

Abstract
Inflamed synovium of rheumatoid arthritis (RA) has been associated with a T helper (Th)1 cytokine profile but the blood situation remains to be clarified. We studied the differential IFN-γ producing activity of peripheral blood mononuclear cells (PBMCs) from RA patients (RA-PBMCs) and from healthy controls (H-PBMCs) in response to IL-12 and IL-18. RA-PBMCs had a decreased IFN-γ production in response to IL-12 and IL-18 when compared with H-PBMCs. RA-PBMCs activated with phytohemagglutinin and phorbol 12-myristate 13-acetate showed an increased sensitivity to IL-12 and IL-18, but still the RA-PBMC response was lower. IL-18 increased IL-12-stimulated IFN-γ production from RA synovium cells obtained after collagenase digestion more effectively than that of RA- or H-PBMCs. A specific inhibitor of IL-18 bioactivity, IL-18-binding protein (IL-18BP), down-regulated IL-12-induced IFN-γ production by RA- or H-PBMCs and had a remarkable effect on RA synovium cells. In conclusion, RA disease combines a polarized immune response with an active Th1 in inflamed joints and a reduced Th1 pattern in peripheral circulation.
Keywords: interferon-γ, interleukin-12, interleukin-18, interleukin-18-binding protein, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic disorder of unknown aetiology primarily affecting joints and leading to their progressive destruction. The chronically inflamed synovium of RA is characterized by a massive infiltration of lymphocytes and macrophages [1] and by an extensive proliferation of fibroblast-like synoviocytes [2]. CD4+ CD45+memory T cells are the major cellular component and show signs of activation [3], but their exact role in the pathogenesis of RA remains controversial [3,4]. In particular, the important biologic mediators produced by activated T cells, such as IL-2, IL-4, and IFN-γ, have been detected only at low levels in RA joints [5-8], in contrast to the abundance of cytokines from macrophages and synoviocytes, such as IL-1, TNF-α, and IL-6 [3]. However, some studies of T-cell cytokine patterns in the RA joint at the mRNA level and others using T-cell clones indicate the predominance of IFN-γ-producing T helper (Th)1 cells [9-11]. By intracellular cytokine staining of peripheral blood and synovial tissue T cells from RA patients, we have confirmed the selective accumulation of Th1 and Th0 cells in the synovium [12]. In addition, IL-12, which plays a critical role in the differentiation of IFN-γ-producing Th1 cells, is produced predominantly by macrophages localized adjacent to lymphatic aggregates. IL-12 can potently and selectively stimulate IFN-γ production by RA synovial tissue, mainly by acting on synovial T cells [13].
IL-18, initially described as an IFN-γ-inducing factor, is a novel cytokine of the IL-1 family [14]. IL-18 stimulates the synthesis of IFN-γ in T cells and natural killer (NK) cells, leading to the development of Th1-type immune responses. In addition, IL-18 also activates the proliferation of activated T cells and their production of IL-2 and granulocyte/macrophage-colony-stimulating factor, and the cytotoxic activity of NK cells through up-regulation of Fas ligand [15]. Early studies suggested that the effects of IL-18 on Th1 differentiation were independent of IL-12. However, later studies showed that exogenous IL-18 in the absence of IL-12 failed to drive the differentiation of naive T cells to Th1 cells [16] and that IL-18 was a potent inducer of IFN-γ from established Th1 cells only in combination with IL-12 [15,17-19].
Accordingly, IL-18 may be involved in various immune-mediated inflammatory conditions. Significant levels of its expression have been demonstrated in the synovium of patients with RA [20,21]. It was suggested that IL-18 in synergy with IL-12 and IL-15 could be involved in both Th1 immune responses and macrophage production of inflammatory cytokines such as TNF-α [20]. To control some of the potentially deleterious properties of IL-18, IL-18-binding protein (IL-18BP) has been identified as a specific inhibitor of its bioactivity. IL-18BP, though it lacks significant homology with IL-18 receptor components, can bind to IL-18 protein with high affinity, thereby acting as a soluble decoy receptor [22,23].
In the present study, we looked at the differential effects of IL-18 alone or in combination with IL-12 on RA cells from blood versus synovium. Our results show that the PBMCs of RA patients produced less IFN-γ in response to IL-12 and IL-18 than those of healthy volunteers. In addition, differences were observed between blood and synovium RA cells. Finally, differential regulatory effects of IL-18BP on these cells were also observed.
Materials and methods
Cytokines and reagents
Recombinant human IL-12 was purchased from R&D Systems (Abingdon, UK). Recombinant human IL-18 was from MBL (Nagoya, Japan). IL-18BP, kindly provided by Dr John Sims, Immunex/Amgen (Seattle, WA, USA), was produced in COS cells as a fusion protein combining IL-18BP and the CH2 and CH3 domains of human IgG1 and purified by protein A affinity. RPMI (Roswell Park Memorial Institute) 1640 culture media was purchased from Invitrogen SARL (Cergy Pontoise, France) and supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (Invitrogen). Phytohemagglutinin (PHA) and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich SARL (St Quentin Fallavier, France).
Preparation of synovium and cell cultures
Peripheral blood samples were obtained from 14 patients (2 men and 12 women) with RA who fulfilled the 1987 revised criteria of the American College of Rheumatology [24], and 12 healthy volunteers (2 men and 10 women). The mean ages ± SEM of RA patients and healthy controls were 52.9 ± 4.8 and 49.3 ± 1.9 years, respectively. The mean disease duration was 12.4 years (range 1–53 years). The majority of patients were being treated with nonsteroidal anti-inflammatory drugs, prednisone (n = 7; range 2–20 mg/day), methotrexate (n = 11) alone or combined with cyclosporin (n = 1) or anti-TNF treatment (n = 1), and cyclophosphamide (n = 2). PBMCs were isolated from heparinized blood by Ficoll density-gradient centrifugation, washed twice with phosphate-buffered serum, and re-suspended in RPMI 1640 medium. Synovium samples were obtained from patients with RA who were undergoing wrist, elbow, or hip synovectomy, or joint replacement.
For the isolation of synovium cells, samples were minced with scissors and digested for 1 hour with collagenase (Sigma, St Louis, MO, USA) and DNase (Invitrogen) in RPMI 1640 medium at 37°C. After removing tissue debris through a cell strainer, the resulting cell suspensions were washed twice with medium. The resultant single-cell suspensions and PBMC suspensions were dispensed into the wells of 96-well plates (Nunc, Roskilde, Denmark) at a density of 1 × 106 cells/ml in 200 μl of RPMI medium. Cultures were at 37°C in 5% CO2/95% humidified air.
Determination of IFN-γ levels by ELISA
IFN-γ levels were measured by quantitative sandwich ELISA, using a commercially available ELISA kit (DuoSet ELISA Development System human IFN-γ, R&D Systems). The detection limit of the assay was 20 pg/ml. IFN-γ values below this limit were regarded as 0.
Statistical analysis
Results were expressed as mean ± SEM of the indicated number of experiments. The statistical significance of differences between two groups was determined by the Mann–Whitney U test. The Wilcoxon signed-rank test was used to analyse matched pairs.
Results
Effect of IL-12 and/or IL-18 on IFN-γ production by normal PBMCs and its modulation by IL-18BP
Peripheral blood mononuclear cells from healthy controls (H-PBMCs) were cultured for 7 days for IFN-γ production with or without IL-12 and IL-18 alone or in combination. As shown in Fig. 1a, IL-18 alone up to 50 ng/ml had only a negligible effect on IFN-γ production. In contrast, IL-12 alone (1 ng/ml) induced a high IFN-γ production from peripheral blood mononuclear cells (PBMCs). However, there was no clear difference between 1 ng/ml vs 5 ng/ml of IL-12 on IFN-γ production (data not shown). IL-12-stimulated IFN-γ production was increased by IL-18 dose dependently in a synergistic fashion (Fig. 1b).
Figure 1.
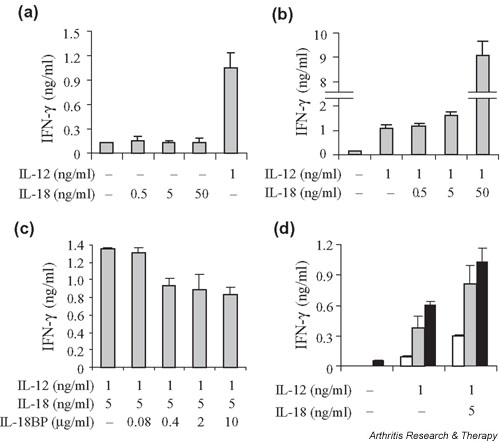
Synergistic effect of IL-12 and IL-18 on IFN-γ production by peripheral blood mononuclear cells (PBMCs) from healthy donors and its modulation by IL-18-binding protein (IL-18BP). PBMCs (1 × 106/ml in RPMI 1640 medium with 10% fetal calf serum) were cultured in triplicate for IFN-γ production with or without IL-12, IL-18, the combination of IL-12 and IL-18, or the combination of IL-12, IL-18, and IL-18BP. (a) PBMCs were cultured for 7 days with 1 ng/ml of IL-12, and various concentrations of IL-18. (b) PBMCs were cultured for 7 days with the combination of IL-12 and IL-18. (c) PBMCs were cultured for 7 days with 1 ng/ml of IL-12 and 5 ng/ml of IL-18, with or without various concentrations of IL-18BP. (d) PBMCs were cultured for 3 days (white bars), 5 days (gray bars), or 7 days (black bars) with or without IL-12 (1 ng/ml) or IL-12 (1 ng/ml) + IL-18 (5 ng/ml). Bars show mean values ± SEM of triplicate cultures.
To study the regulation of IFN-γ production by IL-18BP, IL-18 and IL-18BP were pre-incubated together for 30 minutes at 37°C before addition to the culture. IL-18BP could successfully neutralize IFN-γ production from PBMCs stimulated by IL-12 and IL-18. IFN-γ production induced by IL-12 (1 ng/ml) and IL-18 (5 ng/ml) was reduced by IL-18BP in a dose-dependent manner (Fig. 1c). PBMCs were cultured for 3, 5, and 7 days with or without IL-12 (1 ng/ml), or IL-12 (1 ng/ml) + IL-18 (5 ng/ml). IFN-γ production by spontaneous PBMC culture could not be detected at day 3 or 5 (Fig. 1d). Accordingly, concentrations of 1 ng/ml of IL-12, 5 ng/ml of IL-18, and 2 μg/ml of IL-18BP for 7 days of culture were selected for the following experiments, when these results were extended to more individuals.
Unstimulated H-PBMCs (n = 12) were cultured for 7 days with or without IL-12 and IL-18 alone and in combination with or without IL-18BP, and IFN-γ levels were compared by ELISA. IL-12 induced significant levels of IFN-γ production from H-PBMCs compared with medium alone (P < 0.001). As shown in Fig. 2a (white bars), IL-12-stimulated IFN-γ production from H-PBMCs was significantly augmented by IL-18 (P < 0.01) but not by IL-18BP.
Figure 2.
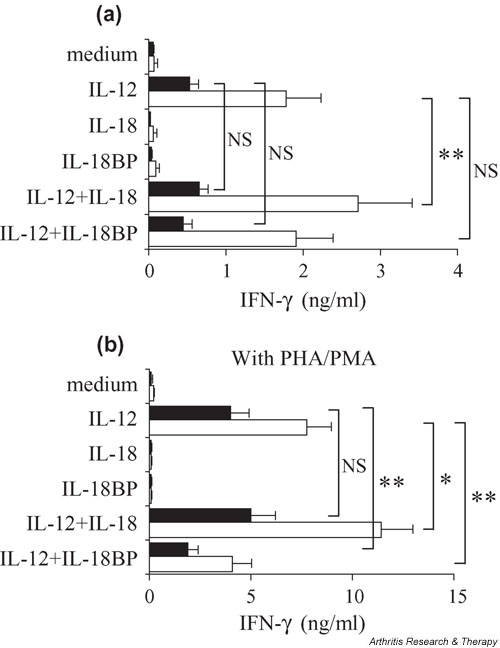
Differences in response to IL-12 or IL-18 between peripheral blood mononuclear cells (PBMCs) from healthy donors (H-PBMCs; n = 15) and from patients with RA (RA-PBMCs; n = 14). (a) H- or RA-PBMCs (1 × 106/ml in RPMI 1640 medium with 10% fetal calf serum) were cultured in triplicate for 7 days with or without 1 ng/ml of IL-12, 5 ng/ml of IL-18, 2 μg/ml of IL-18 binding protein (IL-18BP), or their combination. (b) Cells were activated with 200 ng/ml of phytohemagglutinin (PHA) and 2 ng/ml of phorbol myristate acetate (PMA). IFN-γ concentrations in culture supernatants were measured by ELISA. Black bars represent RA-PBMCs and white bars represent H-PBMCs. *P < 0.05, **P < 0.01. NS, statistically not significant.
Effect of IL-12 and IL-18 on unstimulated PBMCs from RA patients
PBMCs from patients with RA (RA-PBMCs; n = 14) were tested in the same experiments (Fig. 2a, black bars). In culture with medium alone (0.07 ± 0.05 vs 0.05 ± 0.02 ng/ml), H-PBMCs produced very low levels of IFN-γ similar to those produced by RA-PBMCs. IL-12-induced IFN-γ production was reduced in RA-PBMCs, being less than half that of H-PBMCs (P < 0.01). Exogenous IL-18 slightly augmented the IL-12-induced IFN-γ production from RA-PBMCs, but the effect was smaller than that seen in H-PBMCs (RA-PBMCs, +22.6% vs H-PBMCs, +52.3%). In cultures with medium alone, RA-PBMCs produced lower levels of IFN-γ than did H-PBMCs, and this was not corrected by stimulation with IL-12 or IL-18. Although the difference in the percentage increase was not significant, RA-PBMCs produced less IFN-γ than H-PBMCs in response to IL-12 alone or combined with IL-18 when results were expressed as concentrations.
Effect of IL-12 and IL-18 on PHA/PMA-stimulated PBMCs
Because IFN-γ production is associated with T-cell activation, and to further sensitize the cells to the action of IL-12 and IL-18, H- or RA-PBMCs were cultured with the same conditions as above, but in the presence of 200 ng/ml of PHA and 2 ng/ml of PMA. At these suboptimal concentrations, additional PHA and PMA alone had only a small effect on IFN-γ production by PBMCs (Fig. 2b). Despite this stimulation, IL-18 by itself did not induce either H- or RA-PBMCs to produce IFN-γ. In contrast, activated H- or RA-PBMCs (Fig. 2b) produced greater amounts of IFN-γ than resting ones (Fig. 2a) in response to IL-12. The synergistic effect of IL-12 and IL-18 on IFN-γ production by H- or RA-PBMCs was also augmented by PHA and PMA treatment. These findings indicate that activated cells are more sensitive to IL-12 and IL-18 than resting ones. However, even with the stimulation of PHA and PMA, the IFN-γ production by RA-PBMCs in response to IL-12 and IL-18 was lower than that of H-PBMCs (Fig. 2b).
Regulation of IFN-γ production from H- or RA-PBMCs by IL-18BP
While exogenous IL-18 showed a synergistic effect with IL-12 on IFN-γ production, levels of IL-12-stimulated IFN-γ production from resting PBMCs were not influenced by IL-18BP treatment (Fig. 2a). On the other hand, IL-12-stimulated IFN-γ production was decreased by IL-18BP in PBMCs activated by PHA/PMA (reduction with IL-18BP was 47.2% in H-PBMCs [P < 0.01] and 53.0% in RA-PBMCs [P < 0.01]). These observations indicate that the stimulation of PHA/PMA induces PBMCs to produce significant amounts of IL-18. Exogenous IL-18 increased IL-12-induced IFN-γ production by 47% for activated H-PBMCs versus 25% for activated RA-PBMCs. This difference indicated that RA-PBMCs had a defective response to IL-18 even when these cells were activated by PHA/PMA.
Aging of RA PBMCs and sensitivity to Th1-inducing cytokine
Since immune defects have been associated with age, we classified the patients into two groups according to their age and compared the IFN-γ production by their PBMCs in response to IL-12 and IL-18. The mean age, disease duration, and C-reactive-protein levels at the date of the blood sampling are shown in Table 1. The values of C-reactive protein were almost identical in the two groups. The older RA group showed a greater decrease in response to IL-12 and IL-18 than the younger RA group, indicating that this defect was disease related but increased with age, although without reaching significance.
Table 1.
IFN-γ production by PBMCs from patients with rheumatoid arthritis in response to IL-12 and IL-18
| Variable | Younger patients | Older patients |
| No. of patients | 7 | 7 |
| Mean age (years) | 40.1 ± 5.0 | 65.7 ± 4.6 |
| Disease duration (years) | 5.5 ± 2.3 | 17.7 ± 6.4 |
| C-reactive protein (mg/l) | 13.4 ± 2.0 | 13.9 ± 5.0 |
| IFN-γ production (ng/ml) | ||
| In medium alone | 0.05 ± 0.03 | 0.05 ± 0.03 |
| With IL-12 | 0.56 ± 0.14 | 0.50 ± 0.17 |
| With IL-12 + IL-18 | 0.83 ± 0.17 | 0.48 ± 0.14 |
| With IL-12 + IL-18BP | 0.53 ± 0.19 | 0.36 ± 0.15 |
Values are means ± SEM. IL-18BP, IL-18-binding protein; PBMCs, peripheral blood mononuclear cells.
IFN-γ production by RA synovium cells in response to IL-12 and IL-18
T cells from RA synovium are characterized by a reduced production of IFN-γ, contrasting with the large production of IFN-γ by T-cell clones isolated from RA synovium. To further explore this contrast, total RA synovial cells (n = 7), obtained after collagenase digestion, were exposed to IL-12 and IL-18 using the same conditions as were used with the PBMCs (Fig. 3a). IL-12 alone but not IL-18 induced total RA synovium cells to produce significant amounts of IFN-γ. However, IL-18 increased IL-12-stimulated IFN-γ production from total RA synovium cells more effectively (+78.4%, P < 0.05) than those of H- or RA-PBMCs. In cells activated with PHA and PMA, IL-18 increased by 71.4% the IL-12-stimulated IFN-γ production from total RA synovium cells, and IL-18BP decreased it by 38.5% (Fig. 3b). Thus, total RA synovium cells showed a greater response to IL-18 than blood cells. Stimulation with PHA and PMA further augmented the magnitude of IFN-γ production, but the percentage increase of IFN-γ production by exogenous IL-18 was not changed by PHA/PMA. Accordingly, total RA synovium cells were considered to respond better to exogenous IL-18 than the blood cells did. IL-18BP decreased IL-12-stimulated IFN-γ production from total RA synovium cells cultured with or without PHA/PMA (-38.5% and -46.2%, respectively).
Figure 3.
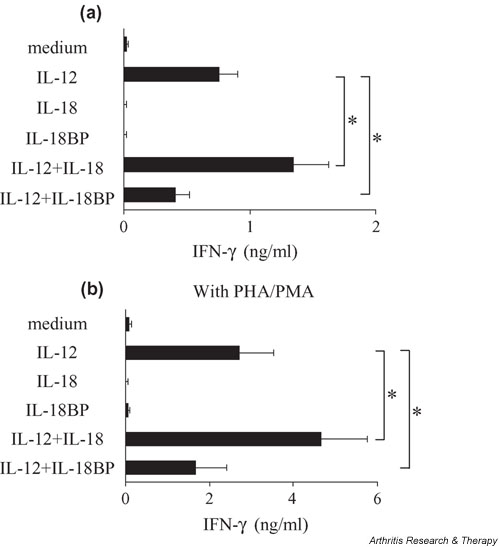
Effect of IL-12 and IL-18 on rheumatoid arthritis synovium cells. (a) Rheumatoid arthritis synovium cells (1 × 106/ml in RPMI 1640 medium with 10% fetal calf serum) were cultured in triplicate for 7 days with or without 1 ng/ml of IL-12, 5 ng/ml of IL-18, 2 μg/ml of IL-18 binding protein (IL-18BP), or a combination of these. (b) Cells were activated with 200 ng/ml of phytohemagglutinin (PHA) and 2 ng/ml of phorbol 12-myristate 13-acetate (PMA). IFN-γ concentrations in culture supernatants were measured by ELISA. *P < 0.05.
RA synovium is known to produce significant amounts of IL-18 spontaneously [21]. Addition of IL-18BP could decrease the IL-12-induced IFN-γ production by these cells by neutralizing the endogenously produced IL-18, indicating the importance of endogenous IL-18 in IFN-γ production by RA synovium.
Discussion
Several lines of evidence have indicated that IFN-γ-producing Th1 cells predominate at the site of chronic inflammation in RA. IL-12 is considered to play a critical role in inducing Th1-cell-mediated organ-specific autoimmune diseases, as shown in several animal models [25-28]. In addition, we have already demonstrated the presence of IL-12 as a contributory factor in inducing the IFN-γ-dominant T-cell cytokine response in joints of patients with longstanding RA [13]. IL-18 is a proinflammatory cytokine that plays an important role in the Th1-type immune response through the induction of IFN-γ synthesis in T cells and NK cells, T-cell proliferation, and cytokine production [15]. Significant levels of expression of IL-18 have been previously demonstrated in the synovium of RA patients [20,21], and the major effect of IL-18 is to increase the IFN-γ-dominant T-cell response induced by IL-12 [21].
T cells and NK cells are the major source of IFN-γ in PBMC cultures, and IL-12 and IL-18 augment IFN-γ production by these cells. Defects in IFN-γ production may result from changes in cell number and/or in cell response. Although differing conclusions have been reached regarding the possible changes in numbers of CD4+ T cells, NK cells, and NK T cells between RA and healthy PBMCs [29], the precise subpopulation with reduced ability to produce IFN-γ remains to be clarified [29-31]. In order to assess overall differences in systemic response to the Th1-inducing cytokines, IL-12 and IL-18, we compared the IFN-γ production by PBMCs from RA patients to that by cells from healthy controls. The IFN-γ production of RA-PBMCs in response to IL-12 and IL-18 was lower than that of H-PBMCs, even with the activation of PHA and PMA. This was still observed when the younger RA patients were compared with age-matched controls. Furthermore, PBMCs from the older RA patients demonstrated a greater decrease in response to IL-12 and IL-18 than that of the younger patients. Indeed, a reduction of the systemic Th1 functions with aging in normal individuals has been reported [32]. The same tendency was observed for the response to Th1-inducing cytokines in RA. These results indicate that RA-PBMCs are defective in their response to Th1-inducing cytokines, with an additional effect related to age. When looking at a possible effect of treatment on these defects, we found no difference between patients treated or not treated with prednisone (data not shown). The finding was similar for patients receiving methotrexate. Preliminary results appear to indicate that PBMCs from RA patients taking methotrexate produced greater amounts of IFN-γ after successful anti-TNF treatment. This result, if it proves to be correct, would further indicate that the defective IFN-γ production by RA-PBMCs might be related to disease activity.
Next, we compared the response to TH1-inducing cytokines of blood versus synovium cells from RA patients. Judging from the effect on IFN-γ production, total RA synovium cells showed a greater response to exogenous IL-18 in the presence of IL-12 than resting PBMCs. The pattern of response to IL-12 and IL-18 of total RA synovium cells was similar to that of activated H-PBMCs, suggesting that these cells had been activated and were more sensitive to TH1-inducing cytokines.
IL-18BP has been identified as a specific inhibitor of IL-18 [22,23]. There is no significant similarity between IL-18BP and IL-18 receptor components. Since it lacks a transmembrane domain, IL-18BP appears to exist only as a soluble, circulating protein. Its major function is to regulate the inflammatory activity of IL-18 by acting as its soluble decoy receptor [22,23]. Our results showed that exogenous IL-18 alone did not induce total RA synovium cells to produce detectable levels of IFN-γ, but the neutralization of endogenous IL-18 by IL-18BP reduced by about 50% the IL-12-induced IFN-γ production by these cells, suggesting a role for endogenously produced IL-18 in IFN-γ production. The effect of IL-18BP was much higher in RA synovium cells than in PBMCs, probably because RA synovium produced more endogenous IL-18 and responsed more strongly to IL-18. Although an IL-18-neutralizing activity was found in RA synovial fluid samples, IL-18 bioactivity was still detectable [21]. These findings indicate that endogenous IL-18 is an important contributory factor to IL-12-induced IFN-γ production in RA synovium.
In a previous study, we used flow cytometry to examine the ability of CD4+ T cells of blood and synovium samples from RA patients to produce IFN-γ and/or IL-4. Total RA synovium cells showed a higher Th1 and a lower Th2 frequency than peripheral blood cells [12]. In addition, Haddad and colleagues reported that activated whole blood cells from RA patients produced higher levels of IL-4 and lower IFN-γ than did cells from healthy controls [33]. Accordingly, the IL-4 : IFN-γ ratio, which reflects the Th2 : Th1 cytokine balance in blood, was higher in RA patients. These findings suggest that in RA the Th1 : Th2 ratio in the blood and that in the synovium are different.
Accumulation of Th2 vs Th1 cells in blood may result from changes in T-cell migration. Indeed, IFN-γ producing T cells were significantly increased in the peripheral blood of RA patients shortly after anti-TNF-α treatment, resulting in a shift of the Th1 : Th2 ratio in favor of Th1 in peripheral blood [34]. Adhesion molecules, such as P- and E-selectin or VCAM-1 (vascular cell adhesion molecule-1), are considered to be important for the selective homing of Th1 cells and not Th2 cells [35]. Expression of E-selectin and VCAM-1 was significantly reduced by anti-TNF therapy [36]. Thus, anti-TNF treatment may suppress the selective migration of Th1 cells into RA synovium through the rapid down-regulation of adhesion molecules. It remains to be clarified whether this is associated with an increased migration of Th2 cells with anti-inflammatory properties.
Recently, cases of severe tuberculosis have been reported in patients with Crohn's disease and RA receiving anti-TNF treatment [37]. Tuberculosis was the most common serious opportunistic infection reported in those patients. Similar observations were made in an HIV population where a secondary cell-mediated immune defect is present, as demonstrated by a defect in IFN-γ production. It is noteworthy that tuberculosis developed in RA patients shortly after the beginning of anti-TNF treatment. Our results showed a decreased response of RA peripheral blood to IL-12 and IL-18, leading to a reduced production of IFN-γ compared with that of healthy controls. These findings could explain to some extent the occurrence of tuberculosis during anti-TNF treatment.
Conclusion
Our data demonstrate that compared with H-PBMCs, RA-PBMCs have a lower response to IL-12 and IL-18, which are both important in Th1 development via T-bet expression. These results are in contrast with the finding that synovium cells in RA show an increased response to IL-12 and IL-18. Thus, RA patients have a polarized immune response, Th1 being overexpressed in inflamed joints and Th1 defects in peripheral circulation.
Competing interests
None declared.
Abbreviations
ELISA = enzyme-linked immunosorbent assay; H-PBMCs = PBMCs from healthy controls; IFN = interferon; IL = interleukin; IL-18BP = IL-18-binding protein; MTX = methotrexate; NK = natural killer (cells); PBMCs = peripheral blood mononuclear cells; PHA = phytohemagglutinin; PMA = phorbol 12-myristate 13-acetate; RA = rheumatoid arthritis; RA-PBMCs = PBMCs from RA patients; Th = T helper (cells); VCAM-1 = vascular cell adhesion molecule-1.
References
- Janossy G, Panayi G, Duke O, Bofill M, Poulter LW, Goldstein G. Rheumatoid arthritis: a disease of T-lymphocyte/macrophage immunoregulation. Lancet. 1981;2:839–842. doi: 10.1016/S0140-6736(81)91107-7. [DOI] [PubMed] [Google Scholar]
- Qu Z, Garcia CH, O'Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining. Arthritis Rheum. 1994;37:212–220. doi: 10.1002/art.1780370210. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 1990;33:768–773. doi: 10.1002/art.1780330602. [DOI] [PubMed] [Google Scholar]
- Panayi GS, Lanchbury JS, Kingsley GH. The importance of the T cell in initiating and maintaining the chronic synovitis of rheumatoid arthritis. Arthritis Rheum. 1992;35:729–735. doi: 10.1002/art.1780350702. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Xu WD, Townsend K, Broide D, Alvaro-Gracia J, Glasebrook A, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. I. Failure to detect T cell lymphokines (interleukin 2 and interleukin 3) and presence of macrophage colony-stimulating factor (CSF-1) and a novel mast cell growth factor in rheumatoid synovitis. J Exp Med. 1988;168:1573–1586. doi: 10.1084/jem.168.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P, van den Berg W. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum. 1997;40:2105–2115. doi: 10.1002/art.1780401203. [DOI] [PubMed] [Google Scholar]
- Miossec P, Naviliat M, Dupuy d'Angeac A, Sany J, Banchereau J. Low levels of interleukin-4 and high levels of transforming growth factor beta in rheumatoid synovitis. Arthritis Rheum. 1990;33:1180–1187. doi: 10.1002/art.1780330819. [DOI] [PubMed] [Google Scholar]
- Chen E, Keystone EC, Fish EN. Restricted cytokine expression in rheumatoid arthritis. Arthritis Rheum. 1993;36:901–910. doi: 10.1002/art.1780360706. [DOI] [PubMed] [Google Scholar]
- Miltenburg AM, van Laar JM, de Kuiper R, Daha MR, Breedveld FC. T cells cloned from human rheumatoid synovial membrane functionally represent the Th1 subset. Scand J Immunol. 1992;35:603–610. doi: 10.1111/j.1365-3083.1992.tb03260.x. [DOI] [PubMed] [Google Scholar]
- Quayle AJ, Chomarat P, Miossec P, Kjeldsen-Kragh J, Forre O, Natvig JB. Rheumatoid inflammatory T-cell clones express mostly Th1 but also Th2 and mixed (Th0-like) cytokine patterns. Scand J Immunol. 1993;38:75–82. doi: 10.1111/j.1365-3083.1993.tb01696.x. [DOI] [PubMed] [Google Scholar]
- Simon AK, Seipelt E, Sieper J. Divergent T-cell cytokine patterns in inflammatory arthritis. Proc Natl Acad Sci USA. 1994;91:8562–8566. doi: 10.1073/pnas.91.18.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Yamamura M, Kawashima M, Harada S, Tsuji K, Shibuya K, Maruyama K, Makino H. Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:1669–1676. doi: 10.1002/1529-0131(199802)41:2<306::AID-ART15>3.3.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Morita Y, Yamamura M, Nishida K, Harada S, Okamoto H, Inoue H, Ohmoto Y, Modlin RL, Makino H. Expression of interleukin-12 in synovial tissue from patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:306–314. doi: 10.1002/1529-0131(199802)41:2<306::AID-ART15>3.3.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Okamura H, Wada M, Nagata K, Tamura T. Endotoxin-induced serum factor that stimulates gamma interferon production. Infect Immun. 1989;57:590–595. doi: 10.1128/iai.57.2.590-595.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-18. Methods. 1999;19:121–132. doi: 10.1006/meth.1999.0837. [DOI] [PubMed] [Google Scholar]
- Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, Hartley SB, Menon S, Kastelein R, Bazan F, O'Garra A. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7:571–581. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, Ikeda M, Kurimoto M. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158:1541–1550. [PubMed] [Google Scholar]
- Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, Tanimoto T, Torigoe K, Fujii M, Ikeda M, Fukuda S, Kurimoto M. Interferon-gamma-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-gamma production. Eur J Immunol. 1996;26:1647–1651. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- Ahn HJ, Maruo S, Tomura M, Mu J, Hamaoka T, Nakanishi K, Clark S, Kurimoto M, Okamura H, Fujiwara H. A mechanism underlying synergy between IL-12 and IFN-gamma-inducing factor in enhanced production of IFN-gamma. J Immunol. 1997;159:2125–2131. [PubMed] [Google Scholar]
- Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, Kennedy K, Carter R, Wei XQ, Xu D, Field M, Foulis A, Liew FY, McInnes IB. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104:1393–1401. doi: 10.1172/JCI7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura M, Kawashima M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, Morita Y, Ohmoto Y, Makino H. Interferon-gamma-inducing activity of interleukin-18 in the joint with rheumatoid arthritis. Arthritis Rheum. 2001;44:275–285. doi: 10.1002/1529-0131(200102)44:2<275::AID-ANR44>3.3.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Aizawa Y, Akita K, Taniai M, Torigoe K, Mori T, Nishida Y, Ushio S, Nukada Y, Tanimoto T, Ikegami H, Ikeda M, Kurimoto M. Cloning and expression of interleukin-18 binding protein. FEBS Lett. 1999;445:338–342. doi: 10.1016/S0014-5793(99)00148-9. [DOI] [PubMed] [Google Scholar]
- Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–136. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger T Jr, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilden RL, Hunder GG. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Germann T, Szeliga J, Hess H, Storkel S, Podlaski FJ, Gately MK, Schmitt E, Rude E. Administration of interleukin 12 in combination with type II collagen induces severe arthritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–4827. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med. 1995;181:381–386. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara Y, Shiozawa K, Takai M, Kyogoku M, Shiozawa S. Natural killer (NK) T cells are significantly decreased in the peripheral blood of patients with rheumatoid arthritis (RA) Clin Exp Immunol. 1999;118:131–136. doi: 10.1046/j.1365-2249.1999.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado A, Mueller YM, Thomas P, Bojczuk P, O'Connors C, Katsikis PD. Decreased effector memory CD45RA+ CD62L-CD8+ T cells and increased central memory CD45RA-CD62L+ CD8+ T cells in peripheral blood of rheumatoid arthritis patients. Arthritis Res Ther. 2003;5:R91–96. doi: 10.1186/ar619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezawa K, Yamamura M, Matsui H, Ota Z, Makino H. Comparative analysis of CD45RA- and CD45RO-positive CD4+T cells in peripheral blood, synovial fluid, and synovial tissue in patients with rheumatoid arthritis and osteoarthritis. Acta Med Okayama. 1997;51:25–31. doi: 10.18926/AMO/30810. [DOI] [PubMed] [Google Scholar]
- Rink L, Cakman I, Kirchner H. Altered cytokine production in the elderly. Mech Ageing Dev. 1998;102:199–209. doi: 10.1016/S0047-6374(97)00153-X. [DOI] [PubMed] [Google Scholar]
- Haddad A, Bienvenu J, Miossec P. Increased production of a Th2 cytokine profile by activated whole blood cells from rheumatoid arthritis patients. J Clin Immunol. 1998;18:399–403. doi: 10.1023/A:1023278606036. [DOI] [PubMed] [Google Scholar]
- Maurice MM, van der Graaff WL, Leow A, Breedveld FC, van Lier RA, Verweij CL. Treatment with monoclonal anti-tumor necrosis factor alpha antibody results in an accumulation of Th1 CD4+ T cells in the peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:2166–2173. doi: 10.1002/1529-0131(199910)42:10<2166::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Austrup F, Vestweber D, Borges E, Lohning M, Brauer R, Herz U, Renz H, Hallmann R, Scheffold A, Radbruch A, Hamann A. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, Meinders AE, Maini RN. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1077–1081. doi: 10.1002/art.1780390702. [DOI] [PubMed] [Google Scholar]
- Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]