

Abstract
Background:
Sickle cell disease (SCD) is an inherited haemoglobinopathy characterised by recurrent organ hypoxia-reperfusion cycles which may result in repeated organ damage including the lungs and heart. In SCD, pulmonary hypertension is a known complication that may precede or complicate acute chest syndrome which is often fatal. This study seeks to know the prevalence of pulmonary hypertension and its relationship with clinical and laboratory parameters in sickle cell disease patients attending a tertiary hospital in Lagos.
Materials and Methods:
This was a case — control study involving patients with sickle cell disease recruited from adult sickle cell clinic of Lagos State University Teaching Hospital, Ikeja and HbAA controls matched for age and sex from a tertiary educational institution in Lagos. Both the patients and controls were subjected to echocardiography and pulmonary hypertension was deduced from their cardiac tricuspid regurgitant jet velocity. Other parameters measured were age, body mass index, full blood count, red cell indices, foetal haemoglobin, chest X-ray, liver function tests, lactate dehydrogenase and pulmonary function tests. Consenting patients were 56 HbSS in steady state and 28 HbAA controls matched for age and sex. Data was analysed using SPSS version 16.0.
Results:
The mean age of patients was 22 ± 6 years. In two 2 of 56 (3.6%) of the participants with sickle cell disease, the pulmonary artery pressure was > 25mmHg and there was significant difference in the mean of the pulmonary artery pressure of the control and that of the patients (P-value 0.013). Also, using the appropriate correlation tests, there was significant relationship between the pulmonary artery pressure and lactate dehydrogenase, aspartate transferase and haematocrit in patients with sickle cell disease.
Conclusion:
Sickle cell disease is an independent cause of pulmonary artery hypertension. Variation in cardiovascular reactions to recurrent hyperhaemolysis and hyperdynamic state in sickle cell disease may explain differences in the development of cardiac complications. Exploration of these reactions may reveal other therapeutic measures to prevent complications in sickle cell disease. Clinical assessment of adult patients with sickle cell disease should include echocardiography.
Keywords: Lagos Nigeria, pulmonary hypertension, sickle cell disease
INTRODUCTION
In South-west Nigeria, haemoglobin S has a stable gene frequency of 2.4% homozygote (Hb SS) and 25% heterozygote (HbAS).1
The point mutation in position 6 of the beta-globin gene results in the replacement of hydrophilic glutamic acid by a hydrophobic valine. The consequence is haemoglobin S with reduced solubility in deoxygenated state and a low affinity for oxygen. The haemoglobin in low oxygen tension in the capillaries and venules releases its oxygen content and polymerises. The crystals deform the skeletal proteins and the membrane phospholipids such that the cell becomes more rigid and the membrane becomes more adherent to the endothelium and other blood elements.2 The result is blockage of the microvasculature, reduction red cell life span (haemolysis) and release of inflammatory cytokines and mitogens. With haemolysis, there is release of haemoglobin and arginase. The haemoglobin mops up the constitutive nitric acid which reduces vasoconstriction, platelet activation, vascular fibrosis and vascular muscle proliferation while arginase mops up arginine which is a substrate for the production of nitric oxide.2,3,4,5,6 Histopathology findings in pulmonary hypertension are medial and intimal thickening of the small pulmonary vessels.7 This, in association with increased haemodynamic in sickle cell disease, contributes to the genesis of pulmonary hypertension.
Moreover, various studies have shown that there is pulmonary pathology in adult patients with sickle cell disease.5,6 Suggested causes include interstitial abnormalities caused by repeated acute chest syndrome, airway hyperactivity, nocturnal oxyhaemoglobin desaturation and thromboembolism.8,9,10 Chronic lung disease with generalised fibrosis and restrictive lung function defect is established at about the second decade of life and death at about the fourth decade.11,12,13,14,15 The damage to the lung tissue and consequently the pulmonary vessels may explain the histopathology findings in pulmonary hypertension.
The normal end systolic pressure in the right ventricle is less than 15 mmHg. An increase equal to or above 25 mmHg is considered raised.16,17,18,19 The gold standard for the measurement is by catheterisation of the right ventricle which is an invasive procedure that requires special skill. An increase in the tricuspid regurgitant jet velocity over 2.5 m/second is considered as suggestive of pulmonary hypertension.18,19,20,21
The study by Castro et al., Pashankar et al., and Sulton et al., showed a high prevalence of 25-32% in adult and paediatric African-American sickle cell patients.20,22,23 The Castro et al., study showed an increase in tricuspid regurgitant jet velocity with age after the age of 40 years.20 The study by Gladwin et al., showed a significant relationship between pulmonary hypertension and lactate dehydrogenase but there was no significant relationship with foetal haemoglobin nor use of hydroxyurea.21 They also showed a high risk of death (rate ratio of 10.1).21 Sickle cell disease is, therefore, a common cause of secondary pulmonary hypertension and it is associated with high risk of death.
This study was therefore designed to determine the prevalence of pulmonary hypertension in sickle cell disease patients in our sickle cell clinic and to correlate this with other clinical and laboratory parameters.
MATERIALS AND METHODS
This was a case-control study involving patients in steady state attending the adult sickle cell disease clinic of Lagos State University Teaching Hospital and HbAA controls matched for age and sex recruited from a tertiary educational institution in Lagos. Institutional ethical approval was obtained from the hospital research and ethics committee. Informed written and verbal consents were obtained from participants.
The exclusion criteria were history of chronic lung disease like tuberculosis and smoking. Those with history of organ damage like chronic liver diseases, renal failure and heart failure were also excluded. These are disorders that may independently predispose patients to secondary pulmonary hypertension. While inclusion criteria were HbSS patients that were not in any form of sickle cell crisis and controls were apparently healthy individuals with HbAA. The number of eligible participants was 56 patients and 28 controls and the duration of study was 6 months.
Haemoglobin SS was determined by haemoglobin electrophoresis on cellulose acetate paper in alkaline medium. Red cell indices, white cell count and platelet count were done with the Sysmex haematology autoanalyser model KX-21N within 2 hours of sample collection. The foetal haemoglobin was determined using the modified Betke's method.24 All the participants were given appointments for chest X-ray in the radiology department of the hospital. The X-ray films were reported by the consultant radiologist in the team to identify lung lesions. The pulmonary function tests were done using the analogue vitalograph 2150 spirometry. The percentage predicted value for forced expiratory volume in first second (FEV1), forced vital capacity (FVC) and the peak expiratory flow rate in liters per minute were calculated on the basis of body mass index, age and sex. The participants were given bronchodilator (salbutamol 200 ug) and the percentage changes in volume noted. The pulmonary function was considered normal if FEV1 and FVC predicted were at least 80% and FEV1/FVC ratio was at least 70%. Obstructive lesion was considered if FEV1/FVC ratio was less than 70% and FEV1 was less than 80% while restrictive lesion was considered if FEV1/FVC ratio was at least 70% and FVC is reduced. Mixed lesion was considered if FEV1/FVC ratio was <70% while FEV1 and FVC were <80%.
Alanine transaminase, aspartate transaminase, total bilirubin and lactate dehydrogenase tests were done using Randox kits. The reference value are: Alanine transferase <45 i.u./l, aspartate transferase <35 i.u./l, total bilirubin 0.3-1.2 mg/dl, lactate dehydrogenase 180-360 i.u./l. Transthoracic echocardiograms were performed using Esaote Biomedica Sim challenge 7,000CFM challenge and GE Vivid I Echo Color Ultrasound System. Cardiac measurements were performed by the consultant cardiologist in the team. Peak velocities of E to A wave and ratio of E to A wave at mitral inflow were determined in a standard manner.25,26 Isovolumic relaxation time was measured as the time from aortic valve closure to the start of mitral inflow. Tricuspid regurgitation was assessed in the parasternal right ventricular inflow, parasternal short axis and apical four chamber views to determine the highest velocity. Continuous wave Doppler sampling of the peak regurgitant jet velocity was used to estimate the right ventricular to right atrial systolic pressure gradient. The Bernoulli equation was used to calculate the pulmonary artery systolic pressure (pulmonary artery systolic pressure = 4V2 + estimated right atrial pressure, where V is the average peak tricuspid regurgitant velocity).27 Mean right atrial pressure was estimated according to the degree of inferior vena cava collapse with inspiration: 5 mmHg for collapse >50% and 15 mmHg for collapse <50%.28
Statistical analysis
Analysis was done using SPSS version 16.0 (Statistical Package for Social Sciences, Inc., Chicago, IL). Unpaired ‘t’ test for parametric and Mann-Whitney test for nonparametric data were used as appropriate to compare averages and the Spearman's Rho or Pearson correlation test were used as appropriate for assessing relationships. The critical level of significance was set at P < 0.05.
RESULTS
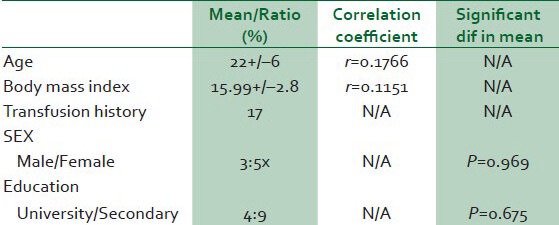
The age range of participants was 14-42 years with a mean of 22 ± 6 years. The male — female ratio was 3:5. All the participants had at least secondary education and 31% of them were either in a University or graduates. The mean body mass index was 15.99 kg/m2 +/-2.8 and 17% of the participants have had blood transfusion. There was no correlation between the pulmonary artery pressure and neither the age nor body mass index. Similarly, there were no significant differences in pulmonary artery pressure of male compared to female nor whether University educated or not [Table 1].
Table 1.
Ociodemographic profile of subjects and relationship with pulmonary hypertension
The mean pulmonary artery pressure in patients with sickle cell disease was 10.46 mmHg +/- 6.93 while it was 7.83 mmHg +/-1.3 in controls. The difference is statistically significant (P = 0.013). Two patients (3.6%) had their pulmonary artery pressure greater than 25 mmHg among the patients with sickle cell disease and 28 patients (52.9%) had evidence of restrictive lung disease.
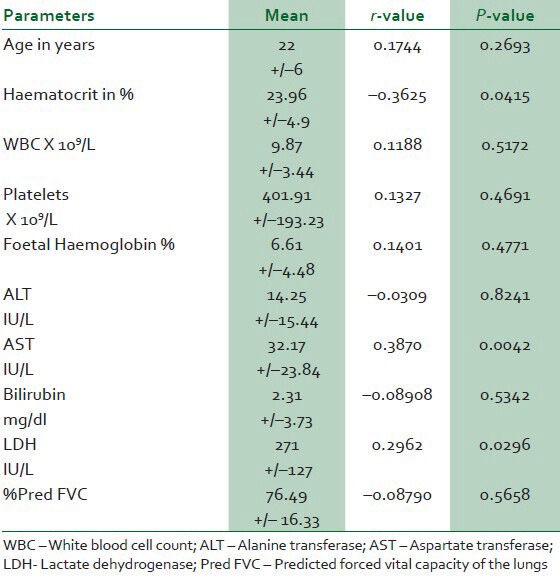
There was negative significant correlation between the estimated pulmonary artery pressure and the haematocrit while the correlation with lactate dehydrogenase and aspartate transferase was positive [Table 2].
Table 2.
Correlation of clinico-laboratory parameters with the mean pulmonary artery pressure in sickle cell disease patients
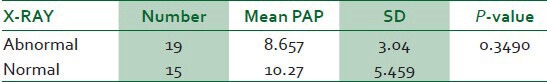
The pathological findings in sickle cell disease on chest X-ray were cardiomegaly, plethora, upper lobe diversion, pulmonary infiltrate and fibrosis. Thirty-four patients had both chest X-ray and echocardiography done and 19 (56%) of them had abnormal chest X-ray findings. There was no significant difference in the means of the pulmonary artery pressure of those with abnormal X-ray findings when compared with that of patients without X-ray lesions (P = 0.3490) [Table 3].
Table 3.
Comparing the echocardiography findings in abnormal and normal x-ray findings in sickle cell patients
DISCUSSION
There was significant statistical difference in the estimated pulmonary artery pressure of our patients compared with that of the controls. However, only 3.6% of these patients with sickle cell disease had evidence of pulmonary hypertension compared to none in controls. The prevalence of 3.6% was low compared to a study in Northern Nigeria where the prevalence in 2008 was 25%.29 Our study population was younger and the cases were a selection (based on exclusion criteria) of a population of patients who are assumed to be better motivated for regular treatment in a tertiary hospital and in a commercial center of the country.
There are reports of pulmonary hypertension in children above 6 years with sickle cell disease.22 In this study, the two patients with raised regurgitant jet velocity had an average age of 25 years. In the Zaria study, the peak was between the age of 18 and 24 years.29 In a study based in the USA, the frequency of pulmonary hypertension increases from the age of 35 years.29 In summary, therefore, pulmonary hypertension develops around the age of 6 and the frequency rises with age; however, the peak age varies from one socioeconomic region to another. The variation may also be a reflection of the different phenotypic expression of haemoglobin SS which presents with various degree of organ damage and survival.
The study demonstrated that with increase in lactate dehydrogenase and aspartate transferase, which increase during haemolysis, there is increase in the pulmonary artery pressure. On the other hand, there is increase in pulmonary artery pressure with reduction in haematocrit. Reduction of haematocrit in sickle cell disease is an indication of hyperhaemolysis. These suggest that haemolysis may contribute significantly to the pathogenesis of pulmonary hypertension in sickle cell anaemia. The differences in vascular reaction to haemolysis, inflammation and increased haemodynamic state, be it genetic or epigenetic, may explain the diversity of phenotypic presentation of patients with sickle cell disease.
In this study, the mean foetal haemoglobin was 6.61 % and there was no correlation with the pulmonary artery pressure. Other studies have also demonstrated that the pulmonary artery pressure increase is independent of the level of foetal haemoglobin or use of hydroxyurea.29 An explanation is that foetal haemoglobin has a threshold effect in preventing sickling.
Pulmonary hypertension has been associated with reduced survival in patients with sickle cell disease.16,17,18,19,20 This study attempted to find a test that may predict pulmonary artery hypertension in sickle cell anaemia. However, it has demonstrated that pulmonary artery pressure is an independent parameter which should be monitored regularly in adult patients with sickle cell anaemia. The study also showed that conditions like chest X-ray abnormalities and restrictive pulmonary function lesions are more common than pulmonary hypertension in adult. They should be included in routine medical checkup in adults with sickle cell disease.
A limitation of this study was the small sample size which is partly based on available fund. The direct measurement of the pulmonary artery pressure by catheterisation which is invasive and which require specialised skill could have been done alongside echocardiography which is a screening tool as it may under or overestimate the pulmonary artery pressure.
CONCLUSION
Conclusion, sickle cell disease is an independent cause of pulmonary artery hypertension. Variation in cardiovascular reactions to recurrent hyperhaemolysis and hyperdynamic state in sickle cell disease may explain differences in the development of cardiac complications. Exploration of these reactions may reveal other therapeutic measures to prevent complications in sickle cell disease. Clinical assessment of patients with sickle cell disease in adults should include echocardiography. The possibility of using the measurement of pulmonary hypertension in assessing prognosis in sickle cell disease will require further studies.
ACKNOWLEGEMENT
This study would not have been possible without the grant from the Lagos State University College of Medicine, Ikeja. We appreciate the devotion of Mr. M.O. Arogundade and Mr. Brodimens in ensuring the quality of the laboratory tests. We thank Miss Ifeoluwa Agboola who provided logistics and Miss Rachel Egede for the data entry.
Footnotes
Source of Support: Nil
Conflict of Interest: The authors are full time staff of the Lagos State University College of Medicine and the teaching hospital. They have no financial commitment with the equipment and reagent manufacturers mentioned in the text.
REFERENCES
- 1.Idowu AT, Oloyede AO, Dosunmu AO. Frequency of Sickle cell genotype among Yorubas in Lagos: Implications of the level of awareness and genetic counseling for sickle cell disease in Nigeria. J Community Genet. 2011;2:13–8. doi: 10.1007/s12687-010-0033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durante W, Johnson FK, Johnson RA. Arginase: A critical regulator of nitric oxide synthetase and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–11. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A, Saldana MJ. Pulmonary hypertension in sickle cell haemoglobinopathy; a clinicopathologic study of 20 cases. Hum Pathol. 2002;33:1037–43. doi: 10.1053/hupa.2002.128059. [DOI] [PubMed] [Google Scholar]
- 4.Liesner RJ, Vandenberghe EA. Sudden death in sickle cell disease. JR Soc Med. 1993;86:484–5. doi: 10.1177/014107689308600822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gladwin MT. Current and future therapies of sickle cell anaemia: An historical perspective. Hematology Am Soc Hematol Educ Program. 2008:176. doi: 10.1182/asheducation-2008.1.176. [DOI] [PubMed] [Google Scholar]
- 6.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis associated pulmonary hypertension and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herve P, Humbert M, Sitbon O, Parent F, Nunes H, Legal C, et al. Patho-biology of pulmonary hypertension. The role of platelets and thrombosis. Clin Chest Med. 2001;22:451–8. doi: 10.1016/s0272-5231(05)70283-5. [DOI] [PubMed] [Google Scholar]
- 8.Young RC, Jr, Rachal RE, Reindorf CA, Armstrong EM, Polk OD, Jr, Hackney RL, Jr, et al. Lung function in sickle cell hemoglobinopathy patients compared with healthy subjects. J Nat Med Assoc. 1988;80:509–14. [PMC free article] [PubMed] [Google Scholar]
- 9.Vichinsky E, Williams R, Das M, Earles AN, Lewis N, Adler A, et al. Pulmonary fat embolism: A distinct cause of severe acute chest syndrome in sickle cell anaemia. Blood. 1994;83:3107–12. [PubMed] [Google Scholar]
- 10.Weil JV, Castro O, Malik AB, Rodgers G, Bonds DR, Jacobs TP. Pathogenesis of lung disease in sickle haemoglobinopathies. Am Rev Respir Dis. 1993;148:249–56. doi: 10.1164/ajrccm/148.1.249. [DOI] [PubMed] [Google Scholar]
- 11.Femi-Pearse D, Gazioglu KM, Yu PN. Pulmonary function studies in sickle cell disease. J Appl Physiol. 1970;28:574–7. doi: 10.1152/jappl.1970.28.5.574. [DOI] [PubMed] [Google Scholar]
- 12.Siddiqui AK, Ahmed S. Pulmonary manifestations of sickle cell disease. Postgrad Med J. 2003;79:384–90. doi: 10.1136/pmj.79.933.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powars D, Weldman JA, Odom-Maryon J, Niland JC, Johnson C. Sickle cell chronic lung disease: Prior morbididty and risk of pulmonary failure. Medicine (Baltimore) 1988;67:66–76. [PubMed] [Google Scholar]
- 14.Klings ES, Wysznski DF, Nolan VJ, Sternberg MH. Abnormal pulmonary function in adult with sickle cell anaemia. Am J Respir Crit Care Med. 2006;173:1264–9. doi: 10.1164/rccm.200601-125OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Field JJ, Glassberg J, Gilmore A, Howard J, Patankar S, Yan Y, et al. Longitudinal analysis of pulmonary function in adult with sickle cell disease. Am J Hematol. 2008;83:574–6. doi: 10.1002/ajh.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fawibe AE, Oluboro PO, Salami AK. Sickle cell chronic lung disease among young adults in Nigeria. West Afr J Med. 2010;29:30–3. doi: 10.4314/wajm.v29i1.55948. [DOI] [PubMed] [Google Scholar]
- 17.Elliot P. Pulmonary hypertension in sickle cell disease. N Engl J Med. 1997;350:857–59. doi: 10.1056/NEJMp038250. [DOI] [PubMed] [Google Scholar]
- 18.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–7. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 19.Simmons BE, Santhanam V, Castaner A, Rao KR, Sachdev N, Cooper R. Sickle cell heart disease; two dimensional echo and Doppler ultrasonographic findings in the heart of adult patients with sickle cell anaemia. Arch Intern Med. 1998;148:1526–8. doi: 10.1001/archinte.148.7.1526. [DOI] [PubMed] [Google Scholar]
- 20.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease; cardiac catherization results and survival. Blood. 2003;101:1257–61. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- 21.Gladwin MT, Sachdev V, Jison ML, Plehn JF, Minter K, Brown B, et al. Pulmonary hypertension as a risk factor for death patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 22.Pashankar FD, Carbonella J, Bazzy-Assad A, Friedman A. Prevalence and risk factors of elevated pulmonary artery pressure in children with sickle cell disease. Pediatrics. 2008;121:777–82. doi: 10.1542/peds.2007-0730. [DOI] [PubMed] [Google Scholar]
- 23.Sulton LL, Castro O, Cross DJ, Spencer JE, Lewis JF. Pulmonary hypertension in sickle cell disease. Am J Cardiol. 1994;74:626–8. doi: 10.1016/0002-9149(94)90760-9. [DOI] [PubMed] [Google Scholar]
- 24.Lewis SM, Bain BJ, Bates I. Practical Haematology. 10th ed. Churchill Livingstone; 2006. Dacie and Lewis; pp. 302–4. [Google Scholar]
- 25.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Chamber Quantification Writing Group. American Society of Echocardiography's Guidelines and Standards Committee; European Association of Echocardiography. Recommendations for chamber quantification: A report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification. J Am Soc Echocardiogr. 2005;18:1440–63. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Quinnones MA, Otto CM, Stoddard M, Waggoner A, Zoghbi WA. Recommendations for quantification of doppler echocardiography: A Report from the Doppler quantification Task Force of the Nomenclature and Standards Committee of the American Society of Echocardiography. J Am Soc Echocardogr. 2002;15:167–84. doi: 10.1067/mje.2002.120202. [DOI] [PubMed] [Google Scholar]
- 27.Berger M, Haimowitz A, Van Tosh A, Berdoff RL, Goldberg E. Quantitative assessment of pulmonary hypertension in patients with tricuspid regurgitation using continuous wave Doppler ultrasound. J Am Coll Cardiol. 1985;6:359–65. doi: 10.1016/s0735-1097(85)80172-8. [DOI] [PubMed] [Google Scholar]
- 28.Kircher BJ, Himelman RB, Schiller NB. Noninvasive estimation of right atrial pressure from the inspiratory collapse of the inferior vena cava. Am J Cardiol. 1990;66:493–6. doi: 10.1016/0002-9149(90)90711-9. [DOI] [PubMed] [Google Scholar]
- 29.Zakari YA, Gordeuk V, Sachdev V, Babadoko A, Mamman AI, Akpanpe P, et al. Prevalence and risk factors for pulmonary artery hypertension among sickle cell disease patients in Nigeria. Am J Hematol. 2008;83:485–490. doi: 10.1002/ajh.21162. [DOI] [PMC free article] [PubMed] [Google Scholar]