

Abstract
Changes in the microbial community structure are observed in individuals with intestinal inflammatory disorders. These changes are often characterized by a depletion of obligate anaerobic bacteria, whereas the relative abundance of facultative anaerobic Enterobacteriaceae increases. The mechanisms by which the host response shapes the microbial community structure, however, remain unknown. We show that nitrate generated as a by-product of the inflammatory response conferred a growth advantage to the commensal bacterium Escherichia coli in the large intestine of mice. Mice deficient for inducible nitric oxide synthase (iNOS) did not support growth of E. coli by nitrate respiration, suggesting that nitrate generated during inflammation was host-derived. Thus the inflammatory host response selectively enhances growth of commensal Enterobacteriaceae by generating electron acceptors for anaerobic respiration.
Over 90% of the cells in the human body are microbes, the majority of which reside in the large intestine, where they provide benefit to the host by stimulating the development of the immune system, by supplying nutrients, and by providing niche protection. The lumen of the large bowel is thought to be primarily anaerobic, with traces of oxygen being consumed by facultative anaerobic bacteria (e.g. Enterobacteriaceae), which constitute a small fraction (approximately 0.1%) of the microbial community (microbiota) (1). The vast majority of microbes in the large intestine belong to the phyla Bacteroidetes (class Bacteroidia) and Firmicutes (class Clostridia), two groups of obligate anaerobic bacteria that lack the ability to respire and instead rely on fermentation of amino acids and complex polysaccharides for growth. On the phylum level, this bacterial community structure is conserved between humans and mice (1, 2). Conditions of inflammation in the large bowel are accompanied by a microbial imbalance (dysbiosis), however, which is characterized by a marked decrease in the representation of obligate anaerobic bacteria and an increased relative abundance of facultative anaerobic bacteria belonging to the family Enterobacteriaceae (3-12) (Fig. S1A and B).
An important component of the host inflammatory response is the generation of reactive nitrogen species (RNS) and reactive oxygen species (ROS) (Fig. S1C). For example, iNOS is expressed at high levels during intestinal inflammation and elevated nitric oxide (NO.) concentrations are detected in colonic luminal gas of individuals with inflammatory bowel disease (13-15). Reaction of nitric oxide radicals (NO.) with superoxide radicals (O2.-) yields peroxynitrite (ONOO-), which can either generate nitrate (NO3-) (16) or oxidize organic sulfides and tertiary amines to S-oxides and N-oxides (17, 18). Similarly, inflammation-derived ROS can generate S-oxides and N-oxides (17, 18). Unlike obligate anaerobic members of the gut microbiota, the facultative anaerobic Enterobacteriaceae can use nitrate, S-oxides, and N-oxides as terminal electron acceptors for anaerobic respiration. We thus hypothesized that colitis produces dysbiosis because highly oxidized by-products of intestinal inflammation (e.g. nitrate, S-oxides, and N-oxides) might enable commensal Enterobacteriaceae to edge out fermenting microbes in the gut lumen by using anaerobic respiration for energy production (Fig. S1C).
E. coli, a prototypic member of the Enterobacteriaceae, possesses three nitrate reductases, two S-oxide reductases, and three N-oxide reductases encoded by the narGHJI, narZYWV, napFDAGHBC, dmsABC, ynfDEFGH, torCAD, torYZ, and yedYZ operons, respectively (19). One common feature shared by these terminal reductases is the incorporation of an essential molybdenum cofactor into the active site. To test the idea that anaerobic respiration provides a growth benefit in the inflamed intestine, we generated mutants deficient for the biosynthesis of the molybdenum cofactor (moaA mutant) in the Escherichia coli strains HS and Nissle 1917 (EcN) (Fig. S2A and S2B). These moaA mutants were anaerobically co-cultured with the respective wild-type strains in mucin broth in the presence or absence of nitrate, DMSO (Dimethyl S-oxide), or TMAO (Trimethylamine N-oxide) (Fig. 1A and S2C). Enrichment for the E. coli wild-type strains occurred in the presence of nitrate, DMSO and TMAO, suggesting that anaerobic respiration can provide a growth benefit during the anaerobic growth conditions encountered in the intestinal mucus layer.
Figure 1. Anaerobic respiration enhances luminal growth of E. coli during DSS-induced colitis.
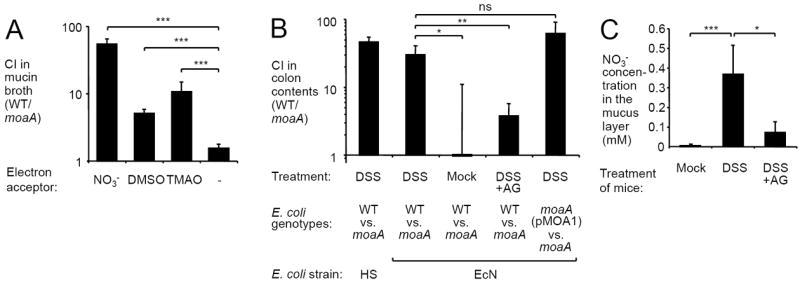
(A) Competitive index (CI) of the EcN wild type (WT) and the moaA mutant after anaerobic growth in mucin broth supplemented with 40 mM of the indicated electron acceptors (N = 3). (B) Mock-treated mice (Mock), DSS-treated mice (DSS) or mice treated with DSS and AG (DSS+AG) were inoculated with the indicated mixtures of E. coli strains and the CI in colon contents determined 5 days after inoculation. A plasmid (pMOA1) carrying the cloned moaA gene was used to complement the moaA mutant (moaA). N is given in Fig. S3C. (C) Concentration of nitrate (NO3-) determined in the cecal mucus layer of mock-treated mice (N = 4), DSS-treated mice (DSS, N = 3) or mice treated with DSS and AG (DSS+AG, N = 4). Bars represent geometric means ± standard error. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significant (Student’s t-test).
We next inoculated untreated mice (C57BL/6) or mice with chemically-induced colitis (dextran sulfate sodium [DSS]-treatment) intragastrically with an equal mixture of EcN and its isogenic moaA mutant (Fig. S1D). Both the wild-type strain and the moaA mutant colonized the intestine of mock-treated mice poorly, but similar numbers of each strain were recovered from colon contents five days after inoculation (Fig. 1B). This result suggested that in the absence of intestinal inflammation, anaerobic respiration did not provide a growth benefit for E. coli. DSS treatment induced inflammation in the colon and increased mRNA levels of pro-inflammatory markers in wild-type mice (Fig. S3). In contrast to mock-treated mice, the EcN wild-type strain was recovered from colon contents of DSS-treated mice in significantly higher numbers than the moaA mutant five days after inoculation. Similar results were observed when DSS-treated mice were inoculated with an equal mixture of the human commensal E. coli strain HS and an isogenic moaA mutant. Expression of moaA from a low copy-number plasmid (pMOA1) in the EcN moaA mutant fully restored the phenotype to wild-type levels. Outgrowth of the EcN wild-type strain over the moaA mutant was also observed in the DSS colitis model when mice were precolonized with E. coli (Fig. S4). These findings supported the idea that anaerobic respiration provided a growth advantage upon commensal E. coli during intestinal inflammation.
While ROS can be generated by several NADPH oxidases, the sole source of NO during inflammation is iNOS. To determine the contribution of RNS to the growth advantage mediated by anaerobic respiration, DSS-treated mice were treated with the iNOS-inhibitor aminoguanidine hydrochloride (AG) and inoculated with a mixture of the EcN wild type and the moaA mutant. Consistent with the idea that RNS are a significant source for the production of terminal electron acceptors during inflammation, the growth advantage of the EcN wild type over the moaA mutant in the DSS-colitis model was significantly (P < 0.01) blunted after AG treatment (Fig. 1B). The nitrate/nitrate redox couple has a greater redox potential than the DMSO/DMS or the TMAO/TMA redox couples, which makes nitrate the preferred respiratory electron acceptor for growth of E. coli under anaerobic conditions (19). Therefore, we next determined if nitrate becomes available in the lumen of the inflamed intestine. To accomplish this objective, the concentration of nitrate was determined in the cecal mucus layer of mock-treated mice or mice with DSS-induced colitis (Fig. 1C). Whereas nitrate levels were at the limit of detection in mock-treated control mice, a significant (P < 0.001) increase in nitrate levels was observed in DSS-treated animals. AG treatment of mice with DSS-induced colitis significantly (P < 0.05) dampened nitrate production, thus supporting the hypothesis that nitrate is generated in the intestinal lumen as part of the host inflammatory response.
We next tested whether nitrate respiration bestows a growth advantage upon E. coli wild-type isolates. To this end, we inactivated the narG, napA, and narZ genes, which encode nitrate reductases, in the probiotic EcN. In contrast to the wild-type strain, the nitrate respiration-deficient narG napA narZ triple mutant lacked nitrate reductase activity and was outcompeted by the wild-type strain during competitive anaerobic growth in mucin broth in the presence of nitrate (Fig. S5). To determine whether nitrate respiration provides a colonization advantage in the intestine, mock-treated and DSS-treated wild-type (C57BL/6) mice were inoculated intragastrically with an equal mixture of the EcN wild type and a narG napA narZ triple mutant (Fig. 2). In the absence of inflammation (mock treatment, Fig. 2A, S6 and S7), both the EcN wild type and the narG napA narZ triple mutant were recovered in similar numbers from colonic (Fig. 2B) and cecal contents (Fig. S8A). In contrast, the EcN wild-type strain was enriched over the narG napA narZ triple mutant when colitis was induced by administration of DSS. Similar results were obtained using an adherent invasive E. coli (AIEC) isolate (LF82) that was isolated from an inflammatory bowel disease patient (Fig. 2C, 2D, S5C, and S5D). To test whether nitrate respiration provides a growth benefit in the absence of iNOS-dependent nitrate production, the competitive colonization experiment was repeated in DSS-treated, iNOS-deficient mice (i.e. mice carrying a mutation in the Nos2 gene) and DSS-treated wild-type mice (C57BL/6) that received AG. The severity of the colitis induced by the DSS treatment was similar among all treatment groups (Fig. 2A and S7) five days after inoculation with E. coli. Remarkably, the EcN wild type and its nitrate respiration-deficient mutant were recovered in equal numbers from DSS-treated iNOS-deficient mice or from DSS+AG-treated wild-type mice. Similar results were obtained using varying concentrations of DSS (Fig. S9) as well as with E. coli K-12 (Fig. S10). Concomitant expression of narG from a low-copy number plasmid (pNARG1) and restoration of the napA mutation to its wild-type allele (napA[restored]) in the narG narZ napA mutant reestablished fitness in the inflamed gut to similar levels observed with the wild-type strain (Fig. 2B and S8A). Collectively, these data suggested that reduction of host-derived nitrate by E. coli confers a growth advantage during gut inflammation.
Figure 2. Wild-type E. coli outcompetes a nitrate respiration-deficient mutant during colitis.
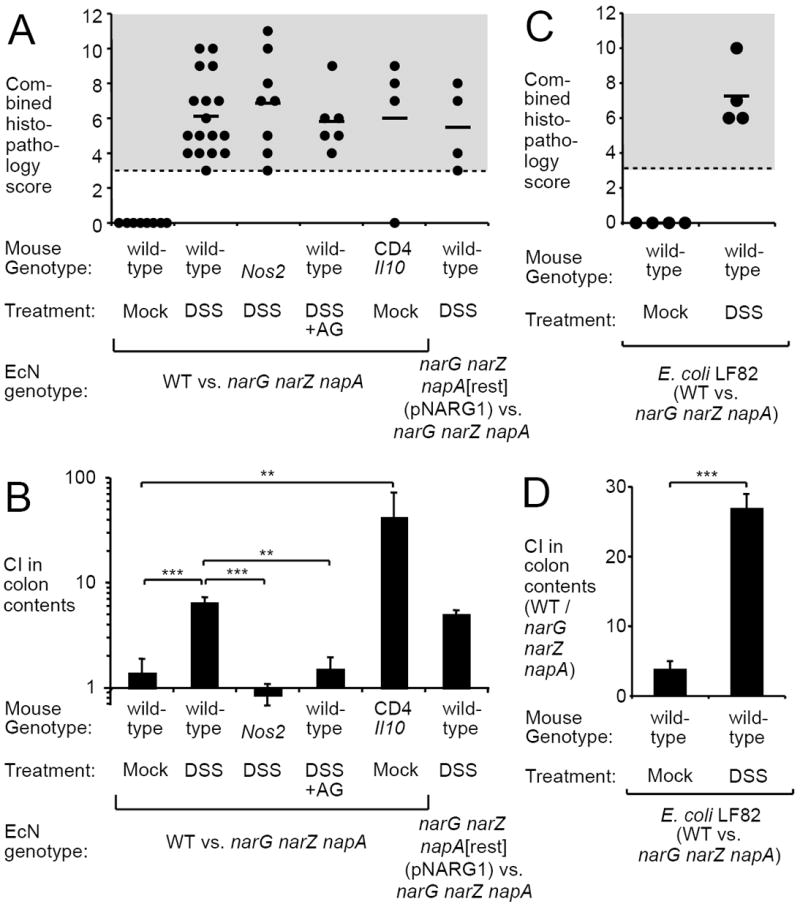
Mock-treated (Mock), DSS-treated (DSS) or DSS+AG-treated (DSS+AG) wild-type mice, Nos2-deficient mice (Nos2) or mice harboring T cells deficient for the production of IL-10 (Cd4 Il10 mice) were inoculated with the indicated mixtures of E. coli strains. WT, E. coli wild type; narG narZ napA, E. coli nitrate respiration-deficient mutant. The narG narZ napA mutant was complemented by introducing a functional chromosomal napA allele and a plasmid (pNARG1) carrying the cloned narG gene (narG narZ napA[rest] [pNARG1]). Pathological changes in the colon (A and C) and the competitive index (CI) recovered from colon contents (B and D) were determined 5 days after inoculation. (A and C) Combined histopathology score in the colon. Each dot represents data from an individual animal. Experiments were performed with EcN (A-B) or E. coli LF82 (C-D). (B and D) Bars represent geometric means ± standard error. **, P < 0.01; ***, P < 0.001 (Student’s t-test). N is given in panels A and C.
To validate our findings in a second murine model of colitis, we generated mice that harbored T cells deficient for the production of the anti-inflammatory cytokine IL-10 (Cd4 Il10 mice [Il10flox/flox Cd4-cre]), a mouse strain that developed spontaneous colitis (Fig. 2A and S7) (20). After the onset of intestinal inflammation, mice were inoculated intragastrically with an equal mixture of the EcN wild type and the narG napA narZ triple mutant (Fig. 2B). The nitrate respiration-proficient wild-type strain outcompeted the narG napA narZ mutant in the colon contents of Cd4 Il10 mice 5 days after inoculation (P < 0.05). To investigate whether growth of E. coli by nitrate respiration can also be observed in an unrelated animal model of intestinal inflammation, bovine ligated ileal loops were inoculated with thapsigargin, a proinflammatory compound, or mock-treated (vehicle control) (Fig. 3A, B and C). At 8 hours after inoculation of loops with a mixture of EcN and the narG napA narZ mutant, significantly (P < 0.05) higher numbers of wild-type EcN were recovered from luminal fluid and mucus (Fig. 3D).
Figure 3. Nitrate respiration enhances luminal growth of EcN during inflammation.
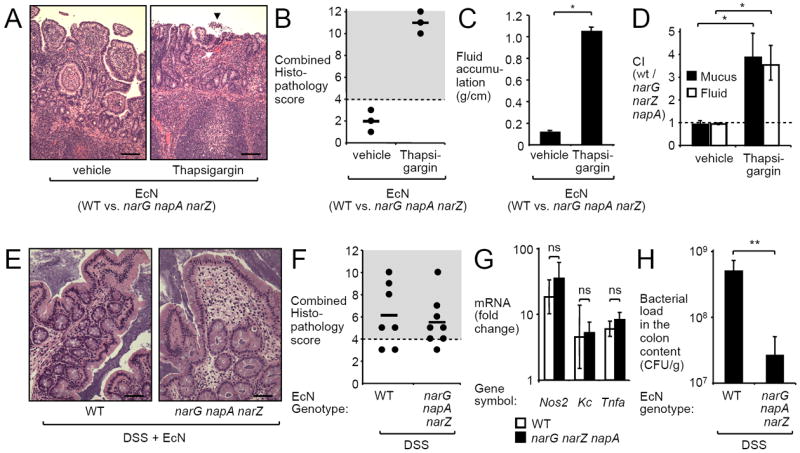
(A-D) Bovine ligated ileal loops treated with thapsigargin or mock-treated (vehicle) were inoculated with a mixture of EcN (WT) and a nitrate respiration-deficient mutant (narG napA narZ). Samples were collected 8 hours after inoculation. (A) Representative H&E stained ileal sections. Scale bar, 200 μm. (B) Combined histopathology score in the ileum. Each dot represents data from an individual animal. (C) Fluid accumulation in ligated ileal loops. (D) Competitive indices (CI) recovered from the luminal fluid (open bars) or mucus scrapings (closed bars). (E-H) DSS-treated mice were inoculated either with EcN (WT) or with the narG napA narZ mutant. Inflammation in the colon (E, F and G) and bacterial numbers recovered from colon contents (E) were determined 5 days after inouclation. (E) Representative H&E stained colonic sections. Scale bar, 100 μm. (F) Combined histopathology score in the colon. (G) Expression of Nos2, Kc and Tnfa in colonic RNA samples using qRT-PCR (fold-increases over mock-treated, mock-treated mice). (H) Bacterial numbers (CFU) recovered from colon contents. (C, D, G, H) Bars represent geometric means ± standard error. *, P < 0.05; **, P < 0.01; ns, not statistically significant (Student’s t-test). N is given in panels B and F.
To determine whether nitrate respiration increases bacterial recovery from the inflamed intestine when mice are inoculated with a single E. coli strain, DSS-treated mice were inoculated either with the EcN wild-type strain or with the narG napA narZ mutant. Mice inoculated with EcN or the narG napA narZ mutant exhibited a similar severity of colonic inflammation (Fig. 3E, F, and G). Importantly, the EcN wild-type strain was recovered in significantly (P < 0.01) higher numbers from colon contents than the nitrate respiration-deficient mutant (Fig. 3H and S8B). Collectively, these data suggested that nitrate respiration conferred a marked growth advantage upon commensal E. coli in the lumen of the inflamed gut.
The picture emerging from this study is that nitrate generated as a by-product of the host inflammatory response can be utilized by E. coli, and likely by other commensal Enterobacteriaceae, to edge out competing microbes that rely on fermentation to generate energy for growth. Obligate anaerobic microbes in the intestine compete for nutrients that are available for fermentation, but cannot utilize non-fermentable nutrients (e.g. fermentation end products.). The ability to degrade non-fermentable substrates likely enables E. coli to sidestep this competition, which explains the fitness advantage conferred by nitrate respiration in the inflamed gut. Through this mechanism, inflammation contributes to a bloom of nitrate-respiration proficient Enterobacteriaceae, providing a plausible explanation for the dysbiosis associated with intestinal inflammation (3-12). This general principle might also influence the dynamics of host-associated bacterial communities outside the large bowel, as nitrate respiration confers a fitness advantage in the oxygen-poor and nitrate-rich environment of the cystic fibrosis airway (21).
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. W. Müller for providing Il10flox/flox Cd4-cre mice and E. Romao for technical assistance. The data reported in the manuscript are tabulated in the main paper and in the supplementary materials. This work was supported by Public Health Service Grants AI076246 and AI088122. P.T. was supported by a scholarship from the Faculty of Medicine, Chiang Mai University, Thailand.
References and notes
- 1.Eckburg PB, et al. Diversity of the human intestinal microbial flora. Science. 2005 Jun 10;308:1635. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley RE, et al. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005 Aug 2;102:11070. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krook A, Lindstrom B, Kjellander J, Jarnerot G, Bodin L. Relation between concentrations of metronidazole and Bacteroides spp in faeces of patients with Crohn’s disease and healthy individuals. Journal of clinical pathology. 1981 Jun;34:645. doi: 10.1136/jcp.34.6.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giaffer MH, Holdsworth CD, Duerden BI. The assessment of faecal flora in patients with inflammatory bowel disease by a simplified bacteriological technique. Journal of medical microbiology. 1991 Oct;35:238. doi: 10.1099/00222615-35-4-238. [DOI] [PubMed] [Google Scholar]
- 5.Seksik P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003 Feb;52:237. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gophna U, Sommerfeld K, Gophna S, Doolittle WF, Veldhuyzen van Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. Journal of clinical microbiology. 2006 Nov;44:4136. doi: 10.1128/JCM.01004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank DN, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007 Aug 21;104:13780. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heimesaat MM, et al. Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via Toll-like receptors 2 and 4. PloS one. 2007;2:e662. doi: 10.1371/journal.pone.0000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007 Aug 16;2:119. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Stecher B, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007 Oct;5:2177. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barman M, et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008 Mar;76:907. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell host & microbe. 2010 Sep 16;8:292. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundberg JO, Hellstrom PM, Lundberg JM, Alving K. Greatly increased luminal nitric oxide in ulcerative colitis. Lancet. 1994 Dec 17;344:1673. doi: 10.1016/s0140-6736(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 14.Singer II, et al. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996 Oct;111:871. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- 15.Enocksson A, Lundberg J, Weitzberg E, Norrby-Teglund A, Svenungsson B. Rectal nitric oxide gas and stool cytokine levels during the course of infectious gastroenteritis. Clinical and diagnostic laboratory immunology. 2004 Mar;11:250. doi: 10.1128/CDLI.11.2.250-254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007 Aug;6:662. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 17.Schoneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochim Biophys Acta. 2005 Jan 17;1703:111. doi: 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Balagam B, Richardson DE. The mechanism of carbon dioxide catalysis in the hydrogen peroxide N-oxidation of amines. Inorg Chem. 2008 Feb 4;47:1173. doi: 10.1021/ic701402h. [DOI] [PubMed] [Google Scholar]
- 19.Gennis RB, Stewart V. In: Escherichia coli and Salmonella. Cellular and molecular biology. Neidhardt FC, et al., editors. Vol. 1. ASM Press; Washington, D.C.: 1996. pp. 217–261. [Google Scholar]
- 20.Pils MC, et al. Commensal gut flora reduces susceptibility to experimentally induced colitis via T-cell-derived interleukin-10. Inflamm Bowel Dis. 2011 Oct;17:2038. doi: 10.1002/ibd.21587. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman LR, et al. Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog. 2010 Jan;6:e1000712. doi: 10.1371/journal.ppat.1000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- 23.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000 Jun 6;97:6640. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis RW, B D, R JR. Advanced bacterial genetics. Cold Spring Harbor Laboratory Press; New York: 1980. [Google Scholar]
- 25.Price-Carter M, Tingey J, Bobik TA, Roth JR. The alternative electron acceptor tetrathionate supports B12-dependent anaerobic growth of Salmonella enterica serovar typhimurium on ethanolamine or 1,2-propanediol. J Bacteriol. 2001 Apr;183:2463. doi: 10.1128/JB.183.8.2463-2475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart V, Parales J., Jr Identification and expression of genes narL and narX of the nar (nitrate reductase) locus in Escherichia coli K-12. J Bacteriol. 1988 Apr;170:1589. doi: 10.1128/jb.170.4.1589-1597.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doane TA, Horwath WR. Spectrophotometric Determination of Nitrate with a Single Reagent. Analytical Letters. 2003;36:2713. [Google Scholar]
- 28.Roers A, et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004 Nov 15;200:1289. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santos RL, et al. Salmonella-induced cell death is not required for enteritis in calves. Infect Immun. 2001;69:4610. doi: 10.1128/IAI.69.7.4610-4617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winter SE, et al. Contribution of flagellin pattern recognition to intestinal inflammation during Salmonella enterica serotype Typhimurium infection. Infect Immun. 2009 Feb 23;77:1904. doi: 10.1128/IAI.01341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010 Sep 23;467:426. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart V, Lu Y, Darwin AJ. Periplasmic nitrate reductase (NapABC enzyme) supports anaerobic respiration by Escherichia coli K-12. J Bacteriol. 2002 Mar;184:1314. doi: 10.1128/JB.184.5.1314-1323.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grozdanov L, et al. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J Bacteriol. 2004 Aug;186:5432. doi: 10.1128/JB.186.16.5432-5441.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun. 1999 Sep;67:4499. doi: 10.1128/iai.67.9.4499-4509.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine MM, Rennels MB. E. coli colonisation factor antigen in diarrhoea. Lancet. 1978 Sep 2;2:534. doi: 10.1016/s0140-6736(78)92268-7. [DOI] [PubMed] [Google Scholar]
- 36.Pal D, Venkova-Canova T, Srivastava P, Chattoraj DK. Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol. 2005 Nov;187:7167. doi: 10.1128/JB.187.21.7167-7175.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon R, Priefer U, Puhler A. A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nat Biotech. 1983 Nov;1:784. [Google Scholar]
- 38.Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991 Apr;100:195. [PubMed] [Google Scholar]
- 39.Kingsley RA, et al. Ferrioxamine-mediated Iron(III) utilization by Salmonella enterica. Appl Environ Microbiol. 1999 Apr;65:1610. doi: 10.1128/aem.65.4.1610-1618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.