

Abstract
Inhibitors of p38 mitogen-activated protein kinase (MAPK) diminish inflammatory arthritis in experimental animals. This may be effected by diminishing the production of inflammatory mediators, but this kinase is also part of the IL-1 signal pathway in articular chondrocytes. We determined the effect of p38 MAPK inhibition on proliferative and synthetic responses of lapine chondrocytes, cartilage, and synovial fibroblasts under basal and IL-1-activated conditions.
Basal and growth factor-stimulated proliferation and proteoglycan synthesis were determined in primary cultures of rabbit articular chondrocytes, first-passage synovial fibroblasts, and cartilage organ cultures. Studies were performed with or without p38 MAPK inhibitors, in IL-1-activated and control cultures. Media nitric oxide and prostaglandin E2 were assayed.
p38 MAPK inhibitors blunt chondrocyte and cartilage proteoglycan synthesis in response to transforming growth factor beta; responses to insulin-like growth factor 1 (IGF-1) and fetal calf serum (FCS) are unaffected. p38 MAPK inhibitors significantly reverse inhibition of cartilage organ culture proteoglycan synthesis by IL-1. p38 MAPK inhibition potentiated basal, IGF-1-stimulated and FCS-stimulated chondrocyte proliferation, and reversed IL-1 inhibition of IGF-1-stimulated and FCS-stimulated DNA synthesis. Decreases in nitric oxide but not prostaglandin E2 synthesis in IL-1-activated chondrocytes treated with p38 MAPK inhibitors are partly responsible for this restoration of response. Synovial fibroblast proliferation is minimally affected by p38 MAPK inhibition.
p38 MAPK activity modulates chondrocyte proliferation under basal and IL-1-activated conditions. Inhibition of p38 MAPK enhances the ability of growth factors to overcome the inhibitory actions of IL-1 on proliferation, and thus could facilitate restoration and repair of diseased and damaged cartilage.
Keywords: chondrocytes, interleukin-1, nitric oxide, p38 mitogen-activated protein kinase, transforming growth factor beta
Introduction
Proinflammatory cytokines are responsible for much of the pathophysiology of both osteoarthritis and rheumatoid arthritis [1]. Activation of p38 mitogen-activated protein kinase (MAPK) has been implicated in the catabolic and anti-anabolic actions of both IL-1 and tumor necrosis factor alpha [2]. These cytokines are also induced in mechanically stressed [3,4] and damaged cartilage. The signal pathways they activate, including p38 MAPK, may thus influence the course of cartilage repair. It is therefore important to understand the consequences of p38 MAPK inhibition on cartilage/chondrocyte responses to the anabolic effectors, which stimulate the repair processes of proliferation and cartilage matrix protein synthesis.
Members of the pyridinyl imidazole class of compounds that inhibit p38 MAPK have been developed, and their potential as therapeutic agents in inflammation, arthritis, septic shock, and myocardial injury is currently being explored [5]. One of these compounds, SB 203580 (SB), is a potent inhibitor of cytokine production in mice and rats, and decreases paw inflammation in collagen-induced arthritis in mice [6]. A second related compound, SB 242235, decreases adjuvant-induced arthritis in rats [7]. SB also inhibits IL-1 induction of inducible nitric oxide synthase (iNOS) in bovine chondrocytes [8], and thus blocks nitric oxide (NO) synthesis. This effect may also protect cartilage from the damaging actions of NO [9].
p38 MAPK was recently identified, however, as part of the signal transduction pathway effecting transforming growth factor beta (TGF-β) stimulation of aggrecan gene expression by the chondrogenic cell line ATDC5 [10,11]. The relationship between TGF-β signaling through p38 MAPK and the Smad family was characterized in C2C12 cells. The conclusion was that the nuclear target of p38, ATF-2, becomes phosphorylated in response to TGF-β and forms a complex with Smad 4 [12]. Similar signal synergy studies have not been carried out for chondrocytes. However, given the important anabolic and anticatabolic [13] actions of TGF-β, any maneuver that modifies responses to TGF-β and other anabolic growth factors could have critical consequences for maintenance and repair of cartilage. These studies were thus initiated to determine whether p38 MAPK inhibition affects chondrocyte responses to TGF-β, insulin-like growth factor 1 (IGF-1), and serum, and also whether p38 MAPK inhibition reverses the anti-anabolic actions of IL-1 on proliferative and synthetic responses of rabbit articular chondrocytes, cartilage, and synovial fibroblasts.
Materials and methods
Materials were obtained from the following suppliers: New Zealand White rabbits, 5–6 lb (Myrtle's Rabbitry, Thompson Station, TN, USA); modified Eagle's medium (MEM), fetal calf serum (FCS), antibiotics, other tissue culture supplies, and protease inhibitor cocktail for use with mammalian cells (Sigma Chemical, St Louis, MO, USA); DuoSet IC for phospho-p38alpha (R&D Systems, Minneapolis, MN, USA); protein assay reagent (Bio-Rad, Hercules, CA, USA); type 1 collagenase and trypsin (Worthington Biochemical, Freehold, NJ, USA); 35S-sodium sulfate, 1 Ci/mmol (NEN, Boston, MA, USA); [methyl-3H]thymidine and prostaglandin E2 (PGE2) enzyme immunoassay kits (Amersham Pharmacia Biotech, Piscataway, NJ, USA); human TGF-β1, human IGF-1, recombinant human IL-1β (R&D Systems, Minneapolis, MN, USA); Sc-58125 (Cayman Chemical, Ann Arbor MI, USA); SB 203580 (SB) and SB 202190, hydrochloride (Calbiochem, San Diego, CA, USA); and N-monomethyl-L-arginine (L-NMA) was synthesized by Dr Paul Dowd and Dr Wei-Zhang (Department of Chemistry, University of Pittsburgh, PA, USA). All other reagents were obtained from Sigma Chemical.
Rabbits were euthanized using a protocol approved by the IACUC of the Pittsburgh, Pennsylvania VA Healthcare System. Chondrocytes were isolated from knee and shoulder joints of mature New Zealand white rabbits and the cells were cultured as previously described [14]. Cartilage slices from the same joints were used in some experiments. Fibroblasts were cultured from synovial membranes of lapine knee joints using a procedure similar to that for chondrocytes as previously reported [15]. Cells were grown to 80% confluence in Falcon Multiwell, 48-well plates, and medium serum was reduced to 1% 24 hours before addition of dimethylsulfoxide vehicle at < 0.5% or the specific p38 MAPK inhibitors SB 203580 (SB) or SB 202190, hydrochloride, at 1 μM. The inhibitor concentrations used were kept below 2 μM as effects not related to p38 MAPK inhibition have been seen in other cell types with higher concentrations [16]. Seven hours later, 50 ng/ml IGF-1, 50 pM TGF-β or 5–10% FCS was added. Proteoglycan synthesis or proliferation was determined 24 hours after addition of growth factors.
Phosphorylated p38 MAPK was determined as an index of activation using a commercially available kit (R&D systems) on cell lysates collected 30 min after activation with IL-1 (2 ng/ml) and 60–120 min after addition of TGF-β (100 pM). Chondrocytes were grown to confluence in six-well plates, the serum reduced for 24 hours, fresh medium added, and the cells lysed after activation with IL-1 or TGF-β. After treatment the cells were washed twice with phosphate-buffered saline, lysed, the lysates analyzed as per kit instructions and the results normalized to the average protein content of 33 μg/ml (determined on 10-fold dilution of lysates as per Bio-Rad protein assay instructions).
Proteoglycan synthesis was measured as the incorporation of 35S-sulfate (6 hour pulse label) into molecules separated from unincorporated label using PD-10 columns as described for this laboratory [14]. Proliferation was measured as the incorporation of [3H]thymidine during a 2 hour pulse label into trichloroacetic acid precipitated material. NO was assayed as the nitrite concentration in conditioned media (CM) using the Griess reaction, and CM PGE2 was assayed using the ELISA kit from Amersham Pharmacia Biotech.
Chondrocytes transduced with an adenoviral vector carrying the human inducible nitric oxide synthase gene (Ad-iNOS) were used in some studies to facilitate evaluation of the effects of NO independent of other actions of iNOS inducing cytokines on the cell. The adenoviral vector, previously described [14] with a titer of 1010 pfu/ml, was prepared by Dr Paul Robbins (University of Pittsburgh School of Medicine Human Gene Therapy Center). Transduction of chondrocytes was carried out as follows: monolayers of chondrocytes were washed with Gey's Balanced Salt Solution, and 1 × 107 pfu virus in 0.2 ml Dulbecco's modified Eagle's medium (DMEM) containing 0.1% bovine serum albumin, with or without 1 mM L-NMA added to each well. The transduction efficiency was 76% under these conditions [14]. The cells were washed after overnight incubation, and the culture continued for 24 hours in MEM, 0.5% fetal bovine serum, with or without L-NMA, agonists added, and conditioned media for determination of NO production collected 24 hours later. Proliferation was also evaluated at this time.
Experiments were performed at least three times, and data are presented as mean ± standard error. Statistically significant differences (P < 0.05) were determined using Student's t test.
Results
Figure 1 shows p38 MAPK is activated, as shown by increased phosphorylation, after exposure of chondrocytes to IL-1 or TGF-β. There is detectable p38 MAPK phosphorylation under basal conditions in normal lapine chondrocytes, and this is consistent with the ability of p38 MAPK inhibition to modulate proliferation in the absence of, as well as in the presence of, cytokine activation. IL-1 increased phosphorylation ninefold after 30 min, consistent with prior observations in rabbit chondrocytes [17] and in human chondrocytes [18,19]. TGF-β caused a persistent activation of sixfold to eightfold, showing that this growth factor can activate this signal pathway in primary chondrocytes as well as chondrogenic ATDC5 cells [10,11].
Figure 1.
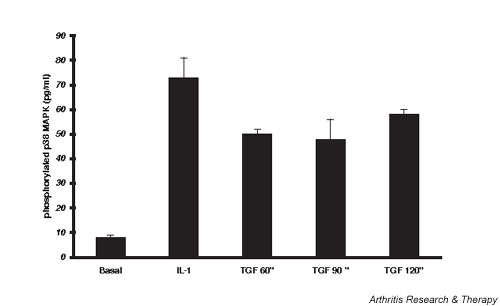
Lapine chondrocyte p38 mitogen-activated protein kinase (MAPK) phosphorylation. Chondrocytes were grown to confluence, medium serum reduced for 24 hours, and the cells lysed 60–120 min after activation with transforming growth factor beta (TGF-β) or 30 min after activation with IL-1. ELISA for phospho-p38 MAPK in the lysates was done as per instructions in the R&D Systems kit. Data were normalized to the average protein content of the lysates of 33 ± 1 μg/ml. Values are plotted as mean pg/ml ± standard error of n = 3–10.
IGF-1 and TGF-β stimulated lapine chondrocyte proteoglycan synthesis from 85 ± 7 pmol/105 cells to 202 ± 15 and 344 ± 50 pmol/105 cells, or by 2.4-fold and 4.0-fold, respectively (Fig. 2). Inhibition of p38 MAPK did not alter basal or IGF-1-stimulated proteoglycan synthesis. However, it did decrease the increase in response to TGF-β to one-half of that seen in the absence of SB. Inhibition of p38 MAPK did not prevent IL-1 inhibition of basal or stimulated proteoglycan synthesis by rabbit chondrocytes: IL-1, 65 ± 10 pmol/105 cells; IL-1 + SB, 65 ± 9 pmol/105 cells; IL-1 + TGF-β, 227 ± 22 pmol/105 cells; and IL-1 + SB + TGF-β, 223 ± 24 pmol/105 cells. Similar results were seen using a second p38 MAPK inhibitor, SB 202190.
Figure 2.
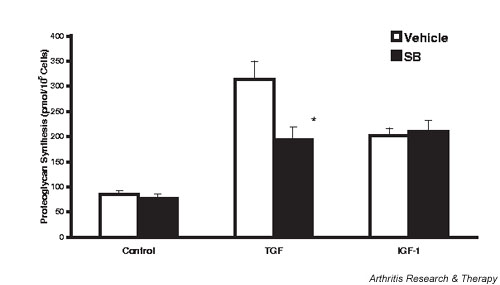
p38 mitogen-activated protein kinase inhibition blunts transforming growth factor beta (TGF-β)-stimulated, but not insulin-like growth factor 1 (IGF-1)-stimulated, proteoglycan synthesis. Rabbit chondrocytes were grown to confluence, the medium serum reduced, and 1 μM SB 203580, and 50 ng/ml IGF-1 or 50-pM TGF-β added. The cells were pulse labeled 24 hours later (for 6 hours) with 35S-sulfate. Conditioned media and cell extracts were assayed for incorporation of label into proteoglycans by chromatography on PD-10 columns, and the data are expressed as pmol/105 cells. Values are the mean ± standard error of n = 6–12. *P < 0.05 versus vehicle. SB, SB 203580.
To confirm the effect of p38 MAPK inhibition on chondrocyte proteoglycan synthesis in situ in the cartilage matrix, experiments using cartilage organ cultures were performed and are reported in Fig. 3. IL-1 at the low, but inhibitory, concentration of 0.1 ng/ml was used, and in this case there is a modest blunting of its action to diminish matrix proteoglycan synthesis by the p38 MAPK inhibitor SB 202190. However, the same concentration of inhibitor also decreased TGF-β-stimulated proteoglycan synthesis by 40%. p38 MAPK inhibition did not affect the ability of 5% FCS (Fig. 3) or of IGF-1 (data not shown) to stimulate cartilage proteoglycan synthesis. Similar results were found using SB 203580 (SB).
Figure 3.
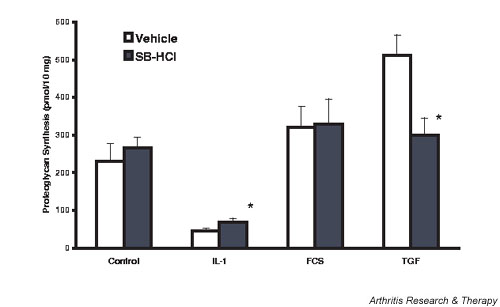
Lapine cartilage: modulation of stimulated and inhibited proteoglycan synthesis by p38 mitogen-activated protein kinase inhibition. Cartilage slices were maintained overnight in Dulbecco's modified Eagle's medium (DMEM)/10% fetal calf serum (FCS), the serum removed, and agonists and inhibitors added 24 hours later. Then 1 μM SB 202190 was added 60 min before 0.1 ng/ml IL-1, and FCS and transforming growth factor beta (TGF-β) were added 6 hours later. Proteoglycan synthesis was evaluated the following day as the amount of 35S incorporated into proteoglycans expressed as pmol/10 mg wet weight. Values are the mean ± standard error of n = 6–12. *P < 0.05 versus vehicle.
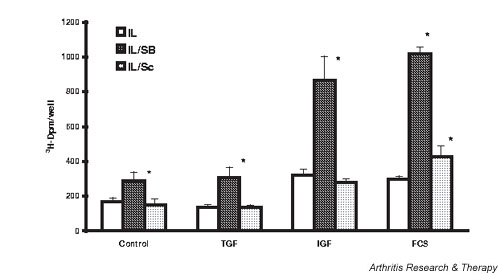
p38 MAPK inhibitors had significant effects on chondrocyte proliferation under basal, IL-1-activated, and growth factor-stimulated conditions. As shown in Fig. 4, IGF-1 (445%) and FCS (978%) stimulated chondrocyte proliferation. SB increased basal, IGF-1-stimulated, and serum-stimulated chondrocyte proliferation by 55%, 73%, and 45%, respectively. The relatively modest stimulation of proliferation by TGF-β (97%) was similar in the presence of and in the absence of p38 MAPK inhibition (data not shown). IL-1 did not significantly inhibit basal chondrocyte proliferation. However, stimulation in response to IGF-1 or FCS was decreased by 54% and 87%, respectively, in IL-1-activated chondrocytes. When p38 MAPK was inhibited, however, growth factor-stimulated proliferation in the presence of IL-1 was significantly increased (IGF-1) or was completely restored (FCS). TGF-β stimulation of IL-1-activated rabbit chondrocyte proliferation (twofold) was less than that seen in response to IGF-1 or to FCS, but the modest increase in the presence of SB (70%) was significant (data not shown).
Figure 4.
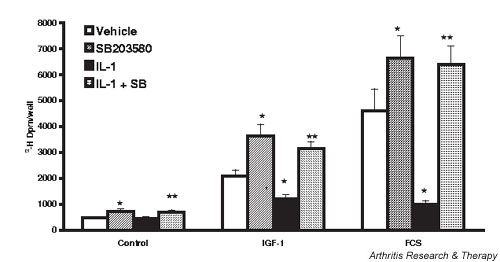
Inhibition of p38 mitogen-activated protein kinase potentiates basal and growth factor-stimulated proliferation of rabbit chondrocytes, and reverses IL-1-inhibited proliferation. Chondrocytes were grown to 80% confluence, the medium serum reduced for 24 hours, SB 203580 (SB) (1 μM) or dimethyl sulfoxide vehicle (0.5%) added, and 2 ng/ml IL-1 added 30 min later. The cells were then stimulated with insulin-like growth factor 1 (IGF-1) (50 ng/ml) or fetal calf serum (FCS) (10%) for 24 hours. Proliferation was assayed as [3H]thymidine incorporation into trichloroacetic acid precipitated material following a 2 hour pulse label. Values are the mean ± standard error of n = 10–20. *P < 0.05 versus vehicle, **P < 0.05 versus IL-1.
IL-1 induces iNOS and cyclooxygenase-2 (COX-2) in chondrocytes. The products of these enzymes, NO and PGE2, have been shown to reduce chondrocyte proliferation [20,21]. SB inhibition of p38 MAPK decreased NO in CM from IL-1-activated cells from 7.5 ± 0.41 μM (IL-1) to 4.7 ± 0.26 μM (IL-1 + SB), a significant 50% inhibition of the increase above control values of 2.4 ± 0.35 μM (vehicle) and 1.97 ± 0.51 μM (SB). A series of experiments was initiated to evaluate the ability of NO alone to modulate chondrocyte proliferation in the absence of other factors present in IL-1-activated cells.
To measure the effects of NO per se on chondrocyte proliferation we transfected chondrocytes with Ad-iNOS vector in the presence of variable concentrations of L-NMA. Figure 5 shows the CM concentration of nitrite as an index of NO synthesis following transfection of chondrocytes with Ad-iNOS as described in Materials and methods. L-NMA (0–0.75 mM) was added to limit NO synthesis and thus to produce conditions of variable NO exposure for these cells. Nitrite ranged from 11 ± 00.54 μM in the absence of L-NMA to 2 ± 00.28 μM with 0.75 μM L-NMA added. In a separate series of experiments, DNA synthesis in sham transfected chondrocytes versus Ad-iNOS transfected chondrocytes with added 0.75 mM L-NMA was evaluated. Basal [3H]thymidine incorporation was the same (sham transfected, 245 ± 010 dpm/well versus Ad-iNOS transfected, 296 ± 021 dpm/well; P = 0.074) as was 10% FCS-stimulated incorporation (sham transfected, 4864 ± 0242 dpm/well versus Ad-iNOS transfected, 4706 ± 0788 dpm/well; P = 0.87), showing that the transfection procedure per se does not affect basal or stimulated chondrocyte proliferation.
Figure 5.
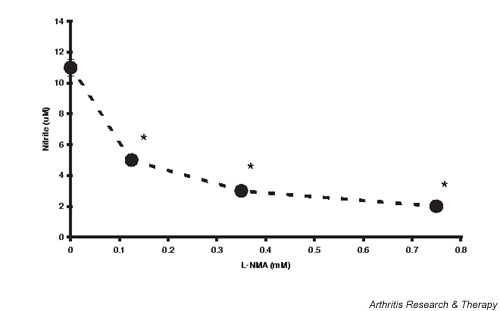
Nitric oxide production by inducible nitric oxide synthase transduced chondrocytes. Chondrocytes were transduced with constant plaque forming units of adenoviral vector carrying the human inducible nitric oxide synthase gene as described in Materials and methods, incubated with variable concentrations of N-monomethyl-L-arginine (L-NMA), and the conditioned media nitrite measured 24 hours later. Values are the mean ± standard error of n = 6–12 (the standard errors lie within the area of the data point symbol). *P < 0.05 versus 0 L-NMA.
A dose response for NO inhibition of proliferation in iNOS transfected rabbit chondrocytes is shown in Fig. 6. Chondrocytes were transfected with Ad-iNOS and incubated with L-NMA (0.75–0.125 mM) to allow variable synthesis of NO. Ad-iNOS transfection and subsequent endogenous production of variable NO inhibits both IGF-1-stimulated and FCS-stimulated chondrocyte proliferation.
Figure 6.
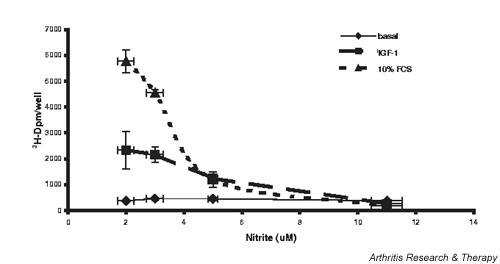
Nitric oxide (NO) inhibits chondrocyte proliferation. Chondrocytes were grown to 80% confluence and transduced with adenoviral vector carrying the human inducible nitric oxide synthase gene as described in Materials and methods. Variable concentrations of N-monomethyl-L-arginine were added to modulate NO synthesis, the cells stimulated with 10% fetal calf serum (FCS) or 50 ng/ml insulin-like growth factor 1 (IGF-1), and proliferation evaluated after 24 hours. Values are the mean ± standard error of n = 6–12.
Figure 4 documented IL-1 inhibition of both IGF-1-stimulated and FCS-stimulated chondrocyte proliferation (by 54% and 87%, respectively). Figure 7 compares the ability of SB inhibition of p38 MAPK and L-NMA inhibition of NO synthesis to restore the proliferative response to growth factors in IL-1-activated cells. When NO synthesis in IL-1-activated cells was inhibited with 0.5 mM L-NMA, the restoration of basal and IGF-1-stimulated proliferation was similar to that seen in the presence of SB. The response to TGF-β was again potentiated by SB but not by L-NMA inhibition of NO synthesis. There was a significant 105% increase in FCS-stimulated proliferation in L-NMA-treated chondrocytes that was less than the 202% increase seen with p38 MAPK inhibition. The NO levels in CM in these experiments were: IL-1, 5 ± 0.2 μM; IL-1 + SB, 2.6 ± 0.13 μM; IL-1 + L-NMA, 1.4 ± 0.10 μM. These values were all significantly different from each other. The results suggest that the decrease in NO production by p38 MAPK inhibitors is responsible for some, but not all, of the restoration of proliferation in response to growth factors.
Figure 7.
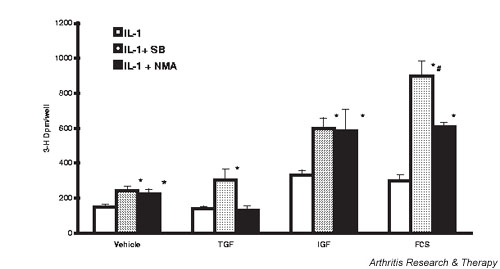
Inhibition of nitric oxide (NO) synthesis restores proliferation in IL-1-activated chondrocytes. Cells were treated as for Fig. 2, but with the addition of N-monomethyl-L-arginine (L-NMA) at a final concentration of 0.5 mM 30 min before IL-1. Values are the mean ± standard error of n = 9–-12. *P < 0.05 versus IL-1 alone, #P < 0.05 versus IL + L-NMA. FCS, fetal calf serum; IGF, insulin-like growth factor 1; SB, SB 203580; TGF, transforming growth factor beta.
p38 MAPK inhibition also blunted IL-1-stimulated PGE2 synthesis (Fig. 8). TGF-β alone significantly increased PGE2 accumulation approximately threefold, from 47 to 170 pg/105 cells per 24 hours. Inhibition of p38 MAPK completely blocked this increase. IL-1 increased PGE2 to values 10-fold higher than did TGF-β, and the combination of IL-1 + TGF-β showed a striking synergy to increase PGE2 accumulation to concentrations threefold higher that with IL-1 alone. In these later cases, p38 MAPK inhibition diminished PGE2 but the medium concentrations were still greater than under basal conditions.
Figure 8.
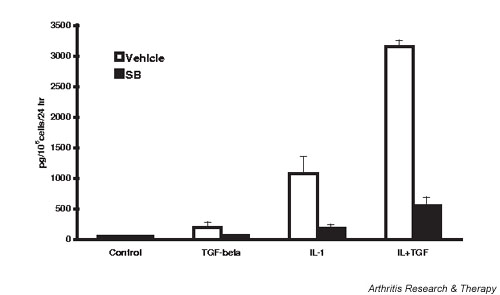
SB 203580 (SB) inhibits transforming growth factor beta (TGF-β)-stimulated and IL-1-stimulated prostaglandin E2 (PGE2) synthesis by lapine chondrocytes. Chondrocytes were treated as in Fig. 4 and conditioned media collected for assay of PGE2 24 hours after activation. Values are the mean ± standard error of n = 6–9. SB 203580 significantly inhibited PGE2 production in all conditions except 'control'.
To test whether these concomitant changes in PGE2 could contribute to the restoration of proliferative response in lapine chondrocytes, we compared the ability of Sc-58125, the specific COX-2 inhibitor, and SB to blunt the inhibitory actions of IL-1. Data from this series of experiments are shown in Fig. 9. As seen before, SB potentiates all growth factor-stimulated proliferation. However, 0.5 μM Sc-58125, which decreased IL-1-stimulated PGE2 in chondrocyte CM from 1073 ± 278 to 30 ± 13 pg/24 hours or in IL-1 + TGF-β-activated chondrocytes from 3153 ± 106 to 27 ± 16 pg/24 hours, failed to enhance basal, IGF-1-stimulated or TGF-β-stimulated proliferation. There was a modest but significant 43% potentiation of proliferation in the presence of FCS, which was far less than the 240% seen with p38 MAPK inhibition. In a separate series of experiments we compared the effect of SB alone with that of SB + Sc-58125 in IL-1-activated cells. Consistent with the data in Fig. 9, complete inhibition of PGE2 synthesis with Sc-58125 in conjunction with SB inhibition of p38 MAPK effected no significant differences in chondrocyte proliferation from those seen with SB alone (data not shown).
Figure 9.
Comparison of the effect of p38 mitogen-activated protein kinase (SB 203580 [SB]) with cyclooxygenase-2 (Sc-58125 [Sc]) inhibition on IL-1 action to diminish chondrocyte proliferation. Cells were treated as for Fig. 4, but with the addition of Sc-58125 at a final concentration of 0.5 μM 30 min before IL-1. Values are the mean ± standard error of n = 6–12. *P < 0.05 versus IL-1 alone. FCS, fetal calf serum; IGF, insulin-like growth factor 1; TGF, transforming growth factor beta.
We also tested the effects on lapine chondrocyte proliferation of exogenous PGE2 at concentrations generated in the previous experiments. Consistent with the results in Fig. 9, there were no significant effects of PGE2 in this concentration range (0.1–6 ng/ml) on chondrocyte proliferation (data not shown).
Cartilage/chondrocyte metabolism may also be affected by synovial hyperplasia and by the products secreted by the synovial fibroblasts [22]. We therefore evaluated the effects of inhibition of p38 MAPK on basal, growth factor-stimulated, and IL-1-activated proliferation of lapine synovial fibroblasts (passage 1). The effects of p38 MAPK inhibition in lapine synovial fibroblasts were modest in comparison with those found in chondrocytes. SB had no effect on basal proliferation and the 29% stimulation in the presence of FCS is less than that seen in chondrocytes. SB had no effect on proliferation in the presence of IGF-1 or TGF-β. SB did significantly stimulate proliferation of IL-1-activated fibroblasts under both basal (1629 ± 115 dpm/well versus 2970 ± 803 dpm/well) and FCS-stimulated conditions (8014 ± 449 dpm/well versus 10372 ± 1104 dpm/well). IL-1 did not increase NO synthesis in these preparations, and there were only modest changes in PGE2 synthesis under the conditions evaluated (data not shown).
Discussion
These studies evaluated the potential of p38 MAPK inhibition to modulate the response of lapine chondrocytes to growth factors under basal and cytokine-activated conditions. p38 MAPK activation (phosphorylation) in primary lapine chondrocytes is documented. The studies show that p38 MAPK inhibition can modestly enhance proteoglycan synthesis in cartilage organ cultures inhibited by low concentrations of IL-1. On the contrary, p38 MAPK inhibition blunted the synthetic response to TGF-β in both isolated chondrocytes and cartilage organ cultures. However, the response to IGF-1 and FCS is not affected. Table 1 summarizes these findings.
Table 1.
Summary of effects of p38 MAPK inhibition on proteoglycan synthesis
| Proteoglycan synthesis | |||
| Basal | + TGF-β | + IGF-1/FCS | |
| SB | No change | - | No change |
| IL-1 | - - | - - | - - |
| IL-1 + SB* | + | No change | No change |
*Relative to IL-1 alone. FCS, fetal calf serum; IGF-1, insulin-like growth factor 1; SB, SB 203580; TGF-β, transforming growth factor beta.
The net effect of p38 MAPK inhibition on matrix protein (proteoglycan) synthesis will thus depend on the growth factor milieu effecting cartilage homeostasis. The partial reversal of IL-1 inhibition in cartilage, and the lack of an effect on the response to the complex mix of factors contained in FCS, suggests that p38 MAPK inhibition in vivo would positively affect cartilage proteoglycan synthesis.
p38 MAPK inhibition potentiates basal, IGF-1-stimulated and FCS-stimulated chondrocyte proliferation with minimal effect on the response to TGF-β. The inhibition modestly increases proliferation of IL-1-activated chondrocytes, enhances the response to IGF-1 in the presence of IL-1, and restores the response to FCS completely. SB inhibition of IL-1-stimulated NO production accounts for some of the restoration of response, but the action of SB to diminish PGE2 is not critical to its effects on proliferation. Table 2 summarizes these findings.
Table 2.
Summary of effects of p38 MAPK and COX-2 inhibition on chondrocyte proliferation
| Lapine chondrocyte proliferation | ||
| Basal | + IGF-1/FCS | |
| SB | + | + |
| IL-1 | No change | - - |
| IL-1 + SB* | + | + + |
| IL-1 + L-NMA* | No change | + |
| IL-1 + Sc-58125* | No change | No change |
*Relative to IL-1 alone. FCS, fetal calf serum; IGF-1, insulin-like growth factor 1; L-NMA, N-monomethyl-L-arginine; SB, SB 203580.
p38 MAPK inhibition had little effect on lapine fibroblast proliferation under basal and IL-1-activated conditions, precluding a concomitant synovial hyperplasia and potential increases in catabolic factors thereby secreted.
The data suggest a significant component of IL-1 inhibition of rabbit chondrocyte proliferation is effected through p38 MAPK-mediated actions. The role of p38 MAPK in the regulation of proliferation has been extensively studied [23]; however, the relationship varies with cell type. For example, p38 MAPK activation is linked with increased proliferation in vascular smooth muscle cells [24], and is necessary for the fibroblast growth factor 2 stimulation of fibroblasts [25], but it arrests proliferation of thymocytes [26]. The current studies show that, in lapine chondrocytes, inhibition of p38 MAPK enhances basal and growth factor-stimulated proliferation, and can restore proliferation in IL-1-activated cells. These data suggest that, in this cell type, p38 MAPK activation is associated with decreased DNA synthesis.
L-NMA inhibition of NO synthesis in IL-1-activated cells did increase IGF-1-stimulated and FCS-stimulated proliferation, but not as effectively as SB under some conditions. This suggests that some, but not all, of the potentiation of proliferation by SB in IL-1-activated cells may be secondary to the decrease in NO synthesis when p38 MAPK is blocked. The NO dose response (Fig. 6) shows that lapine chondrocyte proliferation is sensitive to NO over the concentration range found in the conditioned media of IL-1-activated cells, and is modulated by SB. The effects of NO in the context of IL-1-activated chondrocytes where multiple factors are altered may be different from that in cells where NO synthesis is enhanced in isolation from these factors. The data do suggest, however, that the diminution of NO synthesis by p38 MAPK inhibitors may contribute to their ability to blunt the anti-anabolic actions of IL-1. This pathway may or may not be relevant to human disease, as Badger and colleagues [27] found that SB 242235, another selective p38 MAPK inhibitor, did not decrease IL-1 induction of iNOS and NO synthesis in human chondrocyte cultures. However, p38 MAPK activation by NO has been demonstrated in several cell types [28,29] and has been linked with NO induction of heme oxygenase 1 in HeLa cells [30]. The possibility that some of the pathophysiologic actions of NO in human chondrocytes may be mediated via p38 MAPK activation has not been evaluated, and thus remains a potential point of therapy by inhibitors of p38 MAPK in cytokine-activated human cartilage/chondrocytes.
SB also inhibited IL-1-stimulated increases in PGE2 synthesis/accumulation (Fig. 8). However, Sc-58125 inhibition of COX-2 and the resulting decreases in PGE2 had little effect on chondrocyte proliferation (Fig. 9). This suggests that the SB inhibition of PGE2 production in IL-1-treated and IL-1 + TGF-β-treated cells contributes minimally to the restoration of proliferation in rabbit chondrocytes. The effects of prostaglandins on chondrocyte proliferation have been variable. For example, Blanco and Lotz [21] concluded that NO inhibition of normal human chondrocyte proliferation was effected by concomitant changes in PGE2. Lowe and colleagues [31] showed that exogenous PGE2 had a dose-dependent, biphasic effect on rat chondrocytes with suppression at the lower concentrations tested (0.1 μM, or 35 ng/ml) and stimulation at higher concentrations (5 μM, or 1760 ng/ml). Schwartz and colleagues [32] found that PGE2 from 0.007 to 15 ng/ml increased the cell number and [3H]thymidine incorporation in chick costochondal cartilage cells.
Our data suggest that normal rabbit articular chondrocyte proliferation is relatively insensitive to the range of CM PGE2 attained subsequent to IL-1 activation. The relationship between chondrocyte proliferation and PGE2 thus seems highly species dependent. We recently reported that human chondrocyte proliferation is inhibited by PGE2 concentrations found in CM following IL-1 activation [33]. The mechanisms by which p38 MAPK inhibition restores proliferation in IL-1-activated/stressed human chondrocytes may thus be different from those described for lapine preparations (current studies) or for bovine preparations [27].
These studies were initiated, in part, to determine whether inhibition of p38 MAPK would blunt the actions of TGF-β on chondrocytes. In the case of proliferation, this was not found to be so. Under basal conditions, SB did not modify the modest TGF-β-stimulated increases in DNA synthesis; in IL-1-activated cells, the response to TGF-β was actually enhanced. In contrast, both SB 203580 (SB) and SB 202190 blunted the ability of TGF-β to stimulate proteoglycan synthesis in chondrocytes in a monolayer culture and in situ in a cartilage organ culture. Ridley and colleagues [34] showed that even low concentrations of SB inhibited proteoglycan synthesis in bovine nasal septum cartilage in organ cultures in the presence of 10% FCS. We did not see a similar inhibition in rabbit cartilage. Furthermore, stimulation of proteoglycan synthesis by IGF-1 in lapine chondrocytes in monolayer culture was also unaffected by SB. Perhaps the difference between these results is related to species differences in the relative importance of the p38 MAPK pathway in maintaining proteoglycan synthesis. Regardless, our data do suggest that the signal pathways that effect TGF-β stimulation of chondrocyte proliferation and chondrocyte proteoglycan synthesis differ; the pathway activating proteoglycan synthesis appears to involve p38 MAPK, while that activating proliferation does not.
The data showing only minor modulation of IL-1-inhibited proteoglycan synthesis by SB (Figs 2 and 3) are consistent with prior studies of bovine cartilage [34]. However, even the ability to modestly reverse the anti-anabolic effects of IL-1 may be therapeutic under conditions of mild inflammation as is often seen in osteoarthritis, and in some stages of the repair of injured cartilage. Although proteoglycan synthesis responses to TGF-β are blunted, the responses to IGF-1 and FCS remain intact in the presence of p38 MAPK inhibitors. Coupled with the ability to reverse effects of low concentrations of IL-1 and the minimal effects on synovial fibroblasts, this suggests that p38 MAPK inhibition could have a positive effect on cartilage maintenance and repair.
Conclusions
Although activation of p38 MAPK has been observed in tissues from arthritic joints [1] and from mechanically stressed cartilage [2,3], and has been shown to be involved in IL-1 inhibition of collagen synthesis [8] and IL-1 induction of collagenases [19], this is the first report showing effects of basal levels of p38 MAPK activity on chondrocyte proliferation. Inhibition of p38 MAPK potentiates basal and stimulated proliferation. It thus appears to have a regulatory function in lapine chondrocytes under both normal and cytokine-activated conditions. p38 MAPK inhibition can partially reverse IL-1 inhibition of proteoglycan synthesis, and thus could contribute to maintenance of matrix proteins in cytokine-activated and stressed cartilage. Whether similar effects of p38 MAPK inhibitors on chondrocyte responses to cytokines and growth factors are found in human cartilage/chondrocyte preparations should be evaluated.
Competing interests
None declared.
Abbreviations
Ad-iNOS = adenoviral vector carrying the human inducible nitric oxide synthase gene; CM = conditioned media; COX-2 = cyclooxygenase-2; DMEM = Dulbecco's modified Eagle's medium; ELISA = enzyme-linked immunosorbent assay; FCS = fetal calf serum; IGF-1 = insulin-like growth factor 1; IL = interleukin; iNOS = inducible nitric oxide synthase; L-NMA = N-monomethyl-L-arginine; MAPK = mitogen-activated protein kinase; MEM = modified Eagle's medium; NO = nitric oxide; pfu = plaque forming units; PGE2 = prostaglandin E2; SB = SB 203580; TGF-β = transforming growth factor beta.
Acknowledgments
Acknowledgements
The Department of Veterans Affairs and the Ferguson Orthopaedic Fund supported this work.
References
- Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner C-W, Bitzan P, Zenz P, Redlich K, Xu Q, Steiner G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–2512. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- van der Kraan PM, van den Berg WB. Anabolic and destructive mediators in osteoarthritis. Curr Opin Clin Nutr Metab Care. 2000;3:205–211. doi: 10.1097/00075197-200005000-00007. [DOI] [PubMed] [Google Scholar]
- Honda K, Ohno S, Tanimoto K, Ijuin C, Tanaka N, Doi T, Kato Y, Tanne K. The effects of high magnitude cyclic tensile load on cartilage matrix metabolism in cultured chondrocytes. Eur J Cell Biol. 2000;79:601–609. doi: 10.1078/0171-9335-00089. [DOI] [PubMed] [Google Scholar]
- Fujisawa T, Hattori T, Takahashi K, Kuboki T, Yamashita A, Takigawa M. Cyclic mechanical stress induces extracellular matrix degradation in cultured chondrocytes via gene expression of MMPs and interleukin-1. J Biochem. 1999;125:966–975. doi: 10.1093/oxfordjournals.jbchem.a022376. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/S0162-3109(00)00206-X. [DOI] [PubMed] [Google Scholar]
- Badger AM, Bradbeer JN, Votta B, Lee JC, Adams J, Griswold DE. Pharmacological profile of SB 20 a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- Badger AM, Griswold DE, Kapadia R, Blake S, Swift BA, Hoffman SJ, Stroup GB, Webb E, Rieman DJ, Gowen M, Boehm JC, Adams JL, Lee JC. Disease-modifying activity of SB 24 a selective inhibitor of p38 MAPK in rat adjuvant-induced arthritis. Arthritis Rheum. 2000;43:175–184. doi: 10.1002/1529-0131(200001)43:1<175::AID-ANR22>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Badger AM, Cook MN, Lark MW, Newman-Tarr TM, Swift BA, Nelson AH, Barone FC, Kumar S. SB 203580 inhibits p38 MAPK, nitric oxide production, and inducible nitric oxide synthase in bovine cartilage-derived chondrocytes. J Immunol. 1998;161:467–473. [PubMed] [Google Scholar]
- Clancy RM, Amin AR, Abramson SB. The role of nitric oxide in inflammation and immunity. Arthritis Rheum. 1998;41:1141–1151. doi: 10.1002/1529-0131(199807)41:7<1141::AID-ART2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross talk between smad, ERK1/2, and p38 MAPK pathways regulates TGF-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–14473. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F. p38 MAPK functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp Cell Res. 1999;250:351–363. doi: 10.1006/excr.1999.4535. [DOI] [PubMed] [Google Scholar]
- Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H, Matsumoto K, Nishida E. Involvement of the p38 MAPK pathway in TGF-β-induced gene expression. J Biol Chem. 1999;274:27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- Frenkel SR, Saadeh PB, Mehrara BJ, Chin GS, Steinbrech DS, Brent B, Gittes GK, Longaker MT. TGF-β superfamily members: role in cartilage modeling. Plast Reconstr Surg. 2000;105:980–990. doi: 10.1097/00006534-200003000-00022. [DOI] [PubMed] [Google Scholar]
- Studer RK, Levicoff E, Georgescu H, Miller L, Jaffurs D, Evans CH. Nitric oxide inhibits chondrocyte response to IGF-1: inhibition of IGF-IRβ tyrosine phosphorylation. Am J Physiol. 2000;279:C961–C969. doi: 10.1152/ajpcell.2000.279.4.C961. [DOI] [PubMed] [Google Scholar]
- Stefanovic-Racic M, Stadler J, Georgescu H, Evans CH. Nitric oxide synthesis and its regulation by rabbit synoviocytes. J Rheumatol. 1994;21:1892–1898. [PubMed] [Google Scholar]
- Lali FV, Hunt AE, Turner SJ, Foxwell BMJ. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in IL-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- Scherle PA, Pratta MA, Feeser WS, Tancula EJ, Arner EC. The effects of IL-1 on mitogen-activated protein kinases in rabbit articular chondrocytes. Biochem Biophys Res Commun. 1997;230:573–577. doi: 10.1006/bbrc.1996.5985. [DOI] [PubMed] [Google Scholar]
- Robbins JR, Thomas B, Tan L, Choy B, Arbiser JL, Berenbaum R, Goldring MB. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1β. Arthritis Rheum. 2000;43:2189–2201. doi: 10.1002/1529-0131(200010)43:10<2189::AID-ANR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (MMP 13) gene expression in chondrocytes requires p38, c-JUN N-terminal kinase, and nuclear factor κB. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Khatib A-M, Siegfried G, Quintero M, Mitrovic DR. The mechanism of inhibition of DNA synthesis in articular chondrocytes from young and old rats by nitric oxide. Nitric Oxide Biol Chem. 1997;1:218–225. doi: 10.1006/niox.1997.0126. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Lotz M. IL-1-induced nitric oxide inhibits chondrocyte proliferation via PGE2. Exp Cell Res. 1995;218:319–325. doi: 10.1006/excr.1995.1161. [DOI] [PubMed] [Google Scholar]
- Pap T, Muller-Ladner UN, Gay RE, Gay S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–367. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- Zhao M, Liu Y, Bao M, Kato Y, Han J, Eaton JW. Vascular smooth muscle cell proliferation requires both p38 and BMK1 MAP kinases. Arch Biochem Biophys. 2002;400:199–207. doi: 10.1016/S0003-9861(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Maher P. Phorbol esters inhibit fibroblast growth factor-2-stimulated fibroblast proliferation by a p38 MAP kinase dependent pathway. Oncogene. 2002;21:1978–1988. doi: 10.1038/sj.onc.1205268. [DOI] [PubMed] [Google Scholar]
- Diehl NL, Enslen H, Fortner KA, Merritt C, Stetson H, Charland C, Flavell RA, Davis RJ, Rinc M. Activation of the p38 MAPK pathway arrests cell cycle progression and differentiation of immature thymocytes in vivo. J Exp Med. 2000;191:321–334. doi: 10.1084/jem.191.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger AM, Roshak AK, Cook MN, Newman-Tarr TM, Swift BA, Carlson K, Connor JR, Lee JC, Gowen M, Lark MW, Kumar S. Differential effects of SB242235, a selective p38 MAPK inhibitor, on IL-1 treated bovine and human cartilage/chondrocyte cultures. Osteoarthritis Cartilage. 2000;8:434–443. doi: 10.1053/joca.1999.0319. [DOI] [PubMed] [Google Scholar]
- Lander HM, Jacovina AT, Davis RJ, Tauras JM. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem. 1996;271:19705–19709. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- Browning DD, McShane MP, Marty C, Ye RD. Nitric oxide activation of p38 MAPK in 293T fibroblasts requires cGMP-dependent protein kinase. J Biol Chem. 2000;275:2811–2816. doi: 10.1074/jbc.275.4.2811. [DOI] [PubMed] [Google Scholar]
- Chen K, Maines MD. Nitric oxide induces heme oxygenase-1 via mitogen-activated protein kinases ERK and p38. Cell Mol Biol. 2000;46:609–617. [PubMed] [Google Scholar]
- Lowe GN, Fu Y-H, McDougall S, Polendo R, Williams A, Benya PE, Hanh TJ. Effects of prostaglandins on DNA and aggrecan synthesis in the RCJ 3.1C5.18 chondrocyte cell line: role of second messengers. Endocrinology. 1996;137:2208–2216. doi: 10.1210/en.137.6.2208. [DOI] [PubMed] [Google Scholar]
- Schwartz Z, Gilley RM, Sylvia VL, Dean DD, Boyan BD. The effect of prostaglandin E2 on costochondral chondrocyte differentiation is mediated by cAMP and PKC. Endocrinology. 1998;139:1825–1834. doi: 10.1210/en.139.4.1825. [DOI] [PubMed] [Google Scholar]
- Studer RK, Decker K, Stubbs T, Chu CR. In Proceedings of the 49th Annual Meeting of the Orthopaedic Research Society: 2–5 February 2003; New Orleans, LA. Vol. 28. Chicago: Orthopaedic Research Society; 2003. p38 MAPK and COX-2 inhibition reverse IL-1 effects on human articular chondrocytes; p. 0017. [Google Scholar]
- Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 MAPK. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]