

Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ) is a key regulator of glucose and lipid metabolism. Agonists of this nuclear receptor are used in the treatment of type 2 diabetes and are also studied as a potential treatment of other metabolic diseases, including nonalcoholic fatty liver disease. Silymarin, a concentrated phenolic mixture from milk thistle (Silybum marianum) seeds, is used widely as a supportive agent in the treatment of a variety of liver diseases. In this study, the PPARγ activation potential of silymarin and its main constituents was investigated. Isosilybin A (3) caused transactivation of a PPARγ-dependent luciferase reporter in a concentration-dependent manner. This effect could be reversed upon co-treatment with the PPARγ antagonist T0070907. In silico docking studies suggested a binding mode for 3 distinct from that of the inactive silymarin constituents, with one additional hydrogen bond to Ser342 in the entrance region of the ligand-binding domain of the receptor. Hence, isosilybin A (3) has been identified as the first flavonolignan PPARγ agonist, suggesting its further investigation as a modulator of this nuclear receptor.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors orchestrating the expression of genes relevant to lipid and glucose metabolism and occur in three isoforms, alpha, beta, and gamma.1−3 PPAR type gamma (PPARγ) is most highly expressed in the adipose tissue, but important functional expression of this receptor has been also allocated to a variety of nonadipose tissues and cell types, such as skeletal muscle, liver, pancreatic beta cells, myeloid dendritic cells, and macrophages.4 PPARγ is the molecular target of thiazolidinediones (e.g., pioglitazone), used clinically as insulin sensitizers to lower blood glucose levels in diabetes type 2 patients.5,6 The thiazolidinedione type of PPARγ ligands are agonists of the receptor with a very high binding affinity. However, this ligand type demonstrates a range of undesirable side effects,7 prompting the search for new PPARγ agonists effective in the context of lipid and glucose metabolism and inflammation. Interestingly, recent studies indicate that partial agonists of PPARγ, inducing submaximal receptor activation, exhibit very promising activity patterns by retaining the positive effects attributed to the full agonists but with reduced side effects.8−10
In recent years, a close interplay between type 2 diabetes and nonalcoholic fatty liver disease (NAFLD) has been discovered.11 NAFLD is currently the most common cause of liver disease in the Western world. NAFLD is commonly associated with insulin resistance, obesity, dyslipidemia, type 2 diabetes, and cardiovascular disease.12 Therefore, NAFLD is thought to represent the hepatic manifestation of the metabolic syndrome. Given the epidemic of obesity and type 2 diabetes, the burden of NAFLD is expected to continue rising in the near future.12−14 There is a growing body of evidence that PPARs play a role in the pathogenesis of NAFLD and can therefore serve as targets for its therapy.14−16 PPARs are key modulators of gene expression and hepatic triglyceride accumulation, and PPARγ agonists have shown promising results in clinical studies dealing with the treatment of NAFLD, although more evidence for their efficacy from larger clinical studies is still needed.11,12,17
Natural products represent an attractive pool for discovery of novel bioactive compounds, since they encompass a high diversity of structural motifs that, as a result of natural selection, are often evolutionarily optimized to bind to diverse biomolecules and thereby serve a variety of functions.18 Several classes of natural products originating from food or medicinal plants have already been described as PPARγ ligands.9,19−22 Silymarin is a phenolic mixture extracted from milk thistle [Silybum marianum (L.) Gaertn., Asteraceae] seeds. The major part (typically 70–80%) of silymarin consists of seven flavonolignans (i.e., silybin A, silybin B, isosilybin A, isosilybin B, silychristin, isosilychristin, and silydianin; 1–6) and the flavonoid taxifolin (7).23 Silybins A and B (1 and 2) and isosilybins A and B (3 and 4) are two regioisomeric pairs of diastereomers. Even though they were first described in the 1950s and the diastereomeric mixtures were isolated and structurally characterized during the 1960s and 1970s,24,25 the complete isolation and structural characterization of the four isomers was only achieved in 2003.26,27 Milk thistle fruits and seeds have been used for more than 2000 years to treat liver and biliary disorders, and milk thistle seed extracts are still used in the treatment of some ailments, for example as supportive agents in hepatitis and cirrhosis therapy.28 Although more evidence is needed to validate their clinical efficacy in liver disorders,29−31 milk thistle seed preparations are among the best-selling herbal products. In the U.S., in 2012, milk thistle seed preparations ranked sixth of all botanical dietary supplements sold in food, drug, and mass market outlets, reaching about $21 million USD in retail sales, a 7.5% increase over 2011.32 Silymarin and its components display diverse biological activities in vitro and in vivo, including antioxidant, membrane-stabilizing, anticholestatic, antifibrotic, antiatherogenic, anti-inflammatory, anticarcinogenic, and antiviral activity (against hepatitis C).23,28,33−35 These biological activities are supposed to be the basis for the therapeutic potential of silymarin in liver diseases caused by oxidative stress, such as alcoholic and nonalcoholic fatty liver disease (steatohepatitis) and drug- and chemical-induced toxicity, in viral-induced chronic hepatitis, and in primary liver cancer.31
Considering the relevant hepatotherapeutic traditional use of silymarin, as well as the existing interest in identification of novel PPARγ ligands, in this study it was aimed to investigate whether silymarin and its purified flavonolignan and flavonoid constituents are able to activate PPARγ.
Results and Discussion
Since PPARγ is a key player in several pathways related to glucose and lipid metabolism, this nuclear receptor constitutes an important target for drugs used in the treatment of type 2 diabetes and other diseases related to metabolic syndrome such as NAFLD. To examine whether silymarin, used traditionally for the treatment of liver disorders, has any PPARγ activation potential, it was tested in a PPARγ-driven luciferase reporter gene assay. It exhibited a small but statistically significant agonistic effect (19% activation at 30 μg/mL, p < 0.05; not shown). The main constituents in silymarin were quantified by HPLC analysis and were found to be as follows: 12.7% silybin A (1), 21.7% silybin B (2), 4.5% isosilybin A (3), 3.1% isosilybin B (4), 16.1% silychristin (5), 7.1% silydianin (6), 2.6% taxifolin (7). These results are in good accordance with data published for other commercially available milk thistle seed extracts.35

When the seven main constituents of silymarin (1–7) were tested individually in this assay, it turned out that, despite the high structural similarity of some of the compounds, only isosilybin A (3) was able to significantly activate PPARγ at a concentration of 30 μM [2.08 ± 0.48-fold activation, p < 0.01], while the other tested constituents were inactive (Figure 1). The fact that the active constituent 3 represents only 4.5% of the total extract is in accordance with the rather weak activity observed for silymarin.
Figure 1.

PPARγ activation by silymarin constituents. HEK-293 cells were co-transfected with a plasmid encoding full-length human PPARγ, a PPAR luciferase reporter plasmid, and EGFP as internal control. After reseeding, cells were treated as indicated for 18 h. Since the positive control pioglitazone (5 μM) and the silymarin constituents (30 μM) were reconstituted in DMSO, cells were treated with an equal amount of the solvent vehicle DMSO (0.1%) as negative control. The luciferase activity was normalized to the EGFP-derived fluorescence, and the result is expressed as fold induction compared to the solvent vehicle control. The data shown are means ± SD of three independent experiments each performed in quadruplicate [**p < 0.01 (compared to the solvent vehicle group; n = 3, ANOVA/Bonferroni)].
In order to explore why only 3, but not its stereo- and regioisomers, was able to activate PPARγ, docking studies of all tested compounds within the receptor binding pocket of the protein were performed (Figure 2). The PPARγ ligand-binding domain (LBD) has been described previously to possess a Y-shaped topology: The entrance bears several polar residues (e.g., Arg288, Glu291, Glu343, and Ser342). The two branches of the binding pocket, i.e., arm I and arm II, are mainly composed of hydrophobic residues, with the exception of some moderately polar residues in arm I (e.g., Cys285, Ser289, His323, Tyr327, His449, and Tyr473).36 In comparison to the inactive compounds, isosilybin A (3) formed additional hydrogen bonds to Ser342 in the entrance and to Tyr327 in arm I (Figure 2). Due to the distinct configuration at position 7″ of 3, the 4″-hydroxy-3″-methoxyphenyl moiety is able to form a hydrogen bond with Ser342 in the entrance region. A hydrophobic moiety and an acceptor site with an appropriate conformation to establish a hydrogen bond with Ser342 or an equivalent residue in this part of the PPARγ LBD are regarded as essential structural features for partial PPARγ agonists that possess high binding affinity but low transactivation activity in order to come into consideration as antidiabetic drugs.37 These modeling results provide a plausible explanation for the fact that PPARγ activation was observed only for isosilybin A (3), but not for its stereo- and regioisomers.
Figure 2.
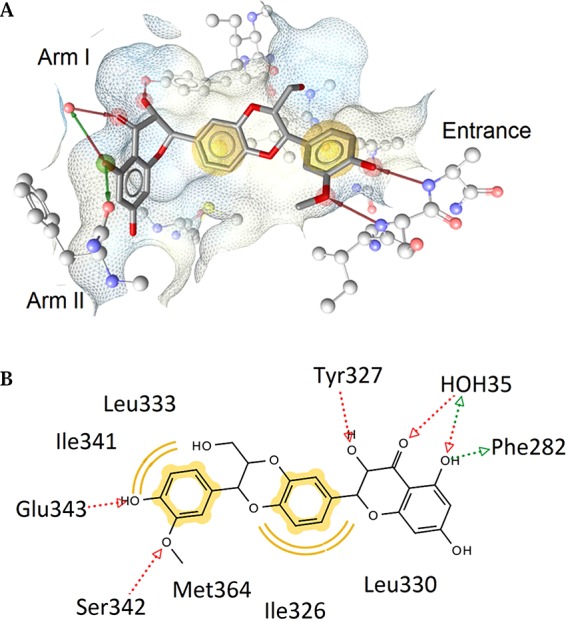
Predicted binding mode of isosilybin A (3), shown in (A) 3D depiction and (B) 2D depiction. Chemical features are color-coded: red/green arrow, hydrogen-bond acceptor/donor; yellow sphere, hydrophobic interaction; surface colored by aggregated lipophilicity (blue)/hydrophobicity (gray). The favorable interactions with the water molecule HOH35 and Ser342, which are not observed in the docking poses for inactive compounds, are suggested to be responsible for the PPARγ partial activation of isosilybin A (3).
Isosilybin A (3) activated the receptor to a smaller extent than pioglitazone, a clinically used PPARγ agonist, even at the highest concentration tested (Figure 3). As can be seen in Figure 4A, the PPARγ activating effect by 3 (30 μM) and pioglitazone (5 μM) were inhibited (p < 0.001) when the PPARγ antagonist T0070907 was added in co-treatment experiments, confirming the PPARγ dependence of the measured effects. It is known that partial receptor agonists often are able to suppress the effects of full agonists upon co-treatment due to competition for receptor binding. To investigate whether 3 is able to reduce the effect of the full PPARγ agonist pioglitazone, the concentration-dependent effect of pioglitazone was tested in the presence or absence of 3 (Figure 4B). Indeed, the pioglitazone-mediated PPARγ activation was clearly reduced in the presence of compound 3.
Figure 3.

Concentration-dependent PPARγ activition by isosilybin A (3) and pioglitazone. HEK-293 cells, transiently transfected with a human PPARγ expression plasmid, a luciferase reporter plasmid (tk-PPREx3-luc), and EGFP as internal control, were treated with different concentrations of pioglitazone or isosilybin A (3) for 18 h. Luciferase activity was normalized by the EGFP-derived fluorescence, and the result is expressed as fold induction compared to the solvent vehicle control (0.1% DMSO). The data points shown are means ± SD of three independent experiments each performed in quadruplicate.
Figure 4.

PPARγ-dependence and co-treatment experiments. (A) HEK-293 cells, transiently transfected with a human PPARγ expression plasmid, a luciferase reporter plasmid (tk-PPREx3-luc), and EGFP as internal control, were treated for 18 h with pioglitazone (5 μM), T0070907 (1 μ M), isosilybin A (3; 30 μM), or combinations as indicated on the x-axis. Luciferase activity was normalized by the EGFP-derived fluorescence, and the results are expressed as fold induction compared to the solvent vehicle control (DMSO, 0.1%). The data points shown are means ± SD of three independent experiments each performed in quadruplicate [**p < 0.01; ***p < 0.001 (n = 3, ANOVA/Bonferroni)]. (B) Cells were transfected as indicated above. Pioglitazone was applied in different concentrations in the presence and absence of 30 μM isosilybin A (3). The data shown are means ± SD of three independent experiments each performed in quadruplicate.
So far, the positive effects observed for silymarin in clinical studies associated with diabetes and NAFLD have mainly been ascribed to its antioxidant and hepatoprotective activity, but PPARγ activation has not been studied before to the best of our knowledge.38−41 When analyzing the silymarin preparation tested by HPLC, it was found that 3 was present in the mixture at a concentration of only 4.5%. Considering that isosilybin A (3) constitutes such a minor fraction of silymarin and that the agonistic properties of this compound seem to be weaker in comparison to pioglitazone (Figure 3), PPARγ activation induced by 3 might not be relevant clinically for the therapeutic use of silymarin. Nevertheless, a contribution of PPARγ activation by 3 to the in vivo action of silymarin cannot be completely ruled out, since several partial agonists activating PPARγ with a weak efficiency in vitro were already demonstrated to display an array of beneficial PPARγ-dependent effects when examined in vivo.8,10,22 Since clinically used PPARγ full agonists of the thiazolidinedione type have a number of undesirable side effects,7 the identification of novel PPARγ activators, including partial agonists, is highly relevant.8−10 While some flavonoids were already reported to activate PPARγ,42,43 this is the first report demonstrating PPARγ activation by a flavonolignan-type compound.
In summary, it is reported for the first time that the flavonolignan isosilybin A (3) from the milk thistle seed extract silymarin acts as a partial PPARγ agonist. Being a new-scaffold PPARγ activator, 3 might serve as a lead for future development of new PPARγ agonists. The question as to whether PPARγ activation by 3 might be clinically relevant for the use of silymarin as an herbal remedy cannot be conclusively answered yet and deserves further investigation.
Experimental Section
Chemicals, Cell Culture Reagents, and Plasmids
Dulbecco’s modified Eagle’s medium (DMEM), containing 4.5 g/L glucose, and l-glutamine were purchased from Lonza (Basel, Switzerland). Fetal bovine serum (FBS) was from Gibco (Lofer, Austria). Silymarin was purchased from Sigma (SO-292-10g). Taxifolin (7) was purchased from Roth, Karlsruhe, Germany (5797.2). Compounds 1, 2, 3, and 4 were isolated and structurally identified as described previously.44 Silydianin (6) was isolated and structurally identified as described.45 The isolation of silychristin (5) is described below. The PPARγ antagonist T0070907 was purchased from Cayman (Ann Arbor, MI, USA), and pioglitazone was from Molekula Ltd. (Shaftesbury, UK). Solvents used for HPLC analyses were of gradient grade. The investigated compounds or dried extracts were dissolved in dimethyl sulfoxide (DMSO), aliquoted, and stored at −20 °C for further use. The final concentration of the solvent vehicle DMSO was 0.1% or lower in all performed experiments. The expression plasmid with human PPARγ (pSG5-PL-hPPAR-gamma1)46 was provided by Prof. Beatrice Desvergne and Prof. Walter Wahli (Center for Integrative Genomics, University of Lausanne, Switzerland), and the luciferase reporter plasmid (tk-PPREx3-luc)47 was kindly supplied by Prof. Ronald M. Evans (Salk Institute for Biological Studies, San Diego, CA, USA). All other chemicals were obtained from Sigma–Aldrich (Vienna, Austria).
Isolation of Silychristin (5)
Isolation was accomplished by preparative HPLC separation of silymarin on a Varian Prep Star SD-1 solvent delivery system equipped with a Dynamax UV-1 absorbance detector, which was set to 280 nm. A 100 mg aliquot of silymarin was dissolved in 0.5 mL of DMSO and 2 mL of methanol and sonicated, and the solution was centrifuged. An Ultra SEP ES RP-18 column (250 × 20 mm, 10 μm) was used as a stationary phase, and a gradient of methanol in water (0–40 min: methanol–water 30:70–55:45; 40–50 min 55:45–100:0) was used as mobile phase (flow rate: 6 mL/min). The peak of 5 (tR 38 min) was collected, and the solvent was evaporated. A yellowish powder (12 mg) was obtained. Its structure was confirmed by NMR spectroscopy in DMSO-d6 on a Varian Unity Inova (600 MHz) spectrometer at 25 °C using the parameters described by Seebacher et al.48 NMR data were found to be in accordance with those values published for 5 by Kim et al.49
HPLC Quantification of Silymarin Constituents
HPLC measurements were performed on an Agilent 1260 HPLC-DAD instrument. As a stationary phase, a Zorbax SB-C18 column (2.1 × 150 mm, 3.5 μm) protected by a Zorbax SB-C8 guard column (2.1 × 12.5 mm, 5 μm) (both Agilent Technologies) was used. The mobile phase consisted of water + 0.1% HCOOH (solvent A) and methanol + 0.1% HCOOH (solvent B). The following gradient was used: 0–30 min A:B 70:30–45:55; 30–40 min A:B 45:55–20:80; 40–45 min A:B 20:80–0:100; 45–46 min A:B 0:100–70:30; 46–55 min A:B 70:30. The flow rate was 230 μL/min, the column temperature was 20 °C, and a detector wavelength of 280 nm was used for quantification.
Silymarin was dissolved in methanol (2.5 mg/mL). For HPLC analysis, 5 μL was injected. For the preparation of calibration curves, reference compounds were dissolved in methanol (1 mg/mL) and serially diluted (0.002 to 1 mg/mL; five concentrations for each compound). From each concentration, 5 μL was injected for HPLC analysis. The purity of the reference compounds used was in the range 93.4–95.8%. All calibration curves showed good linearity (R2 > 0.994).
PPARγ Luciferase Reporter Transactivation
HEK-293 cells (ATCC, Rockville, MD, USA) were maintained in 75 cm2 flasks at 37 °C and 5% CO2, in DMEM with phenol red, with 100 U/mL benzylpenicillin, 100 μg/mL streptomycin, 10% FBS, and 2 mM l-glutamine. The assay was performed as described previously.9,20 Briefly, cells were seeded in 10 cm dishes and transiently transfected by the calcium phosphate precipitation method50 with 4 μg of the reporter plasmid (tk-PPREx3-luc), 4 μg of the PPARγ expression plasmid (pSG5-PL-hPPAR-gamma1), and 2 μg of green fluorescent protein plasmid (pEGFP-N1, Clontech, Mountain View, CA, USA) as internal control. Transfected cells were reseeded in 96-well plates, treated with the indicated compounds or the solvent vehicle (0.1% DMSO), and incubated for 18 h. The cells were then lysed with a reporter lysis buffer (E3971, Promega, Madison, WI, USA). Luciferase activity was evaluated using a GeniosPro plate reader (Tecan, Grödig, Austria), and EGFP-derived fluorescence was used for normalization, to account for differences in the transfection efficiency or cell number.
Docking Study
In order to surmise the binding mode of the main silymarin constituents, a docking study was performed. Basically, in this docking study the quantum mechanics-polarized ligand docking (QPLD) workflow,51 available within the Maestro suite version 9.2.112 (Schrödinger, LLC, New York, NY, 2011, http://schrodinger.com), was employed. Briefly, the ligand and protein preparation as well as the protein–ligand docking were performed in a comparable manner to a previous investigation, which was conducted to propose the binding modes of polyacetylene-type partial PPARγ agonists from natural sources.20 For the docking, the X-ray crystal structure of the LBD of PPARγ in complex with two copies of magnolol, a natural product PPARγ partial agonist, was used (Protein Data Bank52 entry: 3r5n53). The docking poses were postprocessed by (i) the insertion of the water molecule HOH35 into the LBD (which to our best knowledge occasionally has a critical role by mediating interactions from this nuclear receptor to ligands, especially when partial agonists are involved) and (ii) the MMFF94-based minimization within LigandScout 3.1 (Inte:Ligand, GmbH, Maria Enzersdorf, Austria, 2012, http://www.inteligand.com), which was also used for visualization purposes.
Statistical Methods and Data Analysis
All statistical analyses were done with the GraphPad Prism software version 4.03. At least three independent experiments were performed, and one-way analysis of variance (ANOVA) with a Bonferroni post hoc test was used to determine statistical significance. Data with p < 0.05 were considered as significantly different.
Acknowledgments
The expert technical assistance by M. Gössinger, D. Schachner, and H. Beres is gratefully acknowledged. This work was supported financially by the Austrian Science Fund (FWF): S10704, S10705, and S10711 (NFN-project “Drugs from Nature Targeting Inflammation”). The isolation of compounds 1, 2, 3, and 4 was supported in part by the National Institutes of Health/National Center for Complementary and Alternative Medicine via grant R01 AT006842. We acknowledge Inte:Ligand GmbH for providing LigandScout free of charge for this study. We thank Prof. W. Wahli and Prof. B. Desvergne (Center for Integrative Genomics, University of Lausanne, Switzerland), as well as Prof. R. M. Evans (Howard Hughes Medical Institute, California), for providing us with the PPAR expression plasmids and the PPRE luciferase reporter construct, respectively.
Author Contributions
# E.-M. Pferschy-Wenzig and A. G. Atanasov contributed equally to this study.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Evans R. M.; Barish G. D.; Wang Y. X. Nat. Med. 2004, 10, 355–361. [DOI] [PubMed] [Google Scholar]
- Vacca M.; Degirolamo C.; Mariani-Costantini R.; Palasciano G.; Moschetta A. Wiley Interdiscipl. Rev. Syst. Biol. Med. 2011, 3, 562–587. [DOI] [PubMed] [Google Scholar]
- Desvergne B.; Michalik L.; Wahli W. Physiol. Rev. 2006, 86, 465–514. [DOI] [PubMed] [Google Scholar]
- Ahmadian M.; Suh J. M.; Hah N.; Liddle C.; Atkins A. R.; Downes M.; Evans R. M. Nat. Med. 2013, 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J. M.; Moore L. B.; Smith-Oliver T. A.; Wilkison W. O.; Willson T. M.; Kliewer S. A. J. Biol. Chem. 1995, 270, 12953–12956. [DOI] [PubMed] [Google Scholar]
- Lalloyer F.; Staels B. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstrunk A.; Hanf R.; Hum D. W.; Fruchart J. C.; Staels B. Biochim. Biophys. Acta 2007, 1771, 1065–1081. [DOI] [PubMed] [Google Scholar]
- Agrawal R.; Jain P.; Dikshit S. N. Mini Rev. Med. Chem. 2012, 12, 87–97. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G.; Wang J. N.; Gu S. P.; Bu J.; Kramer M. P.; Baumgartner L.; Fakhrudin N.; Ladurner A.; Malainer C.; Vuorinen A.; Noha S. M.; Schwaiger S.; Rollinger J. M.; Schuster D.; Stuppner H.; Dirsch V. M.; Heiss E. H. Biochim. Biophys. Acta 2013, 1830, 4813–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla K.; Hwang B. J.; Choi J. H.; Dewi R.; Ou L.; McLenithan J.; Twadell W.; Pozharskiy E.; Stock J.; Girnun G. J. Biol. Chem. 2011, 286, 41626–41635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. H.; Shackel N. A.; Gorrell M. D.; McLennan S. V.; Twigg S. M. Endocr. Rev. 2013, 34, 84–129. [DOI] [PubMed] [Google Scholar]
- Lomonaco R.; Sunny N. E.; Bril F.; Cusi K. Drugs 2013, 73, 1–14. [DOI] [PubMed] [Google Scholar]
- Tarantino G.; Saldalamacchia G.; Conca P.; Arena A. J. Gastroenterol. Hepatol. 2007, 22, 293–303. [DOI] [PubMed] [Google Scholar]
- Kallwitz E. R.; McLachlan A.; Cotler S. J. World J. Gastroenterol. 2008, 14, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. S.; Kim J. H.; Jo N. Y.; Choi K. M.; Baik S. H.; Park J. J.; Kim J. S.; Byun K. S.; Bak Y. T.; Lee C. H.; Kim A.; Yeon J. E. J. Gastroenterol. Hepatol. 2008, 23, 102–109. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Kung S.; Grewal T.; Roufogalis B. D. Curr. Pharm. Biotechnol. 2012, 13, 278–291. [DOI] [PubMed] [Google Scholar]
- Smith B. W.; Adams L. A. Nat. Rev. Endocrinol. 2011, 7, 456–465. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion-Letellier R.; Dechelotte P.; Iacucci M.; Ghosh S. Gut 2009, 58, 586–593. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G.; Blunder M.; Fakhrudin N.; Liu X.; Noha S. M.; Malainer C.; Kramer M. P.; Cocic A.; Kunert O.; Schinkovitz A.; Heiss E. H.; Schuster D.; Dirsch V. M.; Bauer R. PLoS One 2013, 8, e61755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrudin N.; Ladurner A.; Atanasov A. G.; Heiss E. H.; Baumgartner L.; Markt P.; Schuster D.; Ellmerer E. P.; Wolber G.; Rollinger J. M.; Stuppner H.; Dirsch V. M. Mol. Pharmacol. 2010, 77, 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner C.; de Groot J. C.; Prasad A.; Freiwald A.; Quedenau C.; Kliem M.; Witzke A.; Kodelja V.; Han C. T.; Giegold S.; Baumann M.; Klebl B.; Siems K.; Muller-Kuhrt L.; Schurmann A.; Schuler R.; Pfeiffer A. F.; Schroeder F. C.; Bussow K.; Sauer S. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak S. J.; Ferenci P.; Pawlotsky J. M. Hepatology 2013, 57, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelter A.; Hänsel R. Tetrahedron Lett. 1968, 9, 2911–2916. [Google Scholar]
- Arnone A.; Merlini L.; Zanarotti A. J. Chem. Soc., Chem. Commun. 1979, 696–697. [Google Scholar]
- Kim N. C.; Graf T. N.; Sparacino C. M.; Wani M. C.; Wall M. E. Org. Biomol. Chem. 2003, 1, 1684–1689. [DOI] [PubMed] [Google Scholar]
- Lee D. Y.; Liu Y. J. Nat. Prod. 2003, 66, 1171–1174. [DOI] [PubMed] [Google Scholar]
- Post-White J.; Ladas E. J.; Kelly K. M. Integr. Cancer Ther. 2007, 6, 104–109. [DOI] [PubMed] [Google Scholar]
- Rambaldi A.; Jacobs B. P.; Gluud C. Cochrane Database Syst. Rev. 2007, CD003620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESCOP Monographs, 2nd ed.; 2009; pp 222–248.
- Feher J.; Lengyel G. Curr. Pharm. Biotechnol. 2012, 13, 210–217. [DOI] [PubMed] [Google Scholar]
- Lindstrom A.; Ooyen C.; Lynch M. E.; Blumenthal M. HerbalGram 2013, 99, 60–65. [Google Scholar]
- Polyak S. J.; Morishima C.; Lohmann V.; Pal S.; Lee D. Y.; Liu Y.; Graf T. N.; Oberlies N. H. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 5995–5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saller R.; Meier R.; Brignoli R. Drugs 2001, 61, 2035–2063. [DOI] [PubMed] [Google Scholar]
- Davis-Searles P. R.; Nakanishi Y.; Kim N. C.; Graf T. N.; Oberlies N. H.; Wani M. C.; Wall M. E.; Agarwal R.; Kroll D. J. Cancer Res. 2005, 65, 4448–4457. [DOI] [PubMed] [Google Scholar]
- Zoete V.; Grosdidier A.; Michielin O. Biochim. Biophys. Acta 2007, 1771, 915–925. [DOI] [PubMed] [Google Scholar]
- Guasch L.; Sala E.; Valls C.; Blay M.; Mulero M.; Arola L.; Pujadas G.; Garcia-Vallve S. J. Comput.-Aided Mol. Des. 2011, 25, 717–728. [DOI] [PubMed] [Google Scholar]
- Yeh G. Y.; Eisenberg D. M.; Kaptchuk T. J.; Phillips R. S. Diabetes Care 2003, 26, 1277–1294. [DOI] [PubMed] [Google Scholar]
- Huseini H. F.; Larijani B.; Heshmat R.; Fakhrzadeh H.; Radjabipour B.; Toliat T.; Raza M. Phytother. Res. 2006, 20, 1036–1039. [DOI] [PubMed] [Google Scholar]
- Abenavoli L.; Milic N.; Capasso F. Endocrine 2012, 42, 754–755. [DOI] [PubMed] [Google Scholar]
- Chang C. L.; Chen Y. C.; Chen H. M.; Yang N. S.; Yang W. C. Curr. Med. Chem. 2013, 20, 899–907. [PubMed] [Google Scholar]
- Ortuno-Sahagun D.; Marquez-Aguirre A. L.; Quintero-Fabian S.; Lopez-Roa R. I.; Rojas-Mayorquin A. E. PPAR Res. 2012, 318613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penumetcha M.; Santanam N. PPAR Res. 2012, 858352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T. N.; Wani M. C.; Agarwal R.; Kroll D. J.; Oberlies N. H. Planta Med. 2007, 73, 1495–1501. [DOI] [PubMed] [Google Scholar]
- Wagner H.; Seligmann O.; Seitz M.; Abraham D.; Sonnenbichler J. Z. Naturforsch. B 1976, 31, 876–884. [Google Scholar]
- Peyrin-Biroulet L.; Beisner J.; Wang G.; Nuding S.; Oommen S. T.; Kelly D.; Parmentier-Decrucq E.; Dessein R.; Merour E.; Chavatte P.; Grandjean T.; Bressenot A.; Desreumaux P.; Colombel J. F.; Desvergne B.; Stange E. F.; Wehkamp J.; Chamaillard M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8772–8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S. A.; Umesono K.; Noonan D. J.; Heyman R. A.; Evans R. M. Nature 1992, 358, 771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebacher W.; Simic N.; Weis R.; Saf R.; Kunert O. Magn. Reson. Chem. 2003, 41, 636–638. [Google Scholar]
- Kim N. C.; Graf T. N.; Sparacino C. M.; Wani M. C.; Wall M. E. Org. Biomol. Chem. 2003, 1, 1684–1689. [DOI] [PubMed] [Google Scholar]
- Graham F. L.; van der Eb A. J. Virology 1973, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- Chung J. Y.; Hah J. M.; Cho A. E. J. Chem. Inf. Model. 2009, 49, 2382–2387. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Xu X.; Chen L.; Chen J.; Hu L.; Jiang H.; Shen X. PLoS One 2011, 6, e28253. [DOI] [PMC free article] [PubMed] [Google Scholar]