

Abstract
Tumor antigen-specific CD4+ T cells that directly recognize cancer cells are important for orchestrating antitumor immune responses at the local tumor sites. However, the mechanisms of direct MHC class II (MHC-II) presentation of intracellular tumor antigen by cancer cells are poorly understood. We found that two functionally distinct subsets of CD4+ T cells were expanded after HLA-DPB1*04 (DP04)-binding NY-ESO-1157–170 peptide vaccination in ovarian cancer patients. While both subsets similarly recognized exogenous NY-ESO-1 protein pulsed on DP04+ target cells, only one type recognized target cells with intracellular expression of NY-ESO-1. The tumor-recognizing CD4+ T cells more efficiently recognized the short 8–9-mer peptides than the non-tumor-recognizing CD4+ T cells. In addition to endosomal/lysosomal proteases that are typically involved in MHC-II antigen presentation, several pathways in the MHC class I presentation pathways such as the proteasomal degradation and transporter-associated with antigen-processing (TAP)-mediated peptide transport were also involved in the presentation of intracellular NY-ESO-1 on MHC-II. The presentation was inhibited significantly by primaquine, a small molecule that inhibits endosomal recycling, consistent with findings that pharmacological inhibition of new protein synthesis enhances antigen presentation. Together, our data demonstrated that cancer cells selectively present peptides from intracellular tumor antigens on MHC-II by multiple non-classical antigen-processing pathways. Harnessing direct tumor-recognizing ability of CD4+ T cells could be a promising strategy to enhance antitumor immune responses in the immunosuppressive tumor microenvironment.
Keywords: CD4+ T cells, NY-ESO-1, epitope, endogenous MHC class II presentation, antigen-processing pathway
Introduction
Tumor antigen-specific CD4+ helper T cells play important roles in the induction and maintenance of antitumor immune responses. The roles of antigen-specific CD4+ T cells include provide help to CD8+ T cells during the primary and secondary immune responses, induce the activation/maturation of antigen-presenting cells (APCs), produce cytokines that are essential for differentiation or maintenance of long-lasting T cell responses, and activate B cells to produce tumor antigen-specific antibodies (1, 2). In addition, immune-potentiating cytokines from CD4+ T cells may help other immune cells to overcome the actions of immunosuppressive factors (3, 4). In the classical view of antigen presentation, intracellular and extracellular proteins are presented to CD8+ and CD4+ T cells via the MHC class I (MHC-I) and MHC class II (MHC-II) pathways, respectively (5, 6). Therefore, while tumor antigen-specific CD8+ T cells efficiently recognize intracellular antigen-expressing cancer cells, activation of tumor antigen-specific CD4+ T cells generally requires professional APCs that take up and cross-present tumor antigen proteins. Recently, accumulating evidence demonstrates that APCs at the tumor microenvironment are frequently immunosuppressive and lead to unresponsiveness of T cells (7, 8). The absence of functional APCs that cross-present tumor antigen protein to CD4+ T cells may limit CD4-help at the local tumor sites and could partly explain the rapid exhaustion of tumor antigen-specific CD8+ T cells. An alternative path by which tumor antigen-specific CD4+ T cells could overcome the requirement for APCs within the tumor microenvironment is direct recognition of tumors.
In contrast to murine cancer cells, many types of human cancers constitutively express MHC-II or are induced to express MHC-II in an IFN-γ-dependent manner (9). Tumor antigen-specific CD4+ T cells that directly recognize cancer cells have been described (10–13); however, the mechanism(s) relating to antigen specificity and antigen-processing that the CD4+ T cells use for direct tumor recognition are unknown. Although several distinct non-classical antigen presentation pathways have been identified for the presentation of intracellular proteins on MHC-II of professional APCs (14–17), it is not clear whether these pathways are functional in cancer cells for MHC-II presentation of intracellular tumor antigens. Recently, we have identified a novel non-classical antigen presentation pathway for the presentation of intracellular NY-ESO-1 to HLA-DRB1*01 (DR01)-restricted NY-ESO-1-specific CD4+ T cells by cancer cells (18). Two DR01-restricted CD4+ T cell lines that recognize the NY-ESO-187–98 and NY-ESO-195–106 peptides were studied for the recognition of exogenously pulsed and intracellularly expressed NY-ESO-1 proteins. While both CD4+ T cell lines similarly recognized recombinant NY-ESO-1 protein pulsed on APCs, only the NY-ESO-195–106-specific CD4+ T cells directly recognized NY-ESO-1-expressing DR01+ melanoma cell lines in a heat shock protein 90 (HSP90)-dependent manner. Such tumor-recognizing CD4+ T cell subset is likely selectively activated by the MHC-II-binding peptides that are naturally presented on cancer cells and could have significant potential as a strategy for cancer immunotherapy. In this regard, although DR01 is relatively frequent (8–21% of individuals in population groups in the US (19)), additional MHC-II-binding epitopes for other HLAs will be required for the development of immunotherapeutic strategies using tumor-recognizing CD4+ T cells. In addition, identification of the mechanisms of antigen-processing for endogenous MHC-II presentation is critical for the understanding of direct tumor recognition by CD4+ T cells.
In a previous clinical trial of NY-ESO-1 peptide vaccination (20), patients who were HLA-DPB1*04:01/*04:02 (DP04)+ and had NY-ESO-1-expressing ovarian cancer were repeatedly vaccinated with a DP04-binding peptide, NY-ESO-1157–170. We found, a subset of vaccine-induced CD4+ T cells that secreted cytokines when they were co-cultured with NY-ESO-1+DP04+ cancer cells (20). In the present study, we identified the unique peptide-specificity and antigen-processing pathways that allow direct recognition of cytoplasmic protein presented by MHC-II on cancer cells by the human CD4+ T cell subset. Because of the frequent expression of DP04 (present in 43–70% of Caucasians (21)), our observations will be useful for the development of novel immunotherapies that harness direct tumor-recognizing ability of CD4+ T cells.
METHODS
NY-ESO-1-specific T cells and cell lines
Peripheral blood mononuclear cells (PBMC) and tumor tissues were obtained from patients with epithelial ovarian cancer under an approved protocol from the institutional review board at Roswell Park Cancer Institute, Buffalo, NY. NY-ESO-1 expression in tumor tissues was determined by immunohistochemistry and/or semi-quantitative RT-PCR, and anti-NY-ESO-1 antibody response in serum was analyzed by ELISA, as described previously (22). NY-ESO-1-specific CD4+ and CD8+ T cells in PBMC were amplified by in vitro presensitization from patients who received NY-ESO-1 vaccination (20). NY-ESO-1157–170-specific CD4+ T cells in tumor-infiltrating lymphocytes (TIL) from four patients who were HLA-DP04+ and had spontaneous anti-NY-ESO-1 antibody response were also expanded by stimulation with γ-irradiated and peptide-pulsed CD4−CD8− cells derived from autologous PBMC. HLA-A*02:01 (A02)-restricted NY-ESO-1157–165-specific CD8+ T cells were isolated using a FACSAria instrument (BD Biosciences) with HLA-A02/NY-ESO-1157–165 tetramer. DP04-restricted NY-ESO-1157–170-specific CD4+ T cells were isolated by a FACSAria instrument by gating on IFN-γ+ cells (Miltenyi Biotec) or CD40L+ cells following peptide restimulation (23). For TIL, NY-ESO-1157–170-specific CD4+ T cell lines were established from three patients. Among them, NY-ESO-1-specific CD4+ T cell line from one patient contained TR-CD4. CD4+ T cells derived from PBMC were cloned by limiting dilution and periodic phytohemagglutinin (PHA, Remel) stimulations in the presence of feeder cells (irradiated allogeneic PBMC) and IL-2 (Roche Molecular Biochemicals).
Melanoma cell lines and EBV-transformed B cell lines were from our cell bank. Establishment and characterization of SK-MEL-37 clones-expressing ICP47 were described (18). Cells were cultured in RPMI1640 medium supplemented with 10% FCS, penicillin, streptomycin and L-glutamine.
Generation of monocyte-derived DCs
CD14+ monocytes were magnetically isolated from DP04+ healthy donor PBMCs using anti-CD14 microbeads (Miltenyi Biotech). Monocytes were cultured for 6 days in RPMI1640 medium supplemented with 10% FCS, penicillin, streptomycin and L-glutamine in the presence of 1,000 U/ml GM-CSF and 20 ng/ml IL-4 (CellGenix).
Pretreatment of target cells
Synthetic peptides were pulsed on target cells overnight at 10 μM unless otherwise specified. Recombinant NY-ESO-1 protein was expressed in E. coli and purified by a standard method. NY-ESO-1 protein was pulsed overnight on SK-MEL-29 at a concentration of 10 μg/ml or on DCs at different concentrations. Peptide or recombinant protein-pulsed and -unpulsed target cells were extensively washed before co-culture with T cells. To determine HLA-restriction of T cell recognition, target cells were treated with 10 μg/ml anti-HLA-ABC monoclonal antibody (W6/32; eBioscience), and/or 20 μl of anti-HLA-class II antibody supernatant for one hour before addition of T cells. Culture supernatants from anti-DP (B7/21), anti-DQ (SPV-L3), and anti-DR (L243) hybridomas were used as sources for anti-HLA-class II antibodies. In some experiments, target cells were pre-treated with 1,000 U/ml (50 ng/ml) IFN-γ (Peprotech) for 2 days. Treatment of SK-MEL-37 with inhibitors for the antigen-processing pathway was performed as described (18). All inhibitors were water-soluble except for Lactacystin and Epoxomicin that were dissolved in DMSO. After treatment, SK-MEL-37 was fixed with 1% paraformaldehyde, quenched with glycine and extensively washed in PBS and culture medium. For mRNA electroporation, EBV-transformed B cell line (1 × 106) was mixed with 5 μg in vitro-transcribed mRNA (Ambion) in 50 μl X-Vivo15 (Lonza) and were applied a pulse of 1.25 kV/cm for 700 μsec using the ECM 830 Electroporation system and cuvettes (Harvard apparatus-BTX). Cells were incubated overnight in a culture medium until T cell recognition assays. Electroporation of SK-MEL-37 with synthetic siRNA (Integrated DNA Technologies or Origene) was performed as described (18).
Intracellular cytokine staining
Brefeldin A (BFA) and monensin was added 2 hours after co-culturing T cells and target cells. After 6 hours of co-culture, cells were fixed with 2% paraformaldehyde and permeabilized using FIX & PERM reagents (Invitrogen-CALTAG) according to the manufacturer’s instruction (20). Cytokine production was assessed by intracellular staining measured by flow cytometry. Antibodies were obtained from BD Biosciences.
ELISPOT Assay
The number of IFN-γ-secreting antigen-specific T cells was assessed by ELISPOT assays as described (20). The dark-violet spots were counted by an automated ELISPOT reader (Zeiss or CTL).
Measurement of cytokines
CD4+ T cell (5 × 104) were cultured with SK-MEL-37 (3 × 104), protein-pulsed monocyte-derived DCs (2.5 × 104) or single cell suspension of ovarian tumor tissues (2.5 × 104) in a 96-well culture plate. The culture supernatant was collected 20–24 hours after the co-culture and stored at −20 °C until measurement of cytokines by ELISA according to manufacturer’s instruction. Unconjugated and biotin-labeled antibody pairs for human IFN-γ and GM-CSF were obtained from BD Biosciences, and HRP-labeled avidin D and TMB substrate solution were obtained from eBioscience.
Statistical analyses
Error bars in the graphical data represent means ± s.d. All in vitro experiments were performed at least in duplicate. P values of less than 0.05 were considered statistically significant by Student’s t-test. All statistical analyses were performed using Prism 5 software (GraphPad Software).
RESULTS
Characterization of tumor-recognition by CD4+ T cells
We established DP04-restricted NY-ESO-1157–170 peptide-specific CD4+ T cell clones from an ovarian cancer patient who was repeatedly vaccinated with DP04-binding NY-ESO-1157–170 peptide (20). For this patient, NY-ESO-1157–170 peptide-specific CD4+ T cells were not detected in pre-vaccine PBMCs, but were significantly expanded by the vaccinations (data not shown). One of 4 established NY-ESO-1-specific CD4+ T cell clones produced IFN-γ against a DP04+NY-ESO-1+ melanoma cell line (20). The other 3 clones showed no direct reactivity against the melanoma cell line. To gain insight into the different ability in direct tumor recognition by CD4+ T cells, we characterized target-recognition by representative tumor-recognizing CD4+ T cell clone (TR-CD4) and non-tumor-recognizing CD4+ T cell clone (NTR-CD4). Whereas TR-CD4 specifically recognized NY-ESO-1-expressing DP04+ melanoma cell lines, all DP04+ melanoma cell lines were recognized by both TR-CD4 and NTR-CD4 after pulsing with the NY-ESO-1157–170 vaccine peptide (Fig. 1A and 1B). In subsequent experiments, we selected the NY-ESO-1-expressing melanoma cell line, SK-MEL-37 (SK37) because (i) DR01+DP04+ SK37 was extensively characterized for the recognition by DR01-restricted NY-ESO-1-specific CD4+ T cells in our previous study (18), and (ii) SK37 was efficiently recognized by A02-restricted NY-ESO-1-specific CD8+ T cell clone (18). SK37 expressed both HLA class I and class II molecules and CD40 but no co-stimulatory molecules, CD80 and CD86 (Supplementary Fig. 1). Recognition of SK37 by TR-CD4 was specifically inhibited by anti-HLA-DP blocking antibody, demonstrating DP04-restricted target cell recognition (Fig. 1C). As expected, recognition of SK37 by control NY-ESO-1-specific A02-restricted CD8+ T cells was inhibited by anti-HLA class I blocking mAb. Electroporation of NY-ESO-1-specific siRNA, which efficiently silenced NY-ESO-1 expression in SK37, significantly reduced the recognition by TR-CD4 (Fig. 1D), supporting the NY-ESO-1-specificity in the direct tumor recognition by TR-CD4.
Figure 1.

Characterization of NY-ESO-1-specific tumor-recognizing (TR-CD4) and non-tumor-recognizing (NTR-CD4) CD4+ T cell clones. (A) Recognition of DP04+NY-ESO-1+/− melanoma lines was tested by ELISPOT assays. (B) DP04+ melanoma cells were pulsed overnight with NY-ESO-1157–170 peptide and recognition by T cells was evaluated by intracellular IFN-γ staining. (C) HLA-restriction of SK37-recognition was determined using blocking antibodies by intracellular IFN-γ staining. A02-restricted NY-ESO-1157–165-specific CD8+ T cell clone (ESO-CD8) was used as control tumor-recognizing T cells. (D) Antigen-specificity of tumor recognition. NY-ESO-1 (ESO), pan-MAGE (MAGE) or GFP-specific siRNA was electroporated into SK37. Recognition was evaluated by IFN-γ-ELISPOT assays. (E) Recognition of NY-ESO-1157–170 (Peptide), NY-ESO-1 protein (Protein) and adenovirally-induced NY-ESO-1 (Adeno) in SK29 was tested by IFN-γ ELISPOT assays. (F) Recognition of mRNA-induced intracellular NY-ESO-1. HLA-DP04+ EBV-transformed B cells were electroporated with ESO or GFP mRNA. Reactivity of TR-CD4 was measured by IFN-γ-ELISPOT assays. Statistical significance was calculated by Student’s t-test and is shown as *:P ≤ 0.05; **:P ≤ 0.01; and *** P ≤ 0.001.
We next asked whether the differential ability to recognize NY-ESO-1-expressing cancer cells by TR-CD4 and NTR-CD4 indicates differences in the recognition of intracellular NY-ESO-1 protein in these cells. NY-ESO-1-non-expressing DP04+ melanoma cell line, SK-MEL-29 (SK29), was either infected with adenovirus carrying the NY-ESO-1 gene for intracellular NY-ESO-1 protein expression, or pulsed with recombinant NY-ESO-1 protein or synthetic NY-ESO-1157–170 peptide as a positive control, and the recognition by TR-CD4 and NTR-CD4 was compared. Whereas exogenous NY-ESO-1 protein was efficiently recognized by both TR-CD4 and NTR-CD4, only TR-CD4 recognized intracellular NY-ESO-1 ectopically expressed by adenovirus in SK29 (Fig. 1E). As expected from the classical antigen-processing pathways, CD8+ T cells efficiently recognized intracellularly expressed NY-ESO-1 but not exogenous NY-ESO-1 protein. It is possible that the presentation of adenovirally expressed intracellular NY-ESO-1 may differ from that of the physiologically expressed NY-ESO-1. As an alternative method to induce intracellular NY-ESO-1, EBV-transformed DP04+ B cells were electroporated with in vitro transcribed NY-ESO-1 mRNA. As shown in Fig. 1F, TR-CD4 efficiently recognized target cells electroporated with NY-ESO-1 mRNA, indicating that TR-CD4 can recognize exogenous and endogenous intracellular NY-ESO-1 antigen presented on MHC-II.
Determination of minimal epitopes
We investigated the mechanism for the differential recognition of intracellular NY-ESO-1 by TR-CD4 and NTR-CD4 in terms of peptide-recognition by TCR. The titration curves for TR-CD4 and NTR-CD4 to recognize the vaccine peptide, NY-ESO-1157–170, were similar (Fig. 2A). In addition, two long 20-mer peptides, NY-ESO-1151–170 and NY-ESO-1161–180, were similarly recognized by TR-CD4 and NTR-CD4 (Fig. 2B and 2C). Recognition of naturally processed exogenous NY-ESO-1 protein by TR-CD4 and NTR-CD4 was tested. TR-CD4 more efficiently recognized NY-ESO-1 protein-pulsed monocyte-derived DCs than NTR-CD4 (Fig. 2D), which potentially explains the more efficient tumor recognition by TR-CD4. To investigate the mechanism(s) by which TR-CD4 recognize cancer cells, short overlapping vaccine peptides were tested for recognition by TR-CD4 and NTR-CD4 (Fig. 2E). In contrast to the similar recognition of 10-mer peptide NY-ESO-1160–169 by TR-CD4 and NTR-CD4, recognition of 9-mer peptide NY-ESO-1161–169 by NTR-CD4 was significantly reduced, while it was fully recognized by TR-CD4 (Fig. 2E). In addition, the recognition of 8-mer peptide NY-ESO-1161–168 was more efficient by TR-CD4 than by NTR-CD4 (Fig. 2E). There is a significant difference in the titration curves for the recognition of NY-ESO-1161–169 peptide by TR-CD4 and NTR-CD4 (Fig. 2F). The recognition of NY-ESO-1161–169 by NTR-CD4 was barely detectable at 10 nM, while the recognition by TR-CD4 was still detectable at 0.01 nM concentration. Taken together, these results indicate that naturally processed intracellular and extracellular NY-ESO-1 proteins are loaded on DP04 in a manner which is preferentially recognized by TR-CD4 than NTR-CD4.
Figure 2.

Determination of minimum epitopes for TR-CD4 and NTR-CD4. (A-C) Dose-dependence of NY-ESO-1 peptide recognition by TR-CD4 and NTR-CD4. Indicated peptides were pulsed overnight on SK29 at indicated concentrations. Recognition by TR-CD4 and NTR-CD4 was evaluated by IFN-γ ELISPOT assays. (D) Dose-dependence of NY-ESO-1 protein recognition. NY-ESO-1 protein was pulsed on DP04+ immature monocyte-derived DCs overnight at indicated concentrations. TR-CD4 and NTR-CD4 were stimulated with these DCs for 24 hours. IFN-γ production in the supernatant was measured by ELISA. (E) Recognition of overlapping NY-ESO-1 peptide-pulsed SK29 was evaluated by intracellular IFN-γ-staining. (F) Dose-dependence of NY-ESO-1161–169 recognition. NY-ESO-1161–169 was pulsed on SK29 at indicated concentrations. Recognition was evaluated by intracellular IFN-γ staining. All experiments were repeated at least twice with consistent results. Error bars indicate s.d. from duplicated wells.
We searched for the presence of TR-CD4 in DP04+ ovarian cancer patients who did not receive peptide vaccination but had spontaneous NY-ESO-1-specific serum antibody. As we found previously that ovarian tumors are highly enriched with NY-ESO-1-specific CD8+ T cells (24), ovarian tumor-infiltrating lymphocytes (TIL) were assessed to increase the possibility of detecting NY-ESO-1-specific T cells. We tested TILs from 4 patients and found NY-ESO-1-specific CD4+ T cells that recognize both NY-ESO-1161–169 and SK37 in one TIL-derived NY-ESO-1-specific CD4+ T cell line (Fig. 3A), indicating that TR-CD4 is not induced only by synthetic peptide vaccinations but also spontaneously by the NY-ESO-1 protein expressed in cancer cells and infiltrates in the tumor sites. Next, we tested whether freshly isolated ovarian cancer cells from patients can stimulate TR-CD4 ex-vivo when they express NY-ESO-1 and DP04. Single cell suspensions of 5 NY-ESO-1-expressing and 5 NY-ESO-1-negative tumor specimens from DP04+ patients were tested for recognition by TR-CD4 and NTR-CD4. Notably, TR-CD4 but not NTR-CD4 specifically produced high amounts of IFN-γ when co-cultured with 2 of 5 NY-ESO-1+ ex vivo ovarian cancer cells (Fig. 3B). Together, the infiltration of TR-CD4 at the ovarian tumor site, and the antigen-specific direct stimulation of TR-CD4 by ex vivo ovarian cancer cells, indicate that TR-CD4 plays a role in antitumor immunity in the tumor microenvironment.
Figure 3.

Detection of TR-CD4 at the local tumor site. (A) Tumor-infiltrating lymphocytes from an ovarian cancer patient were stimulated with NY-ESO-1157–170. After 20-days, NY-ESO-1157–170-reactive CD4+ T cells were isolated and expanded. Recognition of SK37, NY-ESO-1157–170, and NY-ESO-1161–169 was tested by intracellular staining. Values in quadrants indicate percentages of cells. (B) TR-CD4 or NTR-CD4 cells were co-cultured with NY-ESO-1+/− tumor single cell suspensions (TSC) obtained from DP04+ patients for 24 hours. IFN-γ level in the supernatant was measured by ELISA.
Antigen-processing pathway of endogenous MHC-II presentation
To elucidate the antigen-processing pathway for the NY-ESO-1/DP04 epitope in SK37, we treated SK37 with inhibitors of antigen-processing at doses that did not inhibit MHC-II exogenous peptide presentation by SK37 (18). As shown in Fig. 4A, treatment of SK37 by a proteasome inhibitor, epoxomicin, substantially inhibited the recognition by TR-CD4. Inhibitory effect by another proteasome inhibitor, lactacystin, was negligible, indicating that the generation of the peptide recognized by TR-CD4 is dependent on the chymotrypsin-like activity of the proteasome (25). Treatment of melanoma cell lines with IFN-γ did not inhibit the recognition by TR-CD4 (Supplementary Fig. S2), indicating both standard- and immuno-proteasomes similarly processed the epitope. In addition, two endosomal/lysosomal protease inhibitors, chloroquine and leupeptin, significantly inhibited the presentation, suggesting the role of the proteases in antigen-processing and/or loading on MHC-II in endosomes/lysosomes (Fig. 4B). As the generation of MHC-II-binding peptide is dependent on proteasome, which is generally involved in the generation of MHC-I-binding peptides, we investigated the involvement of other enzymes that were reported to be involved in the generation of MHC-I-binding peptides. As shown in Fig. 5A, AAF-CMK that inhibits tripeptidyl peptidase II (TPPII) (26, 27) partially inhibited the presentation at the high dose, while aminopeptidase inhibitor (bestatin) (28–30) and metalloprotease inhibitor (1,10-phenanthroline) (31) had no effect. TPPII is a cytosolic peptidase that trims peptides generated by the proteasome. The involvement of TPPII in addition to the cytosolic proteasome in the endogenous MHC-II presentation supports the cytosolic degradation of intracellular NY-ESO-1. In addition, SK37 clones-expressing viral ICP47 which inhibits transporter-associated with antigen-processing (TAP)-mediated peptide transport showed reduced stimulatory activity compared to parental SK37, indicating a role of TAP-mediated peptide transport into the endoplasmic reticulum during the presentation (Fig. 5B).
Figure 4.
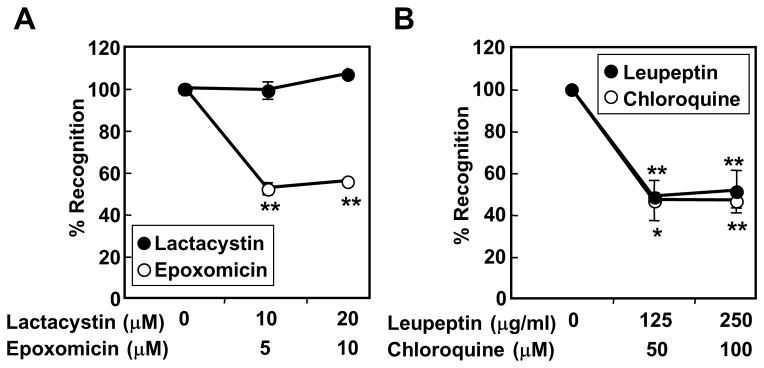
Effect of inhibitors for antigen degradation on the recognition of SK37 by TR-CD4. (A, B) SK37 was cultured for 16–20 hours with or without indicated proteasome inhibitors (A) or protease inhibitors (B) followed by fixing with paraformaldehyde and extensive washes. SK37 was co-cultured for 20 hours with TR-CD4. IFN-γ level in the supernatant was measured by ELISA. Results are shown as % inhibition of IFN-γ production compared to untreated target cells. Statistical significance was calculated by Student’s t-test and is shown as *:P ≤ 0.05 and **:P ≤ 0.01.
Figure 5.

Effect of inhibitors for MHC-I antigen-processing pathways on the recognition of SK37 by TR-CD4. (A) SK37 was treated by the indicated inhibitors for 16–20 hours followed by fixing with paraformaldehyde and extensive washes. SK37 was co-cultured for 20 hours with TR-CD4. IFN-γ level in the supernatant was measured by ELISA. (B) Role of TAP in the presentation. Untreated parental SK37 and SK37 clones stably expressing ICP47 gene were used as stimulator cells. GM-CSF level in the supernatant was measured by ELISA. Statistical significance was calculated by Student’s t-test and is shown as *:P ≤ 0.05; **:P ≤ 0.01; and *** P ≤ 0.001.
Next, we investigated the involvement of non-classical antigen presentation pathways for the presentation of intracellular protein to CD4+ T cells by professional APCs and B cells. We found that macroautophagy and chaperone-mediated autophagy were not involved in this presentation by treatment with macroautophagy inhibitor, 3-methyladenine, and by siRNA-mediated silencing of LAMP-2, respectively (Fig. 6A and 6B) (14, 16). Intracellular viral protein has been presented on recycled MHC-II, which is efficiently inhibited by primaquine (15, 32). As shown in Fig. 6C, treatment with primaquine significantly inhibited the presentation to TR-CD4, indicating the involvement of endosomal recycling, presumably the MHC-II molecules. No significant inhibition by cycloheximide and brefeldin A that inhibit new protein synthesis and vesicular trafficking, respectively, supporting the presentation by recycled but not newly synthesized MHC-II (Fig. 6D). Significant enhancement of presentation by treatment with cycloheximide suggests that inhibiting MHC-II synthesis enhanced cell surface expression of recycled MHC-II loaded with peptides from intracellular NY-ESO-1. Treatment with selective inhibitors (17-DMAG and radicicol) and siRNA-mediated silencing indicated that the presentation did not require chaperoning by heat shock protein (HSP)-90 (Supplementary Fig. S3A and S3B), that played a critical role in the presentation of NY-ESO-195–106 to HLA-DR01-restricted TR-CD4 by SK37 (18, 33, 34). In addition, treatment with PFT-μ and quercetin, that inhibit HSP70 chaperoning and stress-induced HSP expression (35, 36), respectively, enhanced the presentation, suggesting an inhibitory role of HSP70 in this presentation (Supplementary Fig. S3A).
Figure 6.

Effect of inhibitors for previously characterized endogenous MHC-II presentation pathways on the recognition of SK37 by TR-CD4. SK37 was treated by the indicated inhibitors for 40–44 hours (A, C) or 16–20 hours (D) followed by fixing with paraformaldehyde and extensive washes. SK37 was co-cultured for 20 hours with TR-CD4. GM-CSF level in the supernatant was measured by ELISA. (A) Effect of an inhibitor for macroautophagy. (B) Effect of siRNA-mediated silencing of LAMP2. SK37 was electroporated with indicated siRNA and cultured for 3 days. (C) Effect of an endosomal/lysosomal recycling inhibitor. (D) Effect of vesicular transport and protein synthesis inhibitors. All experiments were repeated at least three times with consistent results. Error bars indicate s.d. from duplicated wells. Statistical significance was calculated by Student’s t-test and is shown as *:P ≤ 0.05 and **:P ≤ 0.01.
DISCUSSION
In classical antigen-processing pathways, intracellular proteins are not efficiently loaded onto MHC-II for presentation to CD4+ T cells unless the protein is localized in or targeted to endosomal or lysosomal compartments (37–40). Because most immunogenic tumor antigens including NY-ESO-1 are intracellularly expressed, tumor antigen-specific CD4+ T cells generally are not considered to recognize MHC-II-expressing cancer cells efficiently. Nevertheless, many tumor antigen-specific CD4+ T cells have been reported to directly recognize cancer cells (10–13). However, in contrast to the detailed studies of endogenous MHC-II presentation pathways in experimental systems using model antigens such as viral antigens and ectopically expressed self-antigens and professional APCs (14–17), mechanisms of endogenous MHC-II presentation by cancer cells are poorly understood. It is important to characterize antigen specificity and antigen-processing pathways that are responsible for the direct recognition of cancer cells by CD4+ T cells.
In the present study, we provide several lines of evidence that indicate the requirements for antigen (NY-ESO-1)-specificity and MHC-II (DP04)-restriction for direct tumor recognition by TR-CD4. Although both TR-CD4 and NTR-CD4 similarly recognized the 14-mer NY-ESO-1157–170 vaccine peptide, TR-CD4 efficiently (103–104 fold) recognized 9-mer NY-ESO-1161–169 peptide. The differences in the minimal epitope for TR-CD4 and NTR-CD4 suggest that the unique peptide-TCR interaction is responsible for the tumor-recognizing ability by TR-CD4. Indeed, the sequences of the genes encoding the TCR α and β chains for TR-CD4 and NTR-CD4 are different ((20) and data not shown). A future approach to test whether the TCR is solely responsible for the ability of TR-CD4 to recognize tumor cells will be to retroviral transduce TCR genes from TR-CD4 and NTR-CD4 into polyclonal expanded T cells, and test for the recognition of intracellular NY-ESO-1.
We have characterized the antigen-processing mechanisms for the endogenous MHC-II presentation of intracellular NY-ESO-1 to DR01-restricted NY-ESO-195–106-specific CD4+ T cells by SK37 (18). The use of the same cancer cell line (SK37) as well as pharmacological inhibitors for antigen-processing enabled us to directly compare pathways for DR01- and DP04-restricted endogenous MHC-II presentation of intracellular NY-ESO-1. Both presentations were efficiently inhibited by epoxomicin, an inhibitor of proteasome. Interestingly, another proteasome inhibitor, lactacystin, only inhibited DR01-restricted presentation, potentially indicating that there are differences in the enzymatic activities required for the generation of the DR01- and DP04-binding epitopes (25). The cytosolic proteasome is generally involved in the generation of MHC-I-binding short peptides, which supports our finding that TR-CD4 cells efficiently recognize short 8–9-mer peptides. Both presentations were efficiently decreased by inhibitors for endosomal/lysosomal proteases, indicating that the peptides are loaded on MHC-II in the endosomal or lysosomal compartments.
Although there are multiple shared processing pathways between DR01- and DP04-restricted endogenous MHC-II presentation of intracellular NY-ESO-1, there are also distinct differences. DP04-restricted presentation required TAP-mediated peptide transport into endoplasmic reticulum and endosomal recycling; whereas DR01-restricted presentation required vesicular trafficking through trans-golgi network and chaperoning by HSP90. As HSP90 is known to facilitate the translocation from endosomal/cytosomal compartments to cytosol (41), it is likely that HSP90 plays a role in the reverse translocation of proteasome-dependent cytosolic peptides to endosomal/lysosomal compartments for the loading on MHC-II as seen for constitutive HSP70 (HSC70)-dependent chaperone-mediated autophagy (16). However, in the case of DP04-restricted presentation, the route for the cytosolic peptides to endosomal/lysosomal compartments is yet to be determined. In the classical MHC-I antigen presentation pathway, peptides generated by the proteasome are translocated into endoplasmic reticulum through TAP and loaded onto MHC-I. Because of the TAP-dependency of the DP04-restricted endogenous MHC-II presentation (Figure 5B), the DP04-binding peptide is considered to enter the endoplasmic reticulum. We propose that there are two potential mechanisms for the peptide in the endoplasmic reticulum to be loaded onto MHC-II in the endosomal/lysosomal compartments: (i) fusion between endosome/lysosome and endoplasmic reticulum (42); and (ii) exchange of MHC-I- and MHC-II-loaded peptides during endosomal recycling (43). Confirmation of these mechanisms will require the development of sensitive imaging experiments using epitope-specific antibodies in order to determine the route.
In summary, we have shown that human cancer cells can present intracellular tumor antigens on MHC-II by multiple non-classical antigen-processing pathways, which results in the direct tumor recognition by tumor antigen-specific CD4+ T cells. It is likely that the use of multiple non-classical processing pathways increase the repertoire of intracellular tumor antigen-derived MHC-II-binding epitopes presented on cancer cells. Although we found only a minor subset of NY-ESO-1-specific CD4+ T cells has direct tumor-recognizing ability and the majority of these CD4+ T cells only recognized exogenous NY-ESO-1 protein pulsed on APCs, the implications are significant. For example, direct tumor recognition by CD4+ T cells may provide “CD4-help” in the tumor microenvironment, where professional APCs are frequently dysfunctional or immunosuppressive. Moreover, strategies for expansion and recruitment of tumor-recognizing CD4+ T cells at the local tumor sites, such as vaccination and adoptive T cell therapy may enhance the therapeutic effect of cancer immunotherapy. Therefore, identification of more MHC-II-binding tumor antigen peptides for other tumor antigens and HLA-types is warranted for the development of more effective immunotherapy that will harness tumor-recognizing CD4+ T cells.
Supplementary Material
Acknowledgments
We thank A. Beck and A. Miliotto for assistance in experiments. This work was supported by a Cancer Vaccine Collaborative grant (Cancer Research Institute/Ludwig Cancer Research), Cancer Research Institute Anna-Maria Kellen Clinical Investigator Award, Ovarian Cancer Research Fund, Roswell Park Alliance Foundation, Ovarian Cancer Research Fund, NIH 1R01CA158318-01A1, NIH 2P30 CA016056-36 and RPCI-UPCI Ovarian Cancer SPORE NIH P50CA159981-01A1.
Footnotes
Disclosure of Potential Conflicts of Interest
T. Tsuji and S. Gnjatic have ownership interest (including patents) and are coinventors on primary affiliation assigned NY-ESO-1 patents. No potential conflicts of interest were disclosed by the other authors.
COMPETING FINANTIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 2.Kumamoto Y, Mattei LM, Sellers S, Payne GW, Iwasaki A. CD4+ T cells support cytotoxic T lymphocyte priming by controlling lymph node input. Proc Natl Acad Sci USA. 2011;108:8749–54. doi: 10.1073/pnas.1100567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci USA. 2011;108:21182–7. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. 1999;11:483–93. doi: 10.1016/s1074-7613(00)80123-5. [DOI] [PubMed] [Google Scholar]
- 5.Pamer E, Cresswell P. Mechanisms of MHC class I--restricted antigen processing. Annu Rev Immunol. 1998;16:323–58. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 6.Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol. 2004;5:685–92. doi: 10.1038/ni1088. [DOI] [PubMed] [Google Scholar]
- 7.Talmadge JE. Immune cell infiltration of primary and metastatic lesions: mechanisms and clinical impact. Semin Cancer Biol. 2011;21:131–8. doi: 10.1016/j.semcancer.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Wilke CM, Kryczek I, Zou W. Antigen-presenting cell (APC) subsets in ovarian cancer. Int Rev Immunol. 2011;30:120–6. doi: 10.3109/08830185.2011.567362. [DOI] [PubMed] [Google Scholar]
- 9.Houghton AN, Thomson TM, Gross D, Oettgen HF, Old LJ. Surface antigens of melanoma and melanocytes. Specificity of induction of Ia antigens by human gamma-interferon. J Exp Med. 1984;160:255–69. doi: 10.1084/jem.160.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dadmarz RD, Ordoubadi A, Mixon A, Thompson CO, Barracchini KC, Hijazi YM, et al. Tumor-infiltrating lymphocytes from human ovarian cancer patients recognize autologous tumor in an MHC class II-restricted fashion. Cancer J Sci Am. 1996;2:263–72. [PubMed] [Google Scholar]
- 11.Friedman KM, Prieto PA, Devillier LE, Gross CA, Yang JC, Wunderlich JR, et al. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J Immunother. 2012;35:400–8. doi: 10.1097/CJI.0b013e31825898c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schultz ES, Lethe B, Cambiaso CL, Van Snick J, Chaux P, Corthals J, et al. A MAGE-A3 peptide presented by HLA-DP4 is recognized on tumor cells by CD4+ cytolytic T lymphocytes. Cancer Res. 2000;60:6272–5. [PubMed] [Google Scholar]
- 13.Neumann F, Wagner C, Kubuschok B, Stevanovic S, Rammensee HG, Pfreundschuh M. Identification of an antigenic peptide derived from the cancer-testis antigen NY-ESO-1 binding to a broad range of HLA-DR subtypes. Cancer Immunol Immunother. 2004;53:589–99. doi: 10.1007/s00262-003-0492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 15.Tewari MK, Sinnathamby G, Rajagopal D, Eisenlohr LC. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nat Immunol. 2005;6:287–94. doi: 10.1038/ni1171. [DOI] [PubMed] [Google Scholar]
- 16.Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22:571–81. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Taylor GS, Long HM, Haigh TA, Larsen M, Brooks J, Rickinson AB. A role for intercellular antigen transfer in the recognition of EBV-transformed B cell lines by EBV nuclear antigen-specific CD4+ T cells. J Immunol. 2006;177:3746–56. doi: 10.4049/jimmunol.177.6.3746. [DOI] [PubMed] [Google Scholar]
- 18.Tsuji T, Matsuzaki J, Caballero OL, Jungbluth AA, Ritter G, Odunsi K, et al. Heat Shock Protein 90-Mediated Peptide-Selective Presentation of Cytosolic Tumor Antigen for Direct Recognition of Tumors by CD4+ T Cells. J Immunol. 2012 doi: 10.4049/jimmunol.1103269. [DOI] [PubMed] [Google Scholar]
- 19.Collins MM, Tang T, Slack R, Sintasath D, Hartzman RJ, Ng J, et al. The relative frequencies of HLA-DRB1*01 alleles in the major US populations. Tissue Antigens. 2000;55:48–52. doi: 10.1034/j.1399-0039.2000.550108.x. [DOI] [PubMed] [Google Scholar]
- 20.Odunsi K, Qian F, Matsuzaki J, Mhawech-Fauceglia P, Andrews C, Hoffman EW, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci USA. 2007;104:12837–42. doi: 10.1073/pnas.0703342104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng G, Wang X, Robbins PF, Rosenberg SA, Wang RF. CD4(+) T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci USA. 2001;98:3964–9. doi: 10.1073/pnas.061507398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Odunsi K, Jungbluth AA, Stockert E, Qian F, Gnjatic S, Tammela J, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer research. 2003;63:6076–83. [PubMed] [Google Scholar]
- 23.Tsuji T, Altorki NK, Ritter G, Old LJ, Gnjatic S. Characterization of preexisting MAGE-A3-specific CD4+ T cells in cancer patients and healthy individuals and their activation by protein vaccination. J Immunol. 2009;183:4800–8. doi: 10.4049/jimmunol.0900903. [DOI] [PubMed] [Google Scholar]
- 24.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci USA. 2010;107:7875–80. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96:10403–8. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seifert U, Maranon C, Shmueli A, Desoutter JF, Wesoloski L, Janek K, et al. An essential role for tripeptidyl peptidase in the generation of an MHC class I epitope. Nat Immunol. 2003;4:375–9. doi: 10.1038/ni905. [DOI] [PubMed] [Google Scholar]
- 27.Reits E, Neijssen J, Herberts C, Benckhuijsen W, Janssen L, Drijfhout JW, et al. A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity. 2004;20:495–506. doi: 10.1016/s1074-7613(04)00074-3. [DOI] [PubMed] [Google Scholar]
- 28.Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–76. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 29.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–3. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 30.York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3:1177–84. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 31.Parmentier N, Stroobant V, Colau D, de Diesbach P, Morel S, Chapiro J, et al. Production of an antigenic peptide by insulin-degrading enzyme. Nat Immunol. 2010;11:449–54. doi: 10.1038/ni.1862. [DOI] [PubMed] [Google Scholar]
- 32.van Weert AW, Geuze HJ, Groothuis B, Stoorvogel W. Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. Eur J Cell Biol. 2000;79:394–9. doi: 10.1078/0171-9335-00062. [DOI] [PubMed] [Google Scholar]
- 33.Smith V, Sausville EA, Camalier RF, Fiebig HH, Burger AM. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother Pharmacol. 2005;56:126–37. doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- 34.Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998;16:2639–45. doi: 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- 35.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen RK, Oesterreich S, Lemieux P, Sarge KD, Fuqua SA. Quercetin inhibits heat shock protein induction but not heat shock factor DNA-binding in human breast carcinoma cells. Biochem Biophys Res Commun. 1997;239:851–6. doi: 10.1006/bbrc.1997.7572. [DOI] [PubMed] [Google Scholar]
- 37.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172:6649–57. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
- 38.Rowell JF, Ruff AL, Guarnieri FG, Staveley-O’Carroll K, Lin X, Tang J, et al. Lysosome-associated membrane protein-1-mediated targeting of the HIV-1 envelope protein to an endosomal/lysosomal compartment enhances its presentation to MHC class II-restricted T cells. J Immunol. 1995;155:1818–28. [PubMed] [Google Scholar]
- 39.Lepage S, Lapointe R. Melanosomal targeting sequences from gp100 are essential for MHC class II-restricted endogenous epitope presentation and mobilization to endosomal compartments. Cancer Res. 2006;66:2423–32. doi: 10.1158/0008-5472.CAN-05-2516. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Bartido S, Yang G, Qin J, Moroi Y, Panageas KS, et al. A role for a melanosome transport signal in accessing the MHC class II presentation pathway and in eliciting CD4+ T cell responses. J Immunol. 1999;163:5820–6. [PubMed] [Google Scholar]
- 41.Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, et al. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci USA. 2011;108:16363–8. doi: 10.1073/pnas.1108372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 43.Pathak SS, Lich JD, Blum JS. Cutting edge: editing of recycling class II:peptide complexes by HLA-DM. J Immunol. 2001;167:632–5. doi: 10.4049/jimmunol.167.2.632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.