

Abstract
To develop a new form of DNA coupling under mild reaction and coupling conditions, DNA oligonucleotides were synthesized containing a 3′ ribonucleotide. Upon reaction with millimolar sodium metaperiodate (NaIO4), the ribose is oxidized to a dialdehyde at pH 6.8. This reaction is complete in 30 min, is quenched with millimolar sodium metabisulfite (Na2S2O5) and is then suitable for coupling to hydrazide-agarose supports. Coupling occurs with a half-time of 27 min and 80% couples in 2 h. The EP18 oligonucleotide which binds to the CAAT enhancer binding protein (C/EBP) was synthesized with a 3′ ribose (rEP18) and coupled to hydrazide-agarose. The columns prepared show no significant loss of the oligonucleotide after 50 days. A crude bacterial extract from cells expressing a chimeric fusion protein of GFP-C/EBP was applied to the columns and eluted with different salt concentrations. The active protein elutes in 0.5 M NaCl and SDS-PAGE/silver stained gels show a single major band which comigrates with GFP-C/EBP as well as three minor contaminants. This provides a new alternative way of coupling DNA to solid supports using mild chemistry which is non-detrimental to the DNA and can be performed if required in the presence of nuclear extract.
Keywords: Affinity chromatography, DNA, transcription factors, chemical coupling
Introduction
DNA has been coupled to solid supports by four methods. In one, a 5′-aminoethyl-DNA was coupled using N-hydroxysuccinimide ester (NHS) chemistry to an activated carboxyl silica [1,2]. This method used mild, physiological pH. A second method again used an 5′-amino-DNA coupled but this time coupled to CNBr-activated agarose or silica [3,4]. This method require coupling in alkaline solution. A third method used a hydrazide phosphoramidite reagent to place hydrazides on one or more phosphoryl groups during oligonucleotide solid phase synthesis. The resulting hydrazide oligonucleotides could then be coupled to a glass surface using NHS-ester coupling chemistry or coupled to aldehydes under mild conditions [5]. Finally, a 5′-thiol- oligonucleotide was coupled to a disulfide agarose using thiol-disulfide exchange again using mild conditions [6]. One problem with all of these coupling chemistries is that proteins would also react and couple with these chemistries. Here, we endeavored to use a different approach, which would allow DNA activation and specific coupling to occur even in the presence of proteins under mild conditions, with coupling specific to DNA alone. Such a technique would allow oligonucleotides to be reacted in the presence of proteins resulting in only the DNA being coupled to a support. Such a method would be useful for the oligonucleotide or promoter trapping version of affinity chromatography developed earlier by this laboratory [3,4]. These trapping methodologies have proved to be a powerful method for purifying DNA-binding proteins [7,8].
To accomplish this mild chemistry, it was necessary to use a chemistry that has little or no effect on proteins. Proteins contain amino-, thiol and carboxylate moieties which would preclude the existing methods. For example, to use thiol-disulfide coupling to purify DNA-binding proteins, it would first be necessary to alkylate protein thiols prior to use [6]. Alkaline buffers would need to be avoided. The amino DNA coupling methods would also couple proteins during DNA coupling. In this regard, it is interesting to note that such a coupling procedure has already been used for antibodies, horseradish peroxidase, and other glycopropteins. When polysaccharides are reacted with NaIO4 (sodium metaperiodate), dialdehydes are produced at any vicinal hydroxyls in the saccharide. These aldehydes can be coupled to hydrazide supports under mild conditions and has been used to couple numerous antibodies [9,10], horseradish peroxidase [11] and other glycoproteins with little or no loss of biological activity. However, such coupling has usually involved a final step of reduction with a borohydride. This would normally be considered undesirable since aldehydes can react with protein amines to form imidates (Schiff base). Such imidate formation is readily reversible and would not be a stable linkage. However, a reductant used in these methods would convert this transient imidate to a stable linkage via a secondary amine.
This chemistry has also been used to couple RNA to agarose [12]. RNA contains vicinal hydroxyls at the 3′ end of the RNA. Reaction with NaIO4 yields the dialdehyde which couples to a hydrazide agarose. The resulting RNA-agarose was used for the affinity purification of complementary DNA. Of particular note, the coupling chemistry did not use reduction with borohydride and yet the RNA-hydrazide agarose linkage proved to be very stable.
We reasoned that producing a DNA with a 3′ ribose nucleotide base would provide a gentle method for coupling DNA to a hydrazide support under conditions where protein activity would be virtually unaffected [9–11]. Here, we produce a DNA- agarose using this coupling protocol and show that the chemical linkage to the support is quite stable and can be used for the affinity chromatography of a DNA-binding protein.
1. Materials and Methods
2.1 Materials
T4 polynucleotides kinase was from New England Biolabs (Ipswich, MA, USA). γ-32P-ATP was from Perkin-Elmer (Waltham, MA, USA). NaIO4 was from Acros Organics (Ceel, Belgium) and Na2S2O5 was from Fisher Scientific (Houston, TX, USA). All other chemicals were of the highest purity available commercially or will be specified in sections below.
2.2 Oligonucleotide modification using NaIO4 and Coupling
All oligonucleotides were synthesized by Integrated DNA Technology (Coralville, IA). EP18 (GCAGATTGCGCAATCTGC) is a DNA oligonucleotide and rEP18 is the same sequence except the 3′-C is a ribose nucleotide. For most experiments, the oligonucleotides were 5′ end labeled using T4 polynucleotide kinase and γ-32P-ATP. The oligonucleotides were desalted into buffer KP (100 mM potassium phosphate, pH 6.8) prior to reaction using a 1 ml spin column of BioGel P6 resin (BioRad Laboratories, Richmond, CA). Unless otherwise stated, rEP18 (20 nM-2 μM) was converted to the aldehyde by adding 1/10 volume of 10 mM NaIO4, freshly prepared in water, and reaction for 30 min. at room temperature (20°) in the dark. The reaction was quenched by adding 1/10 volume of 10 mM Na2S2O5, freshly prepared in water, and reacted for 30 min. in the dark at room temperature. When coupled, the rEP18 was mixed with an equal volume of hydrazide-agarose (UltraLink Hydrazide Resin, Fisher Scientific) which had been washed three times by centrifugation into buffer KP, discarding the supernatant. Coupling was at room temperature on a tube rotator, typically for 2 h.
2.3 GFP-C/EBP Expression and Purification
GFP-C/EBP is a chimeric fusion protein containing a His-Tag at the N-terminus and green fluorescent protein followed by the C-terminal 100 amino acid DNA-binding domain of rat C/EBPα at the C-terminus [13]. It was expressed, some of the crude bacterial extract was saved, and the remainder purified using Ni2+-agarose as previously described [13].
2.4 Gel Shift Assay
Unless otherwise stated, gel shift assays were performed on pre-electrophoresed 5% native acrylamide gels run with 0.25X TBE (22 mM H3BO3, 22 mM Tris, 0.5 mM EDTA). Samples contained 2 nM radiolabeled EP18 oligonucleotide, tested protein (typically 5–10 μg purified GFP-C/EBP or column fractions) prepared in 50 mM Tris, pH 7.5, 5% glycerol, 1 mM EDTA, and 1 mM 2-mercaptoethanol were loaded on the gel and electrophoresis was at 90 V for 50 min. The results were detected by autoradiography or using a phosphorimager.
2.5 Stability of Coupling and Affinity Chromatography
50 μl of 200 nM radiolabeled rEP18 was reacted with NaIO4 and Na2S2O5 as described and mixed with 50 μl of hydrazide-agarose and allowed to couple for 4 h. on a rotating wheel. The resin was packed into a small column and thoroughly washed with TE0.1 buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 0.1 M NaCl), TE0.5 (0.5M NaCl), TE1.0 (1.0 M NaCl), TE1.5 (1.5 M NaCl) and TE2.0 (2.0 M NaCl). The column between uses was stored in TE0.1 containing 10 mM NaN3. The column was then counted for Cerenkov radiation (without scintillant) and counted again at various times to record stability using triplicate columns. For affinity chromatography, all subsequent operations were at 4°. 50 μl of 0.31 mg/ml of purified GFP-C/EBP or a bacterial extract in TE (10 mM Tris, pH 7.5, 1 mM EDTA) was applied to the column and the column washed and eluted with 100 μl each time and the eluate collected in a microtiter plate. The column was washed 10 times with TE0.1, and then 3 times each with TE0.5, TE1.0, TE1.5, and TE2.0. Fractions were detected for GFP fluorescence using λex = 398 nm and λem = 512 nm and for DNA-binding by gel shift assay.
2.6 Electrophoresis
Samples were prepared and loaded on a sodium dodecylsulfate 12% polyacrylamide gel for electrophoresis (SDS-PAGE) by the method of Laemmli [14] and silver stained.
2.7 HEK 293 Nuclear Extract preparation
HEK 293 cells were cultured and nuclear extract was prepared as described previously [7].
3. Results and Discussion
As a model system, we used the green fluorescent protein-CAAT enhancer binding protein (GFP-C/EBP) chimeric fusion protein we described earlier [13]. This protein has been shown previously to bind DNA containing the C/EBP response element and provides a protein which can be easily detected and measured using GFP fluorescence. We produced an EP18 oligonucleotide containing this response element as well as a 3′-ribose version of this oligonucleotide referred to as rEP18. The coupling scheme for rEP18 is shown in Fig. 1. This chemistry is performed in potassium phosphate buffer and without borohydride, which should be quite mild conditions for both DNA and proteins.
Figure 1. Reaction scheme.
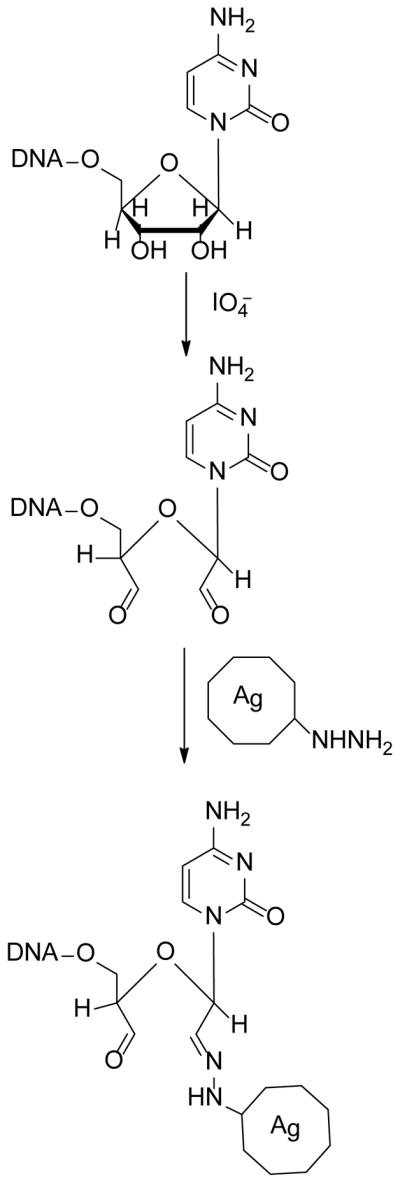
DNA containing a ribose nucleotide at the 3′ end is modified by reaction with sodium meta-periodate (NaIO4) to produce the dialdehyde. The reaction can be quenched by the addition of sodium meta-bisulfite (Na2S2O5). The aldehyde then reacts with a hydrazide-agarose bead to produce coupled DNA.
Fig. 2 shows the results of coupling radiolabeled rEP18 to either Sepharose or hydrazide-agarose, with and without reaction and quenching. After coupling to the resin, the resin is removed and the supernatant was examined by electrophoresis and autoradiography. Regardless of the chemical reaction occuring, rEP18 does not couple to Sepharose, it is all in the supernatants, thus providing a negative control. However, when the rEP18 is reacted with NaIO4, or reacted and quenched with Na2S2O5 (Both), all of it couples to hydrazide-agarose and is no longer in the supernatant (Fig. 2). Also, rEP18 does not couple to hydrazide-agarose without reaction (None), or when the DNA is added to the quench alone (S2O5). When this experiment was repeated with the DNA EP18 (lacking ribose), coupling does not occur with either support (data not shown), confirming that coupling is through the 3′ ribose.
Figure 2. NaIO4 reacted 3′-ribose DNA couples to hydrazide-agarose but not to Sepharose.
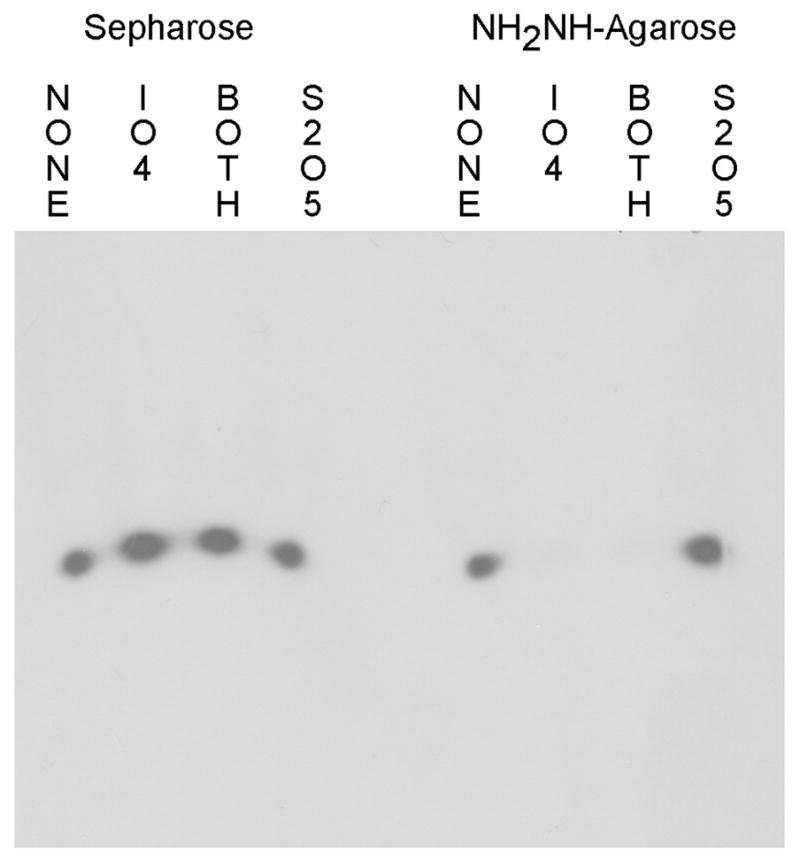
rEP18 was reacted by mixing 5 μl 20 nM radiolabeled rEP18 in 100 mM potassium phosphate, pH 6.8 (buffer KP) with 1 μl of either water (NONE) or 50 mM NaIO4 and incubated in the dark at 20°C for 1 h. The samples were then incubated with 1 μl water or 100 mM Na2S2O5 for an additional 30 min in the dark. 5 μl of each mixture was added to 10 μl of packed Sepharose 4B (Sepharose) or hydrazide-agarose washed into buffer KP. This was allowed to react overnight and the resins were mixed with 5 μl of 0.015% Bromophenol Blue in 50% glycerol (BPB-Glyc) and 20 μl buffer GS (50 mM Tris-HCl, pH 7.5, 5% glycerol, 1 mM EDTA, 1 mM 2-mercaptoethanol), centrifuged and 25 μl of the supernatant was applied to 10% native PAGE for electrophoresis. The gel was then dried and exposed to film for autoradiography.
Coupling of rEP18 to hydrazide-agarose also occurs quite rapidly, as shown in Fig. 3. Aniline has been reported to have a catalytic effect in the aldehyde-hydrazide reaction [15] so the effect of 100 mM aniline on coupling was also investigated. In the absence of aniline, the reaction occurs with an apparent half-time of 27 min. while inclusion of 100 mM aniline resulted in a half-time of 23 min. While the difference in half-time is significant (P < 0.05), we conclude that the effect is small and not useful in this application since 85–90% of the rEP18 couples in either case. Thus, coupling for as little as 2 h. (~4 half-time) in the absence of aniline would result in greater than 80% coupling and is preferred. In data not shown, we also found that the presence of Tris interferes with coupling and should be avoided.
Figure 3. The reaction of rEP18 with hydrazide-agarose in the presence or absence of aniline.
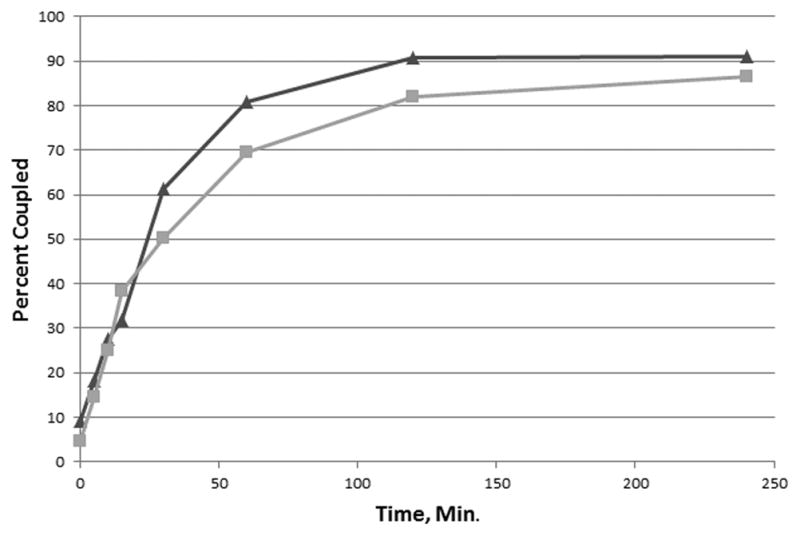
Hydrazide-agarose (200 μl packed resin) was washed with either buffer KP or KP containing 100 mM aniline. To the packed resin was added 200 μl of 20 nM radiolabeled rEP18 reacted in 1 mM NaIO4 for 30 min at 20°C in the dark. The mixtures were stirred magnetically and a portion removed at the times shown. The resin was washed twice with 5 volumes of 1 M NaCl, 50 mM potassium phosphate, pH 6.8 and once with 5 volumes of SDS-PAGE sample buffer. The combined washes and the washed resin were then counted in a scintillation counter without scintillation fluid (Cerenkov radiation). The total counts per minute (washes plus resin) and the counts per minute of the resin were then used to calculate the percentage of the total counts per min coupled to the resin. The results from the semi-logarithmic plot (data not shown) gave a straight line consistent with an apparent first-order reaction with a half-time of 27 min in the absence (squares, grey line) and 23 min in the presence of aniline (triangles, black line).
In Fig. 4, we present the affinity chromatography of partially purified GFP-C/EBP. In Fig. 4A, we follow the chromatography using fluorescence and in Fig. 4B, we show the gel shift of the fractions obtained. The fluorescence shows that the GFP-C/EBP elutes in TE0.5 buffer (containing 0.5 M NaCl). Proteins can be eluted from DNA affinity chromatography using salt and this figure illustrates that elution in this case requires 0.5 M NaCl. Some fluorescence elutes at higher salt concentrations and clearly some active protein elutes in TE2.0 (Fig. 4B). However, the purified (by Ni2+-NTA-agarose) shows some degradation of the GFP-C/EBP, producing a fragment near the mass for GFP alone. This partial degradation is shown in Supplemental Data, Fig. 1. This higher salt eluting fluorescence may not be the intact fusion protein, but rather these degradation products present in the loaded protein, though this was not investigated. The gel shift assay (Fig. 4B) also shows that the DNA-binding activity elutes in TE0.5, with activity also present in the unretained fraction. The column used contained only 200 nM rEP18 and a higher amount of DNA should give higher capacity for GFP-C/EBP. To test this, a column was prepared as in Fig. 4A except using 1 μM DNA. Increasing the amount of DNA coupled five fold (Fig. 4C) lessens the amount of the protein passing through unretained (FT), and more elutes in the high salt buffers (TE0.5–TE2.0); 29% is in FT while 62% in the salt eluates, with 9% in the various wash fractions.
Figure 4. Affinity chromatography of GFP-C/EBP.

50 μl of 0.31 mg/ml purified GFP-C/EBP was mixed with 150 μl TE0.1 and applied to a 50 μl rEP18 column prepared with 200 nM rEP18. All subsequent washes were 0.2 ml of the various buffers collected on a microtiter plate. The column was washed 10 times with TE0.1, and then 3 times each with TE0.5, TE1.0, TE1.5, and TE2.0. A, the fluorescence of each fraction was recorded in epifluorescence mode on a Tecan Infinite M200 fluorescence plate reader with λex = 398 nm, λem = 512 nm. For the TE0.5–TE2.0 fractions, the fluorescence of the 3 fractions was summed. B, The fractions shown were concentrated to 20 μl and added to a gel shift assay using radiolabeled EP18. C, A different column containing 1 μM rEP18 was prepared, loaded and eluted as in panel A. L, Loaded sample; FT, unretained fraction; W1–W10, wash fractions; TE0.5–TE2.0, eluates.
The stability of the column from Fig. 4A&B is shown in Fig. 5. The rEP18 coupled was 5′ end labeled with 32P and simply counting the column without scintillant (Cerenkov radiation) with correction for radioisotope decay can be used to assess the stability of coupling. In each case the column was washed thoroughly before counting and in 3 cases it was used for affinity chromatography such as in Fig. 4. Thus, these columns are stable for at least 50 days with little or no loss of the coupled DNA.
Figure 5. The linkage of rEP18 to hydrazide-agarose is stable for more than a month.

A 50 μl bed column was prepared by coupling 50 μl of 200 nM radiolabeled rEP18 using NaIO4 and Na2S2O5 as described and coupled to hydrazide-agarose for 4h. The column was extensively washed with TE0.1–TE2.0. The column was counted at various times by Cerenkov radiation. Circles show the column after washing and the triangles show the column after its use for affinity chromatography. The experiment was repeated four independent times with similar results.
In Fig. 6 is shown the outcome of loading crude bacterial extract containing GFP-C/EBP onto the column in an analogous experiment to that shown in Fig. 4A&B. Again, the elution profile (Fig. 6A) is similar and the purified protein elutes in TE0.5. When the purity is examined using silver stained SDS-PAGE, the protein elutes at TE0.5 and the major band co-migrates with the GFP-C/EBP purified using Ni2+-agarose affinity chromatography (P). The GFP-C/EBP fusion protein, as previously described [13], contains the enhanced GFP fused at the C-terminus to the 100 amino acid DNA-binding domain of rat C/EBP. The calculated mass is 42.6 kDa., which agrees well with the mass observed in Fig. 6B. The eluted GFP-C/EBP also contained three minor contaminants.
Figure 6. Affinity chromatography of GFP-C/EBP from crude bacterial extract.

A 50 μl column prepared from 100 nM rEP18 was used to purify the GFP-C/EBP from a crude bacterial extract. Elution was as described for Fig. 4. A, Average epifluorescent measurements from two independent experiments are shown ± standard deviation. B, samples were concentrated, applied to 12% SDS-PAGE, and stained with silver. P, GFP-C/EBP purified by Ni2+-agarose affinity chromatography; C, crude bacterial extract; F, flow-through (unretained) fraction; W1–W10, TE0.1 washes.; -, no sample applied.
GFP-C/EBP has previously been purified by affinity chromatography on a column of EP18 coupled to Sepharose using the CNBr coupling method [13]. The conditions used in Fig. 7 of [13] for 4° elution most closely resemble the method used here in Fig. 6. The purity shown in Fig. 6 is considerably higher than that previously reported [13] indicating that the new coupling procedure performs better than that previously used. The same protein was also purified using a different DNA containing the CAAT element in a hairpin DNA with a 5′-thiol. In that method, the DNA couples to a disulfide-agarose by thiol-disulfide exchange. To prevent the protein thiols from also exchanging, these had to be alkylated (with iodoacetamide) prior to application to the column [6]. The DNA- protein complex was then eluted from the column with a thiol. This more complex method gave similar purity to that shown here in Fig. 6. Thus, the new coupling procedure either out-performs one coupling procedure and gives similar performance to a more complex procedure.
To determine if the chemistry was sufficiently mild that it would not affect protein activity even if proteins were present during reaction, the experiments depicted in Fig. 7 were performed. In Fig. 7A, HEK 293 nuclear extract (containing native C/EBP) was mixed with radiolabeled EP18, reacted with NaIO4 and Na2S2O5, and DNA-binding activity measured by a gel shift assay. Nuclear extract after freezing contains some insoluble aggregate (A) which is solubilized by treatment with NaIO4. Regardless of this, the complex (C) formed by C/EBP binding to EP18 is unaffected by either of the chemicals. In other experiments (data not shown), we demonstrated that reaction with NaIO4 does not alter either the excitation or emission spectra of GFP-C/EBP, showing that this protein is also not adversely affected by reaction. This observation was used to design the experiment in panel B. GFP-C/EBP in the presence or absence of radiolabeled rEP18 was either reacted with NaIO4 or not and the fluorescence (input) and radioactivity measured. The resulting mixture was then mixed with hydrazide-agarose, allowed to couple, and the resin then washed thoroughly with TE1.0 to remove any non-covalently bound protein or DNA. In panel B, the fluorescence (GFP-C/EBP) before coupling (input) is compared to the washes after coupling (output) and shows no significant differences caused by either reaction or coupling. Thus, the protein fluorescence is unaffected by reaction and the protein does not couple to hydrazide-agarose. The washes from the resin were also measured for radioactivity (rEP18) and these results are also shown in panel B. The amount of rEP18 recovered in the washes is statistically different (p<0.05) for rEP18 that was reacted and coupled (column C) compared to the same amount of rEP18 that was not reacted but otherwise treated the same (D). Thus, rEP18 couples even in the presence of protein, and in agreement with results in Fig. 2 and 3, rEP18 coupling requires prior reaction. The resins recoverd after washing also confirm that coupling occurred under condition C but not under condition D but because they are counted from a small volume on a solid surface, the cpm measured are not directly comparable to the cpm of the washes and these data are not shown.
Figure 7. The chemical reactions do not affect the gel shift assay for nuclear extract C/EBP, and when mixed, rEP18 couples but GFP-C/EBP does not.

A, 5 μl 20 nM radiolabeled EP18 in buffer KP is mixed with 5 μl 5 mg/ml HEK 293 nuclear extract, and 1 μl 100 mM NaIO4 (or water) is added, the mixture is incubated in the dark for 1 h at 20°C, and then 1 μl 200 mM Na2S2O5 (or water) is added, and further incubated for 30 min. The samples were then mixed with 5 μl BPB-Glyc and 15 μl buffer GS, and 20 μl was loaded on a 5% native PAGE for a gel shift assay. After electrophoresis, the gel was dried and exposed to film for autoradiography. A, an aggregate which didn’t enter the gel; C, the shifted DNA-protein complex; F, free DNA. B, GFP-C/EBP (30 μM) in buffer KP was either mixed with buffer or radiolabeled rEP18 in buffer to give a final concentration of 20 nM DNA as indicated in the figure. The mixture was divided into portions and either reacted with water or NaIO4 and NaS2O5 as indicated at a final concentration of 1 mM each. Portions of each reaction were saved (to determine input, gray bars) or mixed with 10 μl of hydrazide-agarose and allowed to couple for 2 h at 20°C. After coupling, the resin was washed 3 times with 10 volumes of TE1.0 buffer. The washes were combined and either scintillation counted (Cerenkov radiation, CPM, black bars) or the fluorescence was determined (output, white bars) as indicated. All determinations are in triplicate and error bars show standard deviation. The only statistical difference between these data are reacted rEP18 (C) versus unreacted (D, p<0.05).
Thus, Fig. 7 demonstrates that proteins were unaffected by the chemistry used and that rEP18 couples even in the presence of nuclear extract. This is unusual since most coupling chemistries can react with proteins or be detrimental to them. For example, CNBr used to activate Sepharose for coupling must be carefully removed prior to addition of protein since it can modify and digest proteins. In the case of this aldehyde chemistry, even if the reagents are not removed, protein activity will be retained. This is in agreement with previous reports using a similar chemistry for the coupling of antibodies and enzymes [9–11]. However, it should be pointed out that while protein itself is unaffected by the chemistry, this would not be true for saccharides; thus, while non-glycosylated proteins such as bacterial GFP-C/EBP or nuclear C/EBP are unaffected, this would probably not be the case for glycoproteins or proteoglycans.
Linking by way of the 3′ end has certain advantages and disadvantages. The main advantage is that the 5′ end is available for other modifications such as 5′ phoshorylation with 32P for detection. If it were necessary for a particular purpose, a 5′ end ribose nucleotide could be added to an oligonucleotide during synthesis. Normally, oligonucleotides are linked 5′-3′ but they can also be added in a 5′-5′ phosphodiester linkage, commonly referred to as “reverse” orientation, at the 5′ end using a ribose nucleotide. This would leave a ribose at the 5′ end for modification and subsequent coupling. This approach would also allow PCR products to be coupled if the primers used for PCR were synthesized with a reverse ribose nucleotide. Another approach could be used for either native DNA or PCR products. T4 RNA ligase normally adds ribose nucleotides to the 3′ end of RNA but the enzyme also will add one or more ribonucleotides to the 3′ end of DNA, albeit at 200-fold reduction in rate [16]. This has been used to incorporate a single ribose nucleotide by using prCp. The 3′,5′-rCDP (prCp) can contain an α-32P to incorporate a 3′-end label. The incorporated 3′,5′-rCDP (i.e., DNA-prCp) at the 3′ end of the DNA can then be treated with a phosphoesterase (e.g., alkaline phosphatase) to yield vicinal hydroxyls on the ribose for chemical modification with NaIO4. Although these potential chemistries have not yet been explored, we will do so in future work.
4. Conclusions
Coupling DNA by way of a 3′ ribose nucleotide occurs rapidly and under mild conditions. Coupling to hydrazide-agarose, even without using borohydride reduction, provides a stable linkage. Affinity chromatography with resins prepared by this method yields high activity and purity.
Supplementary Material
Highlights.
By incorporating a 3′ ribose nucleotide, DNA can be coupled to solid supports in less than 3 h.
The reaction and coupling occurs under mild conditions, 0.1 M potassium phosphate, pH 6.8.
Prepared columns are stable for at least 50 days.
GFP-C/EBP, a transcription factor model, was purified by DNA affinity chromatography.
Acknowledgments
We thank Ms. Linda Nagore for reading and editing the text. This work was supported by the National Institutes of Health (R01GM043609).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goss TA, Bard M, Jarrett HW. J Chromatogr A. 1990;508:279. doi: 10.1016/s0021-9673(00)91270-6. [DOI] [PubMed] [Google Scholar]
- 2.Goss TA, Bard M, Jarrett HW. J Chromatogr A. 1991;588:157. doi: 10.1016/0021-9673(91)85017-a. [DOI] [PubMed] [Google Scholar]
- 3.Gadgil H, Jarrett HW. J Chromatogr A. 2002;966:99. doi: 10.1016/s0021-9673(02)00738-0. [DOI] [PubMed] [Google Scholar]
- 4.Jiang D, Moxley RA, Jarrett HW. J Chromatogr A. 2006;1133:83. doi: 10.1016/j.chroma.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Raddatz S, Mueller-Ibeler J, Kluge J, Wäss L, Burdinski G, Havens JR, Onofrey TJ, Wang D, Schweitzer M. Nucleic Acids Res. 2002;30:4793. doi: 10.1093/nar/gkf594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panda M, Jiang D, Jarrett HW. J Chromatogr A. 2008;1202:75. doi: 10.1016/j.chroma.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang S, Galindo MR, Jarrett HW. Proteomics. 2010;10:203. doi: 10.1002/pmic.200800693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Mol Cell Biol. 2003;23:6049. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman WL, O’Shannessy DJ. J Immunol Methods. 1988;112:113. doi: 10.1016/0022-1759(88)90041-5. [DOI] [PubMed] [Google Scholar]
- 10.HE, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1988. p. 539. [Google Scholar]
- 11.Nakane PK, Kawaoi A. J Histochem Cytochem. 1974;22:1084. doi: 10.1177/22.12.1084. [DOI] [PubMed] [Google Scholar]
- 12.Robberson DL, Davidson N. Biochemistry. 1972;11:533. doi: 10.1021/bi00754a008. [DOI] [PubMed] [Google Scholar]
- 13.Jarrett HW, Taylor WL. J Chromatogr A. 1998;803:131. doi: 10.1016/s0021-9673(97)01257-0. [DOI] [PubMed] [Google Scholar]
- 14.Laemmli UK. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 15.Cordes EH, Jencks WP. J Am Chem Soc. 1962;84:826. [Google Scholar]
- 16.Uhlenbeck OC, Gumport RI. The Enzymes. 1982;15:31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.