

Abstract
Background
The correlations of genotypic and phenotypic tests with treatment, clinical history and the significance of mutations in viruses of HIV-infected patients are used to establish resistance mutations to protease inhibitors (PIs). Emerging mutations in human immunodeficiency virus type 1 (HIV-1) protease confer resistance to PIs by inducing structural changes at the ligand interaction site. The aim of this study was to establish an in silico structural relationship between natural HIV-1 polymorphisms and unusual HIV-1 mutations that confer resistance to PIs.
Results
Protease sequences isolated from 151 Mexican HIV-1 patients that were naïve to, or subjected to antiretroviral therapy, were examined. We identified 41 unrelated resistance mutations with a prevalence greater than 1%. Among these mutations, nine exhibited positive selection, three were natural polymorphisms (L63S/V/H) in a codon associated with drug resistance, and six were unusual mutations (L5F, D29V, L63R/G, P79L and T91V). The D29V mutation, with a prevalence of 1.32% in the studied population, was only found in patients treated with antiretroviral drugs. Using in silico modelling, we observed that D29V formed unstable protease complexes when were docked with lopinavir, saquinavir, darunavir, tipranavir, indinavir and atazanavir.
Conclusions
The structural correlation of natural polymorphisms and unusual mutations with drug resistance is useful for the identification of HIV-1 variants with potential resistance to PIs. The D29V mutation likely confers a selection advantage in viruses; however, in silico, presence of this mutation results in unstable enzyme/PI complexes, that possibly induce resistance to PIs.
Keywords: Antiretroviral agents, Bioinformatics, Molecular docking simulation, Drug resistance, HIV protease, In silico, Polymorphism, Mutations
Background
Diversity of viral populations is a result of sophisticated recombination, replication and/or selection events that induce drug-resistant human immunodeficiency virus type 1 (HIV-1) variants. The lack of reverse transcription corrections, transitional printing and transversion mutations, along with viral recombination, has resulted in the emergence of HIV-1 variants with high resistance to pharmacological stressors [1,2]. These variants form populations that evade antiretroviral agents, due to emerging phenotypic changes within and around the active enzyme site [3]. These mutations, which give rise to drug resistance, result in reduced efficacy of highly active antiretroviral therapy (HAART) [4]. Correlations between genotypic and phenotypic tests with treatment, clinical history, and significance of mutations identified in HIV-1 of infected patients are used to determine the presence of mutations that confer resistance to protease inhibitors (PIs) [1].
Disruption at interaction sites causes an alteration in affinity between proteins and their inhibitors, and has been recognized as a property of drug resistant HIV-1 proteins [5,6]. Protein folding simulation models can create Local Elementary Structures (LES). These secondary structures are stabilized by amino acids that interact with the polypeptide chain [7]. Using the Gromacs software (version 3.0), LES were found to form in protease (PR) regions 23–33, 74–78, and 83–92, and also docked in a folding nucleus [8]. Other studies have shown that mutations further from the active site can alter the flexibility of HIV-1 PR, inducing structural changes that affect the efficacy of most PIs currently used [9]. Theoretical studies, either alone or in combination with experimental methods, have pointed to an increase in the flexibility of mutant enzymes at various sites, including the active site, as a resistance mechanism that causes a decrease in the affinity of PIs [10]. Part of the cause of such flexibility could be the unusual mutations that generally emerge only after "major" and "minor" resistance mutations have been introduced [11]. Other mutations that can affect the interaction between PR and PIs are natural polymorphisms and unusual mutations in positions that confer drug resistance. Although the main mutations associated with drug resistance have been characterized [12,13], little is known about the influence of natural polymorphisms and unusual mutations with respect to the development of drug resistance. The aim of this study was to describe an in silico experiment that showed structural correlations between natural HIV-1 polymorphisms and unusual HIV-1mutations in the PR region of HIV-1 pol with potential PIs resistance.
Methods
Sequence data
We analysed 151 HIV-1 sequences from Mexican patients who had been tested for resistance to antiretroviral drugs between 2005 and 2011 in the Laboratory of Immunodeficiencies and Human Retroviruses, Western Biomedical Research Center, Mexican Institute of Social Security. Sequences were obtained from 22 naïve, and 129 treated patients that were not responsive to drugs. Sequences were registered in the GenBank database [14], with the following accession numbers: [EU045452–EU045489; GU382757–GU382851; GU437199–GU437200; and KC416212–KC416227]. All sequences were analysed for the presence or absence of highly mutated sequences using HYPERMUT software (version 2.0) [15]. For a reference sequence, we used the subtype B consensus sequence, which was derived from an alignment of subtype B sequences maintained at the Los Alamos HIV Sequence Database (LANL), and available from the HIV Drug Resistance Database (HIVDB), Stanford University [16].
Phylogenetic analysis
Nucleotide homology analysis for HIV-1 sequences was conducted using the NCBI Genotyping Tool program [17]. Subtype determinations were further confirmed by phylogenetic analysis performed with the Molecular Evolution Genetics Analysis (MEGA) software package (version 5.0) [18], which includes the recommended reference sequence sets, available from the Los Alamos HIV Sequence Database [19]. Prior to all phylogenetic analyses, HIV-1 pol sequences were aligned using Clustal X (European Bioinformatics Institute, EMBL) [20]. Sequences with 100% homology were excluded from the analysis. The nucleotide distance matrix was generated using the Kimura two-parameter Neighbour-joining method [21]. The statistical robustness of the generated trees was verified by bootstrap analysis of 1000 replicates.
Detection of multidrug resistance phenotypes in HIV-1 protease
The genetic changes associated with drug resistance in viral sequences were established according to HIVdb algorithm version 6.0.9 (http://hivdb.stanford.edu) [22]. The interpretation of drug resistance was performed at various levels of susceptibility for the following USA Food and Drug Administration (FDA)-approved PIs: atazanavir (ATV); darunavir (DRV); amprenavir (APV); indinavir (IDV); lopinavir (LPV); saquinavir (SQV); tipranavir (TPV); nelfinavir (NFV);and ritonavir (RTV). The resistance mutations were classified as major or minor according to HIVdb criteria, or as natural polymorphisms or unusual mutations if they were not associated with resistance [16]. The prevalence (p) for each mutation in the protease region of pol was quantitatively determined as the frequency of the mutation (M) among total sequences evaluated for each position (N), p = M/N, using Microsoft Excel 2010. The genetic variation was calculated as the total number of mutations at a nucleotide position divided by the number of evaluated sequences. The Phenotypic Variation (PV) was defined as the percentage (%) of amino acid substitutions for each position relative to the consensus sequence. For each region, the PV was classified as follows: conserved, <1%; semi-conserved, 1 to <5%; variable, 5 to <10%; and highly variable, ≥10%. Values found below the 15th percentile and above the 75th percentile were not considered. Phenotypic mutations with a prevalence of ≥1.0% among 151 amino acid sequences were compared for each PI against the IAS–USA drug resistance mutations list [12]. The structural conservation of PR was defined in a complementary way to that of PV.
Analysis of selective pressure
The selective pressure and reconstruction of the ancestral state for each PR codon was determined using a maximum likelihood (ML) substitution model and the HyPhy algorithm, included in MEGA5 package [23,24]. The synonymous site divergence (dS) and nonsynonymous site divergence (dN) per branch was estimated using the Muse–Gaut codon model [25]. The values of the ML model were estimated from the topology of the phylogenetic tree. The probability of rejecting the hypothesis of neutral evolution was significant with p ≤ 0.05. The standardized values of dN–dS were obtained by the total number of substitutions in the tree (calculated as substitutions expected by site). To distinguish between drug pressure and immune system pressure, results were compared using the HIV positive selection mutation database [26].
Molecular modelling
Once the natural polymorphisms and unusual mutation codons with positive selection (dN/dS > 1) and prevalence >1% were obtained, homology modelling was used to predict changes in the PR structure. Homology modelling of natural polymorphisms and unusual mutations followed these steps: (i) template selection; (ii) structural alignment; (iii) model construction; and (iv) refinement [27]. To select the template, HIV-1 protease X-ray crystal structure FASTA sequences available from the Protein Data Bank (PDB) [28] and the HIV-1 subtype B consensus sequence available from the HIVdb were aligned using ClustalX [20]. The PR sequence exhibiting greatest identity with HIV-1 subtype B consensus (wild-type template) was chosen as the template for modelling mutant proteases (PRs). The resistant PRs used for reference were modelled with each major PI resistance mutation. Every mutant protein was modelled using a Swiss-Model workspace, which showed the identity (%). The expected alignment value with the template sequence (E) and the Qualitative Model Energy Analysis (QMEAN4), which estimates the absolute quality model, ranged from 0–1 [28,29].
Estimation of the free energy of binding
Using the Autodock/Vina application on a LINUX platform, which had the PyMOL (version 1.4.1) molecular graphics system installed, we estimated the free energy of binding of the complex between mutant PR structures and Pls [30]. Rectangular boxes were used to define the binding sites and these were adjusted by providing specific coordinates of active PR sites before each docking.
Receptor and ligand representations in the Protein Data Bank, Partial Charge & Atom Type formats (pdbqt) containing atomic charges, atom type definitions and topological information, were produced using Autodock/Vina [30]. To determine if the differences caused by natural polymorphisms and unusual mutations had any effect on the free energy of binding of Pls, the free energy values obtained for the resistant protease/ligand complexes were compared. Natural polymorphisms or unusual mutations with lower or equal affinity to PIs compared with reference proteins containing drug resistance mutations indicate positive resistance. Higher affinity was considered to favour susceptibility of the HIV-1 variant to PIs. The coupled proteases included the wild-type PR [PDB: 1GNO], PRs with major drug-resistance mutations and PRs with natural polymorphisms or unusual mutations at codons having positive selection.
Measurement of distances between protease residues and PIs
To evaluate the natural polymorphism and unusual mutation atoms that affect the affinity of PIs, we measured the distances (Å) between the amino acid residue Cα-atoms implicated in drug resistance, and the closest heteroatoms of the PIs. Complexes that showed signs of free energy of binding were analysed, suggesting increased drug resistance because of the presence of natural polymorphisms and unusual mutations. Distances were compared with those obtained for the same pair of atoms in the wild-type and resistant PR structures available from the PDB [28]. All interatomic distances were measured with PyMOL (version 1.4.1) [31].
Results and discussion
Genetic relationships of HIV-1 variants isolated from Mexican patients
Phylogenetic analysis of the 151 HIV-1 protease fragment nucleotide sequences was conducted using a Neighbour-joining tree. Phylogenetic relationships were grouped into the internal nodes of the tree, using subtype B reference sequences [GenBank: U63632 and U21135]. The HIV-1 variants isolated from Mexican patients, and confirmed by analysis with the NCBI Genotyping Tool, were subtype B. This result is consistent with other molecular epidemiology studies of Mexican HIV-1 patients, with or without antiretroviral intervention, where subtype B prevails [32,33].
Drug resistant phenotypes and genotypes of HIV-1 protease
Non-synonymous genetic changes largely contribute to phenotypic changes [34]. Because of degeneracy in the genetic code, transcription and translation errors during the viral replication cycle, along with functional, structural, pharmacological and immunological selection pressures, there is no absolute mathematical relationship between genetic and phenotypic variations [35]. Variations in the primary structure of the 151 PR sequences are presented in Figure 1. Of the 36 codons associated with major or minor resistance, 19 showed PV <5% (L11, L24, D30, V32, L33, E34, K43, I47, G48, I50, F53, D60, G73, T74, L76, N83, I85, N88 and L89). Among these codons, G48, I50 and F53 were present in conserved regions, and L24, D30, L76 and N88 in semi-conserved regions. Six codons (G16, K20, I54, Q58, H69 and I84) displayed PV of 5–10%. Of these, only codons I54 and Q58 were located in conserved and semi-conserved regions, respectively. The remaining 11 codons associated with drug resistance (L10, M36, M46, I62, L63, I64, A71, V77, V82, L90 and I93) had variation >10%, with L10 and I93 present in conserved regions, and M46, V77 and L90 in semi-conserved regions. There was also a variable PV of 5–10%, in the codons neighbouring the drug resistance positions (T12, I15, L19, G68, and K70), with codons E35, N37, R41, R57, and I72 highly variable. Figure 2 illustrates the mutations with prevalence ≥1% found in the protease region of HIV-1 that were present in the 151 PR sequences examined. According to the IAS-USA, the mutations associated with drug resistance, with a p >10%, were L10I, M36I, I62V, L63P, I64V, A71V/T, V77I, L90M, and I93L[12,36,37].
Figure 1.

Genetic and phenotypic representation of primary structure variation within the HIV-1 protease consensus. Codons 1–3 are shown in grey and were not included in our analysis. Conserved regions are shown in yellow, semi-conserved regions in ochre, variable regions in orange and highly variable regions in red. The major (dark green) and minor (light green) resistance mutations are indicated for each codon. aGenetic variation = total number of mutations at the nucleotide position/number of sequences evaluated. bPhenotypic variation = total number of mutations at the amino acid position/number of sequences evaluated.
Figure 2.

Prevalence of mutations within HIV-1 PR pol. Red bars represent mutations associated with drug resistance, and the green bars represent natural polymorphisms and unusual mutations not associated with drug resistance.
Structural studies of PRs have reported a slight widening of the active site due to mutations associated with drug resistance for the majority of PIs [9,10,38]. However for other inhibitors, such as IDV which is characterized by three aromatic rings, structural changes are caused by mutations at the active site and adjacent positions [39].
Prevalence of natural polymorphisms and unusual mutations in PRs without established drug resistance
Table 1 shows the natural polymorphisms or unusual mutations with a p >1% that were found in the PR sequences of HIV-1 isolated from the Mexican patients. These are weakly associated with PI resistance, but are not included in the IAS–USA guides or the HIVdb as accessory or minor mutations [16,40,41]. Of the 14 mutations, only L63A and H69Y were found in drug resistance positions, and T12A/I, I15V, E35D, N37D/E, R57K, K70E and I72V were contiguous to positions associated with resistance. Overall, these mutations have little effect on drug susceptibility; however, a phenotypic change in any of them could have relevance to the affinity to one or more PIs [6,42]. These mutations, in combination with resistance mutations, might have an effect on the dynamics of the evolution of cross-resistance [43].
Table 1.
Polymorphisms or unusual mutations (p > 1%) weakly associated with PI resistance in HIV-1 protease from treated and naïve individuals according to the HIVdb
| Mutation | p (%) | PV (%) | Region (PV) | Association with drug resistance | Classification |
|---|---|---|---|---|---|
|
W6R |
2.34 |
8.20 |
C (0.98) |
Found in indinavir-resistant PR [44] |
UM |
|
T12A/I |
1.34/1.34 |
9.40 |
V (8.04) |
T12A decreased in patients treated with PIs. [45]T12I appears in cell culture in the presence of SQV [46] |
NP/NP |
|
I13V |
17.33 |
18.33 |
V (8.04) |
Found in isolates from patients treated with NFV [47] |
NP |
|
I15V |
8.28 |
9.27 |
V (8.04) |
Associated with reduced virological response to RTV + SQV therapy [48] |
NP |
|
E35D |
18.21 |
18.54 |
HV(26.05) |
Associated with reduced in vivo virological responses to RTV/AMP [49] |
NP |
|
N37D/E |
9.27/5.96 |
40.07 |
HV(26.05) |
N37D appears together with N37E in patient treated with LPV + RTV [50] |
NP/NP |
|
R41K |
19.00 |
19.00 |
SC(2.39) |
Associated with reduced in vivo virological responses to RTV + APV in PIs experienced patients [49] |
NP |
|
R57K |
17.88 |
18.38 |
* |
Relatively frequent in patients failing treatment with RTV + SQV [51] |
NP |
| L63A |
3.48 |
84.77 |
HV(22.02) |
L63A frequent polymorphism but significantly associated with the antiretroviral treatment [39,52] |
NP |
| H69Y |
3.15 |
7.62 |
V(7.56) |
Appears in viruses selected with LPV [53] |
NP |
|
K70E |
3.31 |
9.11 |
V(7.56) |
K70E appears in virus selected in cell culture with DRV [54] |
NP |
| I72R | 1.32 | 24.50 | HV(27.48) | Associated with viral rebound during therapy with LPV + RTV [50] | UM |
p, prevalence; PV, phenotypic variation; C, conserved; SC, semi-conserved; V; variable; HV, highly variable; APV, amprenavir; ATV, atazanavir; DRV, darunavir; IDV, indinavir; RTV, ritonavir; SQV, saquinavir; PIs, protease inhibitors; NP, natural polymorphisms; UM, unusual mutations.
*Values below the 15th percentile or above the 75th percentile were not considered.
The I13V (17.33%), E35D (18.21%), R41K (19%) and R57K (17.88%) mutations had a p ≥ 10% and were located in polymorphic positions observed in non-B subtypes [35,55,56]. In the HIVdb, W6R and I72R are unusual mutations with a frequency <0.05% that only emerge after multiple major and minor resistance mutations [57]. Table 2 shows 41 mutations with a p >1% that have not been associated with resistance, 25 are natural polymorphisms and the remaining 16 were unusual mutations. According to phenotypic conservation analysis, the L5F and Q7E mutations were within the conserved regions, while D29V, P39S, K43R, Q61E, E65D, C67F, P79L, T91V and Q92G/K were within semi-conserved regions. The T12P/S, K14R, G17D/E, Q18H, L19I/V/T, G68E, H69Q and K70R/T/I mutations were within the variable regions, and N37S/T/C/H/I, L63S/V/R/G/H, I72V/T/E/M and I93F were in highly variable regions.
Table 2.
Natural polymorphisms and unusual mutations of HIV-1 protease (p > 1%) without evidence of resistance to PIs
| Mutation | p (%) | PV (%) | Region (PV) | Classification |
|---|---|---|---|---|
|
L5F |
1.67 |
1.67 |
C (0.95) |
UM |
|
Q7E |
1.52 |
1.89 |
C (0.95) |
UM |
|
T12P/S |
4.03/1.34 |
9.40 |
V (8.04) |
NP/NP |
|
K14R |
9.60 |
11.26 |
V (9.97) |
NP |
|
G17D/E |
1.99/1.32 |
3.31 |
V (9.97) |
UM/UM |
|
Q18H |
1.32 |
3.64 |
V (9.97) |
NP |
|
L19I/V/T |
4.64/1.32/ 1.32 |
7.95 |
V (9.97) |
NP/NP/NP |
|
D29V |
1.32 |
2.65 |
SC(1.24) |
UM |
|
N37S/T/C/H/I |
14.4/2.81/1.99/ 1.66/1.32 |
40.07 |
HV(26.05) |
NP/NP/NP/NP/UM |
|
P39S |
2.98 |
4.97 |
SC(2.09) |
NP |
|
K43R |
3.64 |
3.97 |
SC(2.09) |
NP |
|
Q61E |
2.65 |
3.97 |
SC(4.47) |
NP |
|
L63S/V/R/G/H |
1.99/149/1.99/1.32/1.32 |
84.77 |
HV(22.02) |
NP/NP/UM/UM/NP |
|
E65D |
2.0 |
2.67 |
SC(2.0) |
NP |
|
C67F |
2.0 |
3.33 |
SC(2.0) |
NP |
|
G68E |
4.30 |
5.96 |
V(7.56) |
UM |
|
H69Q |
1.99 |
7.62 |
V(7.56) |
NP |
|
K70R/T/I |
1.99/1.32/1.16 |
9.11 |
V(7.56) |
NP/UM/NP |
|
I72V/T/E/M |
11.26/6.95/2.32/1.32 |
24.50 |
HV(27.48) |
NP/NP/NP/UM |
|
P79L |
1.32 |
2.48 |
SC(1.53) |
UM |
|
T91V |
3.33 |
3.33 |
SC(2.15) |
UM |
|
Q92G/K |
2.03/2.03 |
4.05 |
SC(2.15) |
UM/UM |
| I93F | 1.35 | 47.97 | HV(47.63) | UM |
p, prevalence; PV, phenotypic variation; C, Conserved; SC, semi-conserved; V, variable; HV, highly variable; NP, natural polymorphisms; UM, unusual mutations.
Among the codons presented in the Table 2, the mutations in positions K43, L63, H69 and I93 were located in sites associated with minor resistance, but the distance between its localization and the enzyme’s active site reduces the possibility of the structure contributing to drug resistance. All the described mutations could be due to random transcriptional errors, or positive selection from drug and/or immunological stressors [37,58]. Generally, natural polymorphisms occur in remote regions away from the active site, and form domains that define the shape of the homodimer. However, unusual mutations are found in positions associated with drug resistance and possibly generate allosteric changes in the binding site that favour enzymatic function, or decrease the affinity with certain PIs [59]. Therefore, the study of such structural changes produced by these emerging mutations may help in determining the new effects of PIs with different affinities.
Figure 3 shows PR tertiary structure positions that are: not associated with PI resistance; weakly associated with PI resistance; associated with PI resistance. We have also presented the locations of natural polymorphisms and unusual mutations (Figure 3). The codons T12, N37, L63, H69, K70 and I72 include mutations weakly associated with PI resistance (T12A/I, N37D/E, L63A, H69Y, K70E, and I72R), and mutations lacking evidence of PI resistance (T12P/S, N37S/T/C/H/I, L63S/V/R/G/H, H69Q, K70R/T/I, and I72V/T/E/M).
Figure 3.

Codons with natural polymorphisms and unusual mutations in the HIV-1 PR tertiary structure. Codons in the PR that were not associated with PI resistance (cyan), weakly associated with PI resistance (yellow), and associated with PI resistance (red).
The D29V and P79L mutations are located near the active site of the protease, and therefore possibly contribute to the generation of PI resistance. It is of interest to evaluate these unusual mutations in silico, and establish their association with resistance to PIs.
Phenotypic conservation of HIV-1 protease
Figure 4 shows the conserved, semi-conserved, variable and highly variable regions of PRs according to PV. Mutations were clustered into 15 regions, for amino acids 4–99 of the protease. For average PV calculation, when the asymmetry in the distribution was greater than 1.4 between the 15th and 75th percentiles, the residues were not considered. We found three conserved, three variable, three highly variable and six semi-conserved regions for each chain. The positions excluded from the PV calculated for each region were W6, L10, I13, K14, G17, Q18, E35, G40, R41, M46, I54, V56, R57, I63, V77, N83, L90, Q92, I93 and K97. The PV in these codons had very different values from those presented by the codons in their respective regions. According to our model of protease conservation, the LES formed by fragments 23–33 and 74–78 were in semi-conserved regions (E21–L34 and G73–P81, except for V77). The LES formed by the 83–92 fragment involved two codons with variable PV, I84 (6.29%) and L90 (12.33%), and two codons with semi-conserved PV, T91 (3.33%) and Q92 (4.05%) [8,60]. Codon 90 contained a drug resistance mutation (L90M) common for most PIs, with the exception of DRV and TPV, while T91 and Q92 contained the T91V, Q92G, and Q92K mutations, which are classified in the literature as unusual mutations. The prevalence of the L90M, T91V, Q92G, and Q92K mutations was 12.0, 3.33, 2.03 and 2.03%, respectively. Although the effectiveness and specificity of PR proteolytic activity is determined by its active site (amino acids 25–29), these characteristics are influenced by mutations in neighbouring structures, which mainly affect intramolecular interactions with the active site [5,38,42,61]. Contiguous regions and the active site have a semi-conserved state, with a PV of 1.2%. It has been shown that active sites with poor capacity to carry out structural changes help adjust the specificity of natural substrates without losing proteolytic effectiveness [45]. A study that identified the minimal conserved structure of HIV-1 PR, in the presence or absence of drug stress, showed that most of the PV is a product of pharmacological stress [62]. In contrast, the peripheral structural regions have a relatively high PV (for variable and highly variable regions) courtesy of negative selection, and to a lesser extent through resistance of HIV-1 to immune stress [63,64].
Figure 4.
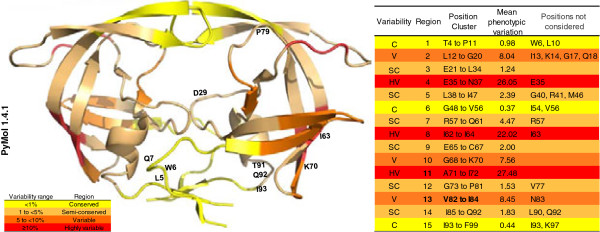
Phenotypic conservation of HIV-1 PR isolated from Mexican patients. A consensus sequence was obtained from 151 individual sequences. Regions are shown from red to yellow in proportion to their phenotypic variation (PV,%). Positions with natural polymorphisms and unusual mutations at drug resistance codons are shown in the protease model. Mutations were clustered into 15 regions between codons 4–99 of the protease.
Selective pressure in the pr fragment of HIV-1 pol
Antiretroviral treatment can exert strong selective pressures within pol, which transcribes PR, reverse transcriptase and integrase [62,65]. We have presented the selection pressure results for 10 codons with natural polymorphisms and unusual mutations (Table 3). According to these results, codons 5, 29, 63, 79, 91 and 93 represent positive pressure (dN–dS > 1) through the ML substitution model using the HyPhy algorithm. When these results are compared with the data available in the UCLA HIV Positive Selection Mutation Database, only codons 63 and 93 were consistent with positive selection under immunological and/or pharmacological stressors [26]. The difference in selection pressure for codons 5, 29, 79 and 91 could be due to variability in the antiretroviral regimen sequences administered to Mexican patients. In addition to positive selection, the aligned sites often evolve at different rates because of other biological factors that include site-specific mutation rates and functional constraints of amino acid substitutions [66].
Table 3.
Selection pressure for codons with unusual mutations and natural polymorphisms
| Codon | Triplet | PV (%) | dN-dS | dN-dS (N) | P value* | Mutations |
|---|---|---|---|---|---|---|
| 5 |
CTT |
1.67 |
2.29 |
0.48 |
0.285 |
L5F |
| 6 |
TGG |
8.20 |
−5.97 |
−1.26 |
0.996 |
W6R |
| 7 |
CAA |
1.89 |
−15.57 |
−3.29 |
1.000 |
Q7E |
| 29 |
GAT |
2.65 |
20.20 |
4.26 |
0.003 |
D29V |
| 63 |
CCC |
84.77 |
2.01 |
0.42 |
0.196 |
L63A/R/S/V/G/H |
| 70 |
AAA |
9.11 |
−1.51 |
−0.32 |
0.964 |
K70I |
| 79 |
CCT |
2.48 |
4.44 |
0.94 |
0.190 |
P79L |
| 91 |
ACT |
3.33 |
5.01 |
1.12 |
0.086 |
T91V |
| 92 |
CAG |
4.05 |
−15.62 |
−3.46 |
0.990 |
Q92G;Q92K |
| 93 | ATT | 47.97 | 28.64 | 6.34 | 1.000 | I93F |
*P < 0.05 was considered statistically significant for positive pressure.
PV, phenotypic variation; dN, number of non-synonymous substitutions per site (n/N); dS, number of synonymous substitutions per site (s/S); N, normalized; P, probability.
The codons that were not associated with resistance due to pharmacological stress, and had PV ≥2% were D29 (2.65%) and P79 (2.48%). These were located near the active site of the enzyme; T91 (3.33%) was also found to be necessary for the establishment of the PR dimer. Codons associated with resistance due to pharmacological stress and PV ≥2% were I47 (2.65%), V82 (10.6%) and I84 (6.29%). Only one of these sequences belonged to a naïve individual, with a mutation at V82; the remaining sequences were from treated individuals (three were treated with reverse transcriptase inhibitors only, the remainder were given reverse transcriptase inhibitors and at least one PI).
Structural prediction of mutant HIV-1 PRs
The molecular structure of all mutant HIV-1 PRs was predicted by comparative homology modelling using the wild-type HIV-1 PR as a template [PDB: 1GNO]. This structure had higher sequence identity compared with the HIV-1 subtype B consensus PR sequence available from the HIVdb. Additional file 1: Table S1 shows the % identity, the expected value of the alignment with the template sequence (E), and the score for the absolute quality of the models. We modelled the proteins with unusual mutations (L5F, D29V, L63G, L63R, P79L and T91V), natural polymorphisms (L63H andL63S), and drug-resistant mutant PRs with single mutations or patterns of mutations (D30N, V32I, M36I, M46I, I47V, G48V, I50V, I50L, I54M, Q58E, T74P, L76V, V82A, V82L, N83D, N88S, I84V, and L90M).
The model’s accuracy was increased because of the identity between the mutant and template sequences; therefore, we concluded that the model was suitable for all structures. The low E values obtained from the modelled proteins indicate template identification, and adequate target template alignment [27]. The reliability of the predicted structures with natural polymorphisms and unusual mutations in drug resistance positions ranged 0.87–0.89, while positions for major mutation proteins ranged 0.83–0.91. The lower QMEAN4 values correlated with mutants containing patterns of resistance, as a result of the reduced identity of these proteins with respect to the template structure [67]. The QMEAN4 values were acceptable for all the modelled structures. Figure 5 shows the overlapping structures of the wild-type PR [PDB: 1GNO] and the D29V mutant, with high similarity between both structures, as well as a difference in the location of the mutation site (position 29).
Figure 5.
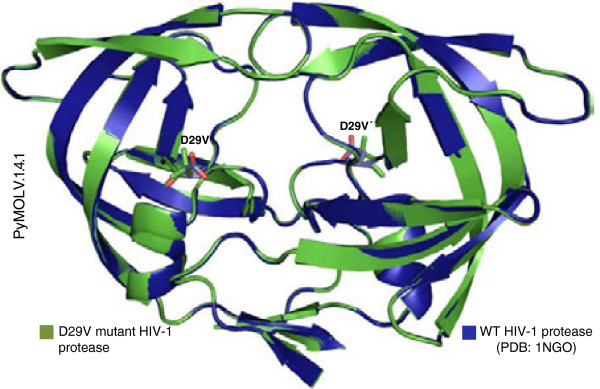
Wild-type and D29V mutant protease structures. The structure of the wild-type HIV-1 protease (WT) was obtained by X-ray crystallography. [PDB: 1GNO] (blue), and the mutant protease (green) can be clearly seen, with the red structures corresponding to the oxygen atoms of D29.
D29 plays a crucial role for the folding of retroviral PRs [38,68]. Using crystallography, it has been shown that D29 forms hydrogen bonds with R87, which partially constitutes the highly conserved triad, G86 − R87 − D88 [62]. The loss of this specific interaction between α − helix 1 (residues 87–91) and D29 destabilizes the dimer interface [69]. The PR structures of related viruses such as HIV − 2, equine infectious anaemia virus (EIAV), feline immunodeficiency virus (FIV), rous sarcoma virus (RSV) and simian immunodeficiency virus (SIV), also demonstrate a proximity between side chains R87 and D29 [70].
The in silico modelling of mutant proteins generated structures very similar to those obtained by X-ray crystallography. The structures with natural polymorphisms, unusual mutations in drug resistance positions, and drug resistance mutations obtained by comparative homology modelling, were appropriate for molecular docking with their respective inhibitors.
Natural polymorphisms and unusual mutations in PRs and their effects on the free energy of binding by PIs
We have presented the free energy of binding (kcal/mol), as well as the average value of the five lowest energy conformations for the complexes formed by PRs and the main PIs (Table 4). The wild-type PR had the lowest free energy of binding for all PIs, except for IDV, compared with mutant PRs containing major and multiple drug resistance mutations. The magnitudes corresponding to the minor values of the free energy of binding to the reference protein were: wild-type protease-1GNO < major drug-resistant mutant proteases < multiple drug-resistant mutant proteases. The PIs had the greatest degree of affinity for PR 1GNO, consistent with the wild-type PR, whereas reduced affinity for mutant PRs was proportional to the number of mutations [46].
Table 4.
Free energy of binding for protease-PI complexes obtained in silico
| Inhibitor | 1GNO-Ligand kcal/mol | Mutant protease with major drug resistance mutation kcal/mol | Multiple mutant kcal/mol | Mutant protease with emergent mutations L5F/D29V/L63G/L63H/L63R/L63S/P79L/T91V kcal/mol |
|---|---|---|---|---|
| Amprenavir |
-8,5(-8,4) |
-8,4(-7,9)/I50V -8,2(-8,12)/I84V |
-8,0(-7,66) |
-8,8(-8,38)/-8,7(-8,28) /-8,8(-8,44)/ -8,8(-8,3)/ -8,8(-8,48)/ -8,7(-8,28)/ -8,8(-8,32)/-8,8(-8,48) |
| Atazanavir |
-8,3(-8,1) |
-7,3(-6,94)/I50L-8,5(-8,14)/I84V -8,2(-7,98)/N88S |
-7,1(-6,86) |
-7,9(-7,82)/ -8,2(-8,14)/ -8(-7,62)/-8,1(-7,96)/ -8,2(-8,04)/-8,2(-8,06)/-9(-8,8)/-8,2(-8) |
| Darunavir |
-9,3(-8,96) |
-9(-8,54)/I47V -9,3(-8,86)/I50V-9,4(-8,84)/I54M -9,1(-8,86)/ I84V |
-8,1(-7,9) |
-9,4(-9)/ -8,8(-8,56)/-9,4(-8,9)/-9,4(-8,92)/ -9,4(-8,92)/-9,4(-8,88) /-9,4(-8,86)/-9,4(-8,88) |
| Indinavir |
-10,4(-10,02) |
-10,8(-10,5)/M46I -10,8(10,4)/V82A-10,5(-10,28)/I84V |
-10,7 (-10,26) |
-10,4(-10,18)/-10,1(-9,92)/-10,8(-10,48)/-10,4(-10)/ -10,4(-10,18)/-10,4(-9,8)/-10,4(-10,02)/-10,7(-10,46) |
| Lopinavir |
-10,3(-9,84) |
-9,9(-9,56)/V32I -9,4(-8,92)/I47V -9,5(-8,62)/L76V -9,7(-9,24)/V82A |
-9,7(-9,14) |
-9,9(-9,56) /-9,7(-9,18) /-9,6(-9,24)/-9,3(-8,86)/ -9(-8,32)/-9,6(-9)/-9,5(-9,2)/-9,6(-9,42) |
| Nelfinavir |
-10,3(-9,46) |
-10(-9,44)/D30N -10,1(-9,8)/L90M |
-9,9(-9,46) |
-10,1(-9,62) /-10,4(-9,8)/-10,1(-9,74)/-10(-9,84)/ -10,1(-9,72)/-10,1(-9,74) /-10(-9,78)/-10,1(-9,7) |
| Saquinavir |
-10,9(-10,46) |
-10,9(-10,6)/G48V -10,4(-10,2)/L90M |
-10,9 (-10,54) |
-9,9(-9,64)/-10,3(-9,62) /-10,4(-10,06)/-10,6(-10,44)/ -10,6(-10,42)/-10,6(-10,42)/ -10,6(-10,5)/-10,5(-10,2) |
| Tipranavir |
-10,6(-10,1) |
-10,4(-9,8)/I47V -10,3(-10,04)/Q58E -10,2(-10,0)/T74P -10,4(-9,9)/V82L -10,2(-9,6)/N83D -10,6(-10,12)/I84V |
-9,9(-9,64) |
-10,2(-9,62) /-10,1(-9,92) /-10,3(-9,98)/-10,2(-9,88)/ -10,2(-9,72)/-10,3(-9,72)/-10,3(-9,72)/-10,2(-9,78) |
| Ritonavir | -8,0(-7,85) | -7,82(-7,44)/I47V -7,8(-7,56)/I50V -7,4(-6,94)/V82L -7,87(-7,58)/I84V -7.9(-7,72)/L90M | -7,26(-6,69) | -8,3(-7,93)/-7,85(-7,54)/ -7,75(-7,52)/-7,9(-7,76)/ -8,3(-8,14)/-8,2(-8,09)/-8,4(-8,8)/-7,79(-7,52) |
The average value of the five conformations with less free energy of binding of the protease-PI complex is presented in parentheses. Values that do not correspond to decreasing order of free energy of binding are presented in boldface type.
Among the PIs, IDV demonstrated a higher affinity for mutant proteins than PR [PDB: 1GNO]. Additionally, a study that correlated the in vivo genetic resistance of HIV-1 to IDV indicated that the development of resistance occurs through the combined effects of several mutations, which do not confer a measurable degree of resistance when they occur on their own [39]. For the other PIs, significant viral resistance has been shown to be a result of the appearance of one or two substitutions in drug-resistance positions [40,71].
The difference between affinities of complexes formed by wild-type and drug-resistant PRs indicates some contribution of phenotypic changes towards PI resistance [72,73]. The complexes with the largest differences involved ATV and DRV, both with a difference of −1.2 kcal/mol. This indicates high susceptibility of both compounds to drug resistance mutations. Lower differences were observed (between −0.7 and −0.3 kcal/mol) for other complexes, indicating these drug resistance mutations have a minor or supplementary effect [72,73].
We obtained a positive value when we calculated the difference of the free energy of binding between the wild-type-IDV complex and the drug-resistant mutant-IDV complex. This is consistent with a high genetic barrier to resistance for IDV, which has lower susceptibility to drug-resistance mutations compared with other PIs [39]. When comparing the free energy of binding between the complexes with drug resistance mutations versus natural polymorphisms and unusual mutation complexes, resistance to ATV, LPV, NFV, and TPV was always observed. The PRs with L5F, D29V, L63G, L63H, L63R, L63S, and P79L mutations had lower or equal free energy of binding to ATV, LPV, NFV and TPV, than those with wild-type or drug-resistant PRs.
The complex formed by the D29V mutant showed considerable differences between the distance of the V29 and D30 Cα -atoms and the heteroatoms closest to the PIs (Table 4). This is probably because of the absence of the Cβ carboxyl group in the valine compared with the wild-type D29. The electrostatic interactions exercised by the D29 carboxyl oxygens provide stronger affinity to PIs in the active site, resulting in greater affinity compared with the V29 mutant [5,74,75]. The absence of V29 carboxyl oxygens decreases the level of interactions, thus decreasing affinity. Such differences can be observed when measuring the distance between the functional groups of the wild-type, resistant and D29V mutant PRs docked to DRV and TPV (Figure 6). For each natural polymorphism and unusual mutation, Table 5 shows the degree of resistance to PIs based on free energy of binding differences when compared with reference PRs.
Figure 6.
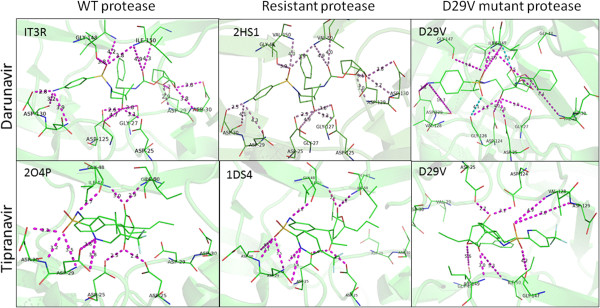
HIV-1 protease structures. Wild-type (WT), resistant and D29V mutant proteases coupled to darunavir (top) and tipranavir (bottom). The numbers in the left upper corner are the PDB ID numbers used to model darunavir and tipranavir with the protease and to measure the distance (Å) between functional groups (purple).
Table 5.
Distances (Å) between the amino acid of the protease and the PI heteroatoms
| |
|
Protease |
|
aa/Chain-PIs | |
Protease |
|
|---|---|---|---|---|---|---|---|
| aa/Chain-PIs | Wild type | Resistant | Emergent | Wild type | Resistant | Emergent | |
|
PR-IDV |
[2R5P] |
[1K6C] |
D29V |
PR-LPV |
[2Q5K] |
[1RV7] |
D29V |
| 29/A-N2 |
6.1 |
6.1 |
5.4 |
25/A-O4 |
5.4 |
6.8 |
9.1 |
| 29/B-O4 |
3.9 |
4.2 |
4.8 |
25/B-O4 |
5.5 |
7.8 |
9.9 |
| 30/A-N2 |
6.5 |
6.6 |
6.3 |
29/A-O1 |
5.8 |
10.8 |
6 |
| 30/B-O4 |
6.3 |
6.4 |
7 |
29/B-O3 |
6 |
10.3 |
5.3 |
| 82/A-O2 |
10.1 |
10.7 |
11.2 |
30/A-O1 |
3.7 |
9.8 |
7.1 |
| 82/B-N1 |
6.1 |
6.2 |
7.5 |
30/B-O3 |
6.7 |
10.1 |
5.3 |
| 84/A-O2 |
8.3 |
8.2 |
9.3 |
84/A-O4 |
8.2 |
10.1 |
10.4 |
| 84/B-N1 |
7.9 |
7.9 |
8.4 |
84/B-O3 |
7.6 |
8.1 |
8.2 |
|
PR-SQV |
[3OXC] |
[3CYW] |
D29V |
PR-RTV |
[3NDX] |
[1RL8] |
D29V |
| 29/A-OD1 |
4.1 |
5.3 |
3.5 |
29/A-O76 |
4 |
3.8 |
4.9 |
| 29/B-N3 |
6.1 |
6 |
7.5 |
29/B-N5 |
5.6 |
4 |
5.1 |
| 30/A-OD1 |
3.8 |
4.2 |
3.8 |
30/A-O76 |
6.1 |
6.1 |
6.9 |
| 30/B-N3 |
6.5 |
6.4 |
7.5 |
30/B-S3 |
4 |
5.5 |
8.8 |
| 48/A-ND2 |
3.4 |
5.6 |
7.9 |
82/A-N11 |
9.4 |
9.5 |
9.6 |
| 48/B-N3 |
4 |
6.3 |
6.3 |
82/B-O7 |
10.5 |
10.3 |
11.6 |
|
PR-APV |
[3NU3] |
[3NU4] |
D29V |
PR-DRV |
[IT3R] |
[2HS1] |
D29V |
| 29/A-O6 |
4.5 |
4.2 |
4 |
29/A-O26 |
3.9 |
4.1 |
10.4 |
| 29/B-N3 |
4.3 |
4.6 |
7.6 |
29/B-N1 |
4.6 |
4.4 |
7.3 |
| 30/A-O6 |
4.5 |
3.8 |
3.8 |
30/A-O26 |
3.8 |
3.8 |
12.3 |
| 30/B-N3 |
3.3 |
3.7 |
10.7 |
30/B-N1 |
3.8 |
3.7 |
9.5 |
| 32/A-O6 |
6.6 |
6.6 |
6.9 |
32/A-O26 |
7.2 |
7.5 |
13.9 |
| 32/B-N3 |
6.3 |
6.3 |
15 |
32/B-N1 |
6.1 |
7.6 |
13.3 |
|
PR-TPV |
[2O4P] |
[1DS4] |
D29V |
PR-ATV |
[3EKY] |
[3OXX] |
D29V |
| 29/A-N28 |
6.1 |
6.2 |
9.6 |
29/A-OAI |
3.9 |
5.1 |
5.6 |
| 29/B-O1 |
9.1 |
9.4 |
8.4 |
29/B-OAJ |
3.9 |
4 |
8.3 |
| 30/A-N28 |
6.2 |
6 |
8.7 |
30/A-OAI |
6.1 |
6 |
8.7 |
| 30/B-O1 |
10 |
10.2 |
8.6 |
30/B-OAJ |
6 |
6 |
8.5 |
| 82/A-O8 |
10.7 |
10 |
10.2 |
50/A-O1 |
5.7 |
5.6 |
4.8 |
| 82/B-O7 |
10.3 |
9.9 |
7.7 |
50/B-O1 |
5.3 |
5.4 |
3.6 |
| 84/A-O8 |
8.3 |
8.1 |
8.5 |
|
|
|
|
| 84/B-O8 |
8.4 |
7.8 |
8.1 |
|
|
|
|
|
PR-NFV |
[3EKX] |
[2PYM] |
D29V |
|
|
|
|
| 29/A-N12 |
5.9 |
6.3 |
5.8 |
|
|
|
|
| 29/B-O38 |
5 |
4.4 |
3.8 |
|
|
|
|
| 30/A-N12 |
6.4 |
6.7 |
6.3 |
|
|
|
|
| 30/B-O38 | 3.9 | 3.7 | 3.7 |
The classification of atoms corresponds to the PDB file visualized in PyMOL. Measurements correspond to the distance between the alpha carbon of the PR amino acids and the heteroatoms of the inhibitor, and are expressed as number of amino acids/chain PR − PI heteroatom.
PIs, protease inhibitors; aa, amino acid; PR, protease; APV, amprenavir; ATV, atazanavir; DRV, darunavir; IDV, indinavir; LPV, lopinavir; RTV, ritonavir; SQV, saquinavir; TPV, tipranavir.
Of the emerging mutations, D29V appears to favour resistance in silico in seven of nine PIs. Designing more effective DRV analogues requires an interaction between D29 and the bis-tetrahydrofuran ring, as this contributes to complex stability [5,42]. All complexes that formed among the PRs with natural polymorphisms, unusual mutations and drug resistance mutations to TPV and LPV had similar free energies of binding. TPV mainly forms hydrogen bonds with residues D25, D29, D30, G48 and I50, while LPV interacts with G27, D29 and D30. A study that elucidated the mechanism by which PIs minimize the harmful effects of resistance mutations, showed that TPV, ATV, LPV, APV, IDV and DRV conserve their antiretroviral activity in the presence of drug resistance mutations. This phenomenon is due to the compensation of the loss of enthalpy (ΔH) with an entropy gain (−TΔS), except in the case of TPV [75]. Our results are consistent with another report that showed isolated strains with a high level of phenotypic resistance to LPV were susceptible to other PIs [76]. This corresponds with PR resistance to TPV and LPV that contain emerging mutations whose free energies of binding were greater than those obtained with wild-type PR.
We found a high prevalence (89%) of L63PGHRS mutations in HIV-1 variants isolated from Mexican individuals, probably because of the prevalence of HIV-1 subtype B [32,33]. In the present study, among the functional groups found at position 63 (L63G, L63S, L63H and L63R), only glycine had hydrophobic characteristics, while serine was hydrophilic, and histidine and arginine were alkaline. These four mutations conferred resistance to NFV, ATV, TPV and LPV, most probably through an allosteric effect, given that the substitutions were not located close to the residues where the PIs bind [10,74]. Few mutations at position 63 have been examined for their resistance effects to PIs. The L63P mutation has a compensatory effect that increases catalytic activity from 110% to 530%; when L63P is associated with M46I, it forms a combination that is resistant to APV, IDV, LPV or NFV [9,77]. Residue 63 provides hydrophobic contacts between the slit of the loop formed by amino acids 38–42 and a β-sheet (residues 59–63) [74]. Although the study of mutations in this position has been limited to L63P to assess the effect of mutations that provide non-hydrophobic characteristics, alternative mechanisms could be shown by which HIV-1 PR compensates for pharmacological stressors.
Clinical characteristics of patients with unusual mutations at resistance sites and/or natural polymorphisms
Of the participating individuals, 48 of 151 (31.8%) showed resistance to at least one PI. Of these, 34 (70.8%) showed a high level of resistance, four (8.3%) showed intermediate levels of resistance, and 10 (20.8%) showed low level resistance.
Of the 151 sequences, 24 (15.9%) had one or more unusual mutations at resistance sites and/or natural polymorphisms. They were isolated from 21 (87.5%) male and three (12.5%) female patients; 70.8% of whom lived in the central-east of Mexico, and 29.2% in the north-west. Of these 24 patients, 23 (95.8%) received antiretroviral therapy, and one (4.2%) was naïve to treatment. The nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) used, in order of frequency, were AZT, 3TC, ddI, d4T, ddC, NVP, EFV and ABC. The main PIs used were IDV, RTV and SQV. The average viral load in this group of patients was 228,225 virus copies/mL and a mean CD4+ lymphocyte count of 223 cells/μL. According to the case definition of HIV infection and AIDS by the Centers for Disease Control and Prevention (Atlanta, USA) [78], patients were classified as asymptomatic (n = 2, 8.5%), symptomatic (n = 6, 25%), AIDS (n = 12, 50%), and of unknown clinical category (n = 4, 16.5%).
Conclusions
The use of bioinformatics to identify potential mutations that confer resistance to antiretroviral drugs allows researchers to develop realistic three-dimensional models that illustrate the atomic interactions between an enzyme and its substrate. In silico, the structural correlation of natural polymorphisms and unusual mutations of drug resistance codons, allows the identification of HIV-1 variants resistant to PIs. The D29V mutation increases the probability of resistance to PIs as it generates unstable complexes at the HIV-1 protease active site. The prevalence of this mutation in different populations should be further studied, and parallel crystallographic studies are required to confirm our in silico findings.
Among mutant PRs-PIs complexes evaluated, TPV and LPV had free energies of binding greater than those obtained with wild-type PRs.
Furthermore, the presence of a high rate of L63P, I93L, V77I and I62V polymorphisms among the Mexican population is similar to that observed in patients that underwent antiretroviral treatments in other American and western European countries. These data reinforced the knowledge regarding the molecular epidemiology of the HIV-1 subtype B in Mexico through the presence of HIV polymorphisms.
Endnote
The Contents of this publication are the authors responsibility and do not necessarily represent the official views of the Instituto Mexicano del Seguro Social.
Abbreviations
FDA: USA Food and Drug Administration; HAART: Highly Active Antiretroviral Therapy; HIV: Human Immunodeficiency Virus; LES: Local Elementary Structures; MEGA: Molecular Evolution Genetics Analysis; PDB: Protein Date Bank; PIs: Protease Inhibitors; PV: Phenotypic Variation; QMEAN: Qualitative Model Energy Analysis; PR: protease; LANL: Los Alamos HIV sequence Database; HIVDB: HIV Drug Resistance Database; p: prevalence; M: Mutation; ML: Maximun Likelihood; dS: synonimous site divergence; dN: nonsynonimous site divergence.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
CMM, MED, MVT and MFS performed sequences and bioinformatics analyses. EVV designed the study. AVO, FGG, MCM and LGFR provided clinical samples and collected the data. BTM performed the data analysis, CMM and EVV wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Identity value, expected value and QMEAN analyses of mutant proteases models tested to estimate the quality of the predicted structure.
Contributor Information
Carlos Mata-Munguía, Email: qfbcamm@msn.com.
Martha Escoto-Delgadillo, Email: brujamajujja@hotmail.com.
Blanca Torres-Mendoza, Email: blanca.torresm@imss.gob.mx.
Mario Flores-Soto, Email: mario.flores@imss.gob.mx.
Mildred Vázquez-Torres, Email: mildred_salud@hotmail.com.
Francisco Gálvez-Gastelum, Email: galvez1975@hotmail.com.
Arturo Viniegra-Osorio, Email: arturo.viniegra@imss.gob.mx.
Marcelo Castillero-Manzano, Email: marcelo.castillero@imss.gob.mx.
Eduardo Vázquez-Valls, Email: eduardo.vazquez@imss.gob.mx.
Acknowledgements
The study was funded in part by the Instituto Mexicano del Seguro Social, Grant Number FIS/IMSS/G09/752. The authors wish to thank Ms. Luz Segovia-Santos for her editorial services and to Greg Davies for his English revision services.
References
- Shafer RW, Kantor R, Gonzales MJ. The genetic basis of HIV-1 resistance to reverse transcriptase and protease inhibitors. AIDS Rev. 2000;2:211–228. [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nat Rev Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- Ribeiro RM, Bonhoeffer S. Production of resistant HIV mutants during antiretroviral therapy. Proc Natl Acad Sci USA. 2000;97:7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Freed EO. Recent progress in antiretrovirals - lessons from resistance. Drug Discov Today. 2008;13:424–432. doi: 10.1016/j.drudis.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- Ohtaka H, Schon A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42:13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- Broglia RA, Provasi D, Vasile F, Ottolina G, Longhi R, Tiana G. A folding inhibitor of the HIV-1 protease. Proteins. 2006;62:928–933. doi: 10.1002/prot.20849. [DOI] [PubMed] [Google Scholar]
- Broglia RA, Tiana G, Sutto L, Provasi D, Simona F. Design of HIV-1-PR inhibitors that do not create resistance: blocking the folding of single monomers. Protein Sci. 2005;14:2668–2681. doi: 10.1110/ps.051670905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana S, Carloni P, Rothlisberger U. Drug resistance in HIV-1 protease: Flexibility-assisted mechanism of compensatory mutations. Protein Sci. 2002;11:2393–2402. doi: 10.1110/ps.0206702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V, Simmerling C. Targeting structural flexibility in HIV-1 protease inhibitor binding. Drug Discov Today. 2007;12:132–138. doi: 10.1016/j.drudis.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007;21:215–223. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VA, Calvez V, Günthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir Med. 2013;21:4–12. [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Li JF, Wei X, Lipscomb J, Irlbeck D, Craig C, Smith A, Bennett DE, Monsour M, Sandstrom P, Lanier ER, Heneine W. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med. 2008;5:e158. doi: 10.1371/journal.pmed.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2002;30:17–20. doi: 10.1093/nar/30.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PP, Korber BT. Detecting hypermutations in viral sequences with an emphasis on G – > A hypermutation. Bioinformatics. 2000;16:400–401. doi: 10.1093/bioinformatics/16.4.400. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–W659. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken C, Korber B, Shafer RW. HIV sequence databases. AIDS Rev. 2003;5:52–61. [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Kantor R, Katzenstein DA, Camacho R, Morris L, Sirivichayakul S, Jorgensen L, Brigido LF, Schapiro JM, Shafer RW. for the International Non Subtype. HIV-1 pol mutation frequency by subtype and treatment experience: extension of the HIVseq program to seven non-B subtypes. AIDS. 2006;20:643–651. doi: 10.1097/01.aids.0000216363.36786.2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Yang Z, Goldman N, Friday A. Comparison of models for nucleotide substitution used in maximum-likelihood phylogenetic estimation. Mol Biol Evol. 1994;11:316–324. doi: 10.1093/oxfordjournals.molbev.a040112. [DOI] [PubMed] [Google Scholar]
- Muse SV, Gaut BS. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994;11:715–724. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- Pan C, Kim J, Chen L, Wang Q, Lee C. The HIV positive selection mutation database. Nucleic Acids Res. 2007;35:D371–D375. doi: 10.1093/nar/gkl855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protocols. 2008;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P, Tosatto SCE, Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL. The PyMOL Molecular Graphics System. 2002. [ http://www.pymol.org]
- Vazquez-Valls E, Escoto-Delgadillo M, Lopez-Marquez FC, Castillero-Manzano M, Echegaray-Guerrero E, Bitzer-Quintero OK, Kobayashi-Gutierrez A, Torres-Mendoza BM. Molecular epidemiology of HIV type 1 in Mexico: emergence of BG and BF intersubtype recombinants. AIDS Res Hum Retroviruses. 2010;26:777–781. doi: 10.1089/aid.2009.0195. [DOI] [PubMed] [Google Scholar]
- Avila-Ríos S, García-Morales C, Garrido-Rodríguez D, Ormsby CE, Hernández-Juan R, Andrade-Villanueva J, González-Hernández LA, Torres-Escobar I, Navarro-Álvarez S, Reyes-Terán G. Mexican HIV Molecular Epidemiology Project Group. National prevalence and trends of HIV transmitted drug resistance in Mexico. PLoS One. 2011;6:e27812. doi: 10.1371/journal.pone.0027812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman JL, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;9(291):1001–4. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- Bürger R, Willensdorfer M, Nowak MA. Why are phenotypic mutation rates much higher than genotypic mutation rates? Genetics. 2006;172:197–206. doi: 10.1534/genetics.105.046599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Liu TF, Holmes SP, Shafer RW. HIV-1 subtype b protease and reverse transcriptase amino acid covariation. PLoS Comput Biol. 2007;3:e87. doi: 10.1371/journal.pcbi.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lee C. Distinguishing HIV-1 drug resistance, accessory, and viral fitness mutations using conditional selection pressure analysis of treated versus untreated patient samples. Biol Direct. 2006;1:14. doi: 10.1186/1745-6150-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi M, Gervasio FL, Tiana G, Provasi D, Broglia RA, Parrinello M. Insight into the folding inhibition of the HIV-1 protease by a small peptide. Biophys J. 2007;93:2813–2821. doi: 10.1529/biophysj.107.106369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condra JH, Holder DJ, Schleif WA, Blahy OM, Danovich RM, Gabryelski LJ, Graham DJ, Laird D, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Yang T, Chodakewitz JA, Deutsch PJ, Leavitt RY, Massari FE, Mellors JW, Squires KE, Steigbigel RT, Teppler H, Emini EA. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J Virol. 1996;70:8270–8276. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hou T, Wang W, Liu JS. Detecting and understanding combinatorial mutation patterns responsible for HIV drug resistance. Proc Natl Acad Sci USA. 2010;107:1321–1326. doi: 10.1073/pnas.0907304107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer RW, Rhee SY, Bennett DE. Consensus drug resistance mutations for epidemiological surveillance: basic principles and potential controversies. Antivir Ther. 2008;13(Suppl 2):59–68. [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc Chem Res. 2007;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- Haq O, Levy R, Morozov A, Andrec M. Pairwise and higher-order correlations among drug-resistance mutations in HIV-1 subtype B protease. BMC Bioinformatics. 2009;10:S10. doi: 10.1186/1471-2105-10-S8-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick L, Yang SS, Rossi R, Zepp C, Heefner D. An Escherichia coli expression assay and screen for human immunodeficiency virus protease variants with decreased susceptibility to indinavir. Antimicrob Agents Chemother. 1998;42:3256–65. doi: 10.1128/aac.42.12.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svicher V, Ceccherini-Silberstein F, Erba F, Santoro M, Gori C, Bellocchi MC, Giannella S, Trotta MP, Monforte A, Antinori A, Perno CF. Novel human immunodeficiency virus type 1 protease mutations potentially involved in resistance to protease inhibitors. Antimicrob Agents Chemother. 2005;49:2015–2025. doi: 10.1128/AAC.49.5.2015-2025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen H, Yasargil K, Winslow DL, Craig JC, Krohn A, Duncan IB, Mous J. Characterization of human immunodeficiency virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31–8959. Virology. 1995;206:527–534. doi: 10.1016/S0042-6822(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Markowitz M, Conant M, Hurley A, Schluger R, Duran M, Peterkin J, Chapman S, Patick A, Hendricks A, Yuen GJ, Hoskins W, Clendeninn N, Ho DD. A preliminary evaluation of nelfinavir mesylate, an inhibitor of human immunodeficiency virus (HIV)-1 protease, to treat HIV infection. J Infect Dis. 1998;177:1533–40. doi: 10.1086/515312. [DOI] [PubMed] [Google Scholar]
- Marcelin AG, Flandre P, De Mendoza C, Roquebert B, Peytavin G, Valer L, Wirden M, Abbas S, Katlama C, Soriano V, Calvez V. Clinical validation of saquinavir/ritonavir genotypic resistance score in protease-inhibitor-experienced patients. Antivir Ther. 2007;12:247–52. [PubMed] [Google Scholar]
- Marcelin AG, Lamotte C, Delaugerre C, Ktorza N, Ait Mohand H, Cacace R, Bonmarchand M, Wirden M, Simon A, Bossi P, Bricaire F, Costagliola D, Katlama C, Peytavin G, Calvez V. Genophar Study Group. Genotypic inhibitory quotient as predictor of virological response to ritonavir-amprenavir in human immunodeficiency virus type 1 protease inhibitor-experienced patients. Antimicrob Agents Chemother. 2003;47:594–600. doi: 10.1128/AAC.47.2.594-600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla A, Brun S, Mo H, Real K, Podding J, Bernstein B, Hertogs K, Larder B, Lie Y, Hellmann N, Vasavanonda S, Chernyavskiy T, Freimuth W, Japour A, Sun E, Kempf D. Genotypic and phenotypic analysis of viral isolates from subjects with detectable viral load on therapy with ABT-378/ritonavir. Antiviral Ther. 2000;5(Suppl 3):30. [Google Scholar]
- Harrigan PR, Hertogs K, Verbiest W, Pauwels R, Larder B, Kemp S, Bloor S, Yip B, Hogg R, Alexander C, Montaner JS. Baseline HIV drug resistance profile predicts response to ritonavir-saquinavir protease inhibitor therapy in a community setting. AIDS. 1999;13:1863–1871. doi: 10.1097/00002030-199910010-00008. [DOI] [PubMed] [Google Scholar]
- Shafer RW, Hsu P, Patick AK, Craig C, Brendel V. Identification of biased amino acid substitution patterns in human immunodeficiency virus type 1 isolates from patients treated with protease inhibitors. J Virol. 1999;73:6197–6202. doi: 10.1128/jvi.73.7.6197-6202.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo A, Stewart KD, Sham HL, Norbeck DW, Kohlbrenner WE, Leonard JM, Kempf DJ, Molla A. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J Virol. 1998;72:7532–7541. doi: 10.1128/jvi.72.9.7532-7541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer S, Azijn H, Van Ginderen M, De Baere I, Pauwels R, De Béthune MP. In vitro selection experiments demonstrate an increased genetic barrier to resistance development to TMC114 as compared with currently licensed protease inhibitors. Antiviral Ther. 2002;7:S5. [Google Scholar]
- Coman RM, Robbins AH, Fernandez MA, Gilliland CT, Sochet AA, Goodenow MM, McKenna R, Dunn BM. The contribution of naturally occurring polymorphisms in altering the biochemical and structural characteristics of HIV-1 subtype C protease. Biochemistry. 2008;47:731–743. doi: 10.1021/bi7018332. [DOI] [PubMed] [Google Scholar]
- Kantor R, Katzenstein DA, Efron B, Carvalho AP, Wynhoven B, Cane P, Clarke J, Sirivichayakul S, Soares MA, Snoeck J, Pillay C, Rudich H, Rodrigues R, Holguin A, Ariyoshi K, Bouzas MB, Cahn P, Sugiura W, Soriano V, Brigido LF, Grossman Z, Morris L, Vandamme AM, Tanuri A, Phanuphak P, Weber JN, Pillay D, Harrigan PR, Camacho R, Schapiro JM. et al. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: results of a global collaboration. PLoS Med. 2005;2:e112. doi: 10.1371/journal.pmed.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The HIVdb Release Notes - HIV Drug Resistance Database. [ http://hivdb.stanford.edu/DR/asi/releaseNotes/index.html]
- Ali A, Bandaranayake RM, Cai Y, King NM, Kolli M, Mittal S, Murzycki JF, Nalam MN, Nalivaika EA, Ozen A, Prabu-Jeyabalan MM, Thayer K, Schiffer CA. Molecular basis for drug resistance in HIV-1 protease. Viruses. 2010;2:2509–2535. doi: 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozisek M, Bray J, Rezacova P, Saskova K, Brynda J, Pokorna J, Mammano F, Rulisek L, Konvalinka J. Molecular analysis of the HIV-1 resistance development: enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J Mol Biol. 2007;374:1005–1016. doi: 10.1016/j.jmb.2007.09.083. [DOI] [PubMed] [Google Scholar]
- Broglia RA. Learning to design resistance proof drugs from folding. Eur Phys J D At Mol Opt Plasma Phys. 2009;51:137–151. [Google Scholar]
- Parera M, Fernández G, Clotet B, Mart¡nez MA. HIV-1 protease catalytic efficiency effects caused by random single amino acid substitutions. Mol Biol Evol. 2007;24:382–387. doi: 10.1093/molbev/msl168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccherini-Silberstein F, Erba F, Gago F, Bertoli A, Forbici F, Bellocchi MC, Gori C, D'Arrigo R, Marcon L, Balotta C, Antinori A, Monforte A, Perno CF. Identification of the minimal conserved structure of HIV-1 protease in the presence and absence of drug pressure. AIDS. 2004;18:F11–F19. doi: 10.1097/01.aids.0000131394.76221.02. [DOI] [PubMed] [Google Scholar]
- Boeri E, Gianotti N, Canducci F, Hasson H, Giudici B, Castagna A, Lazzarin A, Clementi M. Evolutionary characteristics of HIV type 1 variants resistant to protease inhibitors in the absence of drug-selective pressure. AIDS Res Hum Retroviruses. 2003;19:1151–1153. doi: 10.1089/088922203771881257. [DOI] [PubMed] [Google Scholar]
- Karlsson AC, Deeks SG, Barbour JD, Heiken BD, Younger SR, Hoh R, Lane M, Sällberg M, Ortiz GM, Demarest JF, Liegler T, Grant RM, Martin JN, Nixon D. Dual pressure from antiretroviral therapy and cell-mediated immune response on the human immunodeficiency virus type 1 protease gene. J Virol. 2003;77:6743–6752. doi: 10.1128/JVI.77.12.6743-6752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J Virol. 2003;77:1306–1315. doi: 10.1128/JVI.77.2.1306-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira T, Salemi M, Gordon M, Vandamme AM, Van Rensburg EJ, Engelbrecht S, Coovadia HM, Cassol S. Mapping sites of positive selection and amino acid diversification in the HIV genome. Genetics. 2004;167:1047–1058. doi: 10.1534/genetics.103.018135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J+, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Bonomi M, Barducci A, Gervasio FL, Parrinello M. Multiple routes and milestones in the folding of HIV-1 protease monomer. PLoS ONE. 2010;5:e13208. doi: 10.1371/journal.pone.0013208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y, Caflisch A, Onuchic JN, Wolynes PG. The folding and dimerization of HIV-1 protease: evidence for a stable monomer from simulations. J Mol Biol. 2004;340:67–79. doi: 10.1016/j.jmb.2004.04.028. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Hahn BH. The evolution of HIV-1 and the origin of AIDS. Philos Trans R Soc Lond B Biol Sci. 2010;365:2487–2494. doi: 10.1098/rstb.2010.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinformatics. 2009;10:S8. doi: 10.1186/1471-2105-10-S8-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Park MS, Stern HA. Accounting for ligand conformational restriction in calculations of protein-ligand binding affinities. Biophys J. 2010;98:901–910. doi: 10.1016/j.bpj.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslet H, Rosenfeld R, Giffin M, Lin YC, Tam K, Torbett BE, Elder JH, McRee DE, Stout CD. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr D Biol Crystallogr. 2007;63:866–875. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- Muzammil S, Armstrong AA, Kang LW, Jakalian A, Bonneau PR, Schmelmer V, Amzel LM, Freire E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J Virol. 2007;81:5144–5154. doi: 10.1128/JVI.02706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend J, Parkin N, Liegler T, Martin JN, Deeks SG. Isolated lopinavir resistance after virological rebound of a ritonavir/lopinavir-based regimen. AIDS. 2004;18:1965–1966. doi: 10.1097/00002030-200409240-00016. [DOI] [PubMed] [Google Scholar]
- Weikl TR, Hemmateenejad B. How conformational changes can affect catalysis, inhibition and drug resistance of enzymes with induced-fit binding mechanism such as the HIV-1 protease. Biochim Biophys Acta. 2013;1834:867–873. doi: 10.1016/j.bbapap.2013.01.027. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(RR-17):1–19. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identity value, expected value and QMEAN analyses of mutant proteases models tested to estimate the quality of the predicted structure.