed by including known amounts of N-palmitoyl galactosylsphingoid (GalSpd) and N-(2-hydroxy)-stearoyl GalSpd as internal standards in GalCer containing either 2-hydroxy or non-hydroxy fatty acyl chains, respectively. Following fatty acid release by heating with acidic aqueous acetonitrile and derivatization by the methyl iodide method, GC analysis revealed percentage recoveries of 96.2 ± 9.1% and 70.3 ± 6.2% for GalCer’s non-hydroxy and 2-hydroxy fatty acyl residues, respectively. These recoveries appear to be adequate since the experimental conditions produce no bias amongst different 2-hydroxy fatty acids and agree well with determinations made by more complex procedures (see below).
Complete characterization of bovine brain GalCer’s fatty acyl composition is shown in Fig. 3 and reveals a preponderance of long, saturated or mono-unsaturated acyl chains with a large fraction being hydroxylated at the second carbon atom. In general, these findings agree well with earlier studies in which multi-step derivatization procedures were used and in which identifications were based solely on GC retention times [9, 17–21] rather than on GC–MS analysis. A survey of these earlier studies reveals that our results fall well within the expected range of variation. Most of this variation is probably due to differences in tissue developmental state and/or animal age [22–24] and as well as regional compositional differences within tissue [17].
Fig. 3.
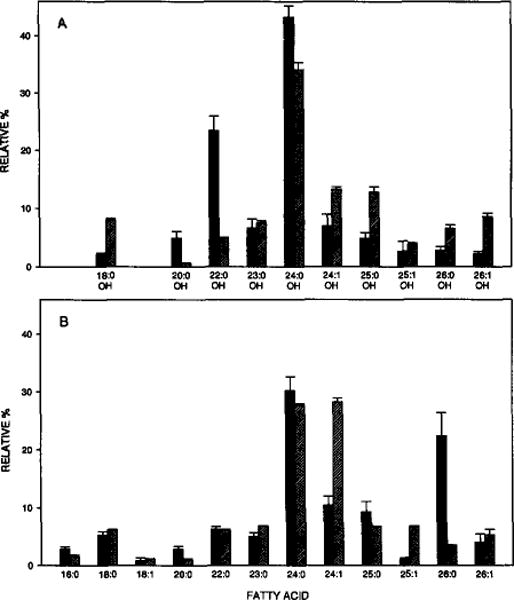
Fatty acyl composition of bovine brain GalCer. Solid bars represent FAMEs derived from Avanti’s bovine brain GalCer. Cross-hatched bars represent FAMEs derived from Sigma’s Type I and Type II bovine brain GalCer (see Methods). Data are % (w/w). A FFAP–fused-silica capillary column was used as described in the Methods section.
One final experiment was performed to determine whether the methyl iodide method can be used directly on intact GalCer to saponify and derivatize the amide-linked fatty acyl chains as has been shown for ester-linked fatty acyl chains in phosphoglycerides, glycerides, cholesteryl esters [14]. After treatment of GalCer (0.5 mg) as described for the free fatty acids (see Methods section), the resulting lipids were analyzed by TLC using light petroleum–diethyl ether–acetic acid (80:20:1, v/v/v) to assess derivatization of released fatty acids. Inspection of TLC plates revealed that both release and derivatization of fatty acids were poor (<50%) and many unidentified side-products were generated. Attempts to increase the yield by raising the incubation temperature (100°C) produced no net improvement. Although fatty acid release increased, the ensuing yield of derivatized hydroxy fatty acids decreased, apparently because of degeneration of the 2-hydroxy group. In contrast, if temperature was lowered, fatty acid release declined and the ensuing derivatization improved only slightly.
In summary, a simple and direct means of analyzing sphingolipids fatty acyl composition has been demonstrated. The method is particularly useful for sphingolipids containing mixtures of non-hydroxy and 2-hydroxy fatty acids since stable derivatives of both fatty acid types can be produced simultaneously and then positively identified by capillary GC–MS.
Acknowledgments
We would like to thank Dr. Shaukat Ali for synthesizing the sphingolipid internal standards, Drs. Hermann Schlenk and Herb Dutton for their helpful comments, Carmen Perleberg for secretarial services, and Core D of USPHS PPG HL08214 for the use of GC–MS equipment. This work was supported by USPHS Grant GM45928 and by the Hormel Foundation.
References
- 1.Kishimoto Y, Radin NS. J Lipid Res. 1959;1:72. [PubMed] [Google Scholar]
- 2.Kishimoto Y, Radin NS. J Lipid Res. 1963;4:130. [PubMed] [Google Scholar]
- 3.Radin NS. J Am Oil Chem Soc. 1965;42:569. doi: 10.1007/BF02541292. [DOI] [PubMed] [Google Scholar]
- 4.Carter HE, Rothfus JA, Gigg R. J Lipid Res. 1961;2:228. [Google Scholar]
- 5.Gaver RC, Sweeley CC. J Am Oil Chem Soc. 1965;42:294. doi: 10.1007/BF02540132. [DOI] [PubMed] [Google Scholar]
- 6.Kadowaki H, Bremer EG, Evans JE, Jungalwala FB, McCluer RH. J Lipid Res. 1983;24:1389. [PubMed] [Google Scholar]
- 7.Kishimoto Y, Radin NS. J Lipid Res. 1963;4:139. [PubMed] [Google Scholar]
- 8.Carroll KK. J Lipid Res. 1962;3:263. [Google Scholar]
- 9.O’Brien JS, Rouser G. J Lipid Res. 1964;5:339. [PubMed] [Google Scholar]
- 10.Scheutwinkel-Reich M, Reindl B, Stan H-J. In: Recent Developments in Mass Spectrometry in Biochemistry, Medicine and Environmental Research, 8. Frigerio A, editor. Elsevier; Amsterdam: 1983. p. 251. [Google Scholar]
- 11.Laine RA, Young ND, Gerber JN, Sweeley CC. Biomed Mass Spectrom. 1974;1:10. doi: 10.1002/bms.1200010105. [DOI] [PubMed] [Google Scholar]
- 12.Poole CF, Zlatkis A. J Chromatogr Sci. 1979;17:115. [Google Scholar]
- 13.Drozo J. J Chromatogr. 1975;113:303. [Google Scholar]
- 14.Ciucanu I, Kerek F. J Chromatogr. 1984;284:179. [Google Scholar]
- 15.Aveldano MI, Horrocks LA. J Lipid Res. 1983;24:1101. [PubMed] [Google Scholar]
- 16.Abe K, Tamai Y. J Chromatogr. 1982;232:400. doi: 10.1016/s0378-4347(00)84180-8. [DOI] [PubMed] [Google Scholar]
- 17.DeVries GH, Norton WT. J Neurochem. 1974;22:251. doi: 10.1111/j.1471-4159.1974.tb11587.x. [DOI] [PubMed] [Google Scholar]
- 18.Catalogue. Avanti Polar Lipids Inc; Birmingham, AL: p. 4. [Google Scholar]
- 19.Ruocco MJ, Shipley GG. Biochim Biophys Acta. 1986;859:246. doi: 10.1016/0005-2736(86)90220-8. [DOI] [PubMed] [Google Scholar]
- 20.Johnston DS, Chapman D. Biochim Biophys Acta. 1988;937:10. doi: 10.1016/0005-2736(88)90222-2. [DOI] [PubMed] [Google Scholar]
- 21.Jones JD, Almeida PF, Thompson TE. Biochemistry. 1990;29:3892. doi: 10.1021/bi00468a015. [DOI] [PubMed] [Google Scholar]
- 22.Kishimoto Y, Radin NS. J Lipid Res. 1959;1:79. [PubMed] [Google Scholar]
- 23.Eng LF, Gerstl B, Hayman RB, Lee YL, Tietsort RW, Smith JK. J Lipid Res. 1965;6:135. [PubMed] [Google Scholar]
- 24.Svennerholm L, Stallberg-Stenhagen S. J Lipid Res. 1968;9:215. [PubMed] [Google Scholar]