

Abstract
We investigated the role of the proinflammatory cytokine TNF-α, the second messenger C2-ceramide, and protein kinase R (PKR) in bovine articular cartilage degradation. Bovine articular cartilage explants were stimulated with C2-ceramide or TNF-α for 24 hours. To inhibit the activation of PKR, 2-aminopurine was added to duplicate cultures. Matrix metalloproteinase (MMP) expression and activation in the medium were analysed by gelatin zymography, proteoglycan release by the dimethylmethylene blue assay, and cell viability by the Cytotox 96® assay. C2-ceramide treatment of cartilage explants resulted in a significant release of both pro- and active MMP-2 into the medium. Small increases were also seen with TNF-α treatment. Incubation of explants with 2-aminopurine before TNF-α or C2-ceramide treatment resulted in a marked reduction in expression and activation of both MMP-2 and MMP-9. TNF-α and C2-ceramide significantly increased proteoglycan release into the medium, which was also inhibited by cotreatment with 2-aminopurine. A loss of cell viability was observed when explants were treated with TNF-α and C2-ceramide, which was found to be regulated by PKR. We have shown that C2-ceramide and TNF-α treatment of articular cartilage result in the increased synthesis and activation of MMPs, increased release of proteoglycan, and increased cell death. These effects are abrogated by treatment with the PKR inhibitor 2-aminopurine. Collectively, these results suggest a novel role for PKR in the synthesis and activation of MMPs and support our hypothesis that PKR and its activator, PACT, are implicated in the cartilage degradation that occurs in arthritic disease.
Keywords: articular cartilage, ceramide, matrix metalloproteinase, PKR, TNF-α
Introduction
Rheumatoid arthritis and osteoarthritis each affects a significant proportion of the population and the resulting loss of articular cartilage and inflammation causes severe pain and disability. There are no effective treatments for repair of the damaged articular cartilage in these diseases and while their likely aetiologies are very different, common pathways of degradation are important in both. Cartilage degradation occurs as a result of an imbalance of extracellular matrix proteinases and their inhibitors, in particular the matrix metalloproteinases (MMPs) and the tissue inhibitors of MMPs (TIMPs). Specifically, MMP-2 and -9 have been reported to be elevated in osteoarthritis cartilage [1,2] and within the synovial fluid of patients with rheumatoid arthritis [3], suggesting critical roles for these degradative enzymes in arthritic disease. In addition to its ability to degrade the cartilage matrix directly, MMP-2 plays a significant role in the activation of collagenases that are also strongly implicated in arthritic disease. MMPs and TIMPs, in turn, are regulated via induction of the early response genes c-fos and c-jun and by proinflammatory cytokines that are known to be involved in arthritic diseases [4,5], such as interleukin-1 and tumour necrosis factor α (TNF-α). TNF-α is capable of inducing cartilage catabolism in vitro [5] via increased MMP expression and activation [4] and is elevated in the synovial fluids from patients with arthritic disease [6,7]. It signals via a number of pathways including activation of sphingomyelinases, which degrade the membrane phospolipid sphingomyelin into phosphocholine and ceramide [8]. In turn, ceramide exerts its effects in a variety of ways depending on cell type (for a review see [9]), but several studies have implicated this second messenger in the regulation of MMPs [10-13]. In fibroblasts, the cell-permeable ceramide analogue C2-ceramide has been shown to stimulate mRNA expression for MMP-1 and MMP-3 through activation of signal pathways that ultimately lead to the induction of c-jun and c-fos and AP-1-dependent transcription of MMP genes [10]. In addition, triggering of the ceramide pathway in human keratinocytes results in overexpression of MMP-9 [11]. Recently it was shown that ceramide stimulates proteoglycan degradation and mRNA expression of MMP-1, -3, and -13 in rabbit articular cartilage [12,13]. This finding is important because it establishes a direct link between cartilage degradation and the ceramide pathway.
Previously, we have shown that the protein kinase R (PKR)-activating protein (PACT) [14] is up-regulated in regions of cartilage that subsequently develop osteoarthritis-like changes in vivo [15] and that PACT and PKR are involved in the TNF-α signalling pathway in articular chondrocytes [16]. The PKR pathway is also known to be activated by the sphingolipid ceramide [17]. This has led us to the hypothesis that TNF-α induces MMP expression in chondrocytes via ceramide-mediated activation of PKR. In the current study we have therefore investigated the role of TNF-α, the cell-permeable ceramide analogue C2-ceramide, and PKR in a well-characterised in vitro model of articular cartilage degradation [18]. We have used this model to activate degradative pathways in otherwise healthy cartilage to reveal potential signalling mechanisms that may be important in arthritic disease.
Materials and methods
Materials
All chemicals were obtained from Sigma (Poole, UK) unless otherwise stated and were of analytical grade. Recombinant human cytokines were purchased from Peprotech EC Ltd (London, UK). Culture medium consisted of Dulbecco's Modified Eagle's Medium with Glutamax-I (DMEM; Gibco BRL, Paisley, UK) containing 10 IU penicillin and 10 μg/ml streptomycin.
In vitro cartilage samples
Cartilage was taken from the metacarpophalangeal joint of 7-day-old bovine calves within 12 hours of slaughter using a scalpel and full-depth cartilage explants (20–70 mg) cultured overnight at 37°C in a humidified atmosphere of 5% CO2, 95% air, in 1 ml of DMEM supplemented with 10% fetal calf serum (FCS). DMEM containing FCS was removed and explants were incubated overnight in serum-free DMEM with insulin–transferrin–sodium selenite liquid supplement; they were then cultured in the presence of TNF-α (20–100 ng/ml) or cell-permeable C2-ceramide (10–100 μM) (Tocris, Bristol, UK) for 24 hours. These concentrations of TNF-α and C2-ceramide are known to stimulate catabolism in cartilage [12,13,18]. To inhibit the activation of PKR, 2-aminopurine (2-AP) (1–10 mM) was added to duplicate cultures 1 hour before the addition of treatments. This range of concentrations has been shown to inhibit activation of PKR in a number of studies [19-21] and did not affect chondrocyte viability in explants over 24 hours of culture (data not shown). Untreated explants or explants treated with 2-AP only served as controls. At the end of each experiment, explants were snap frozen in liquid nitrogen and media collected for analysis as described below.
Gelatin substrate zymography
MMP-2 and MMP-9 activities were detected in media samples by gelatin substrate zymography as described previously [22]. Briefly, gelatin (porcine skin, BDH, Poole. Dorset, UK) was incorporated into 7.5% (w/v) sodium dodecyl sulfate (SDS)–polyacrylamide gels at a final substrate concentration of 1.0 mg/ml. Media samples (20 μl, diluted 1 : 1 with sample buffer (0.12 M Tris/HCl (pH 6.8), containing 4% (w/v) SDS, 20% (v/v) glycerol, and 0.01% (w/v) bromophenol blue), a recombinant MMP-9 standard (1 μl) (Oncogene, Nottingham, UK), bovine-fibroblast-conditioned medium (5 μl) known to contain MMP-2 activity, and protein molecular weight markers (5 μl; molecular weight 14–200 kDa, BioRad, Hertfordshire, UK) were loaded onto the gels and resolved by electrophoresis. Then the gels were washed, with agitation, in 2.5% (v/v) Triton X-100 for 1 hour with at least three changes of solution and subsequently incubated for 16–20 hours at 37°C in 50 mM Tris/HCl (pH 7.8), containing 50 mM CaCl2 and 0.5 M NaCl. Gels were stained with Coomassie® Brilliant Blue R 250 (2 g/l) in distilled water, methanol, and acetic acid (4.5 : 4.5 : 1) for at least 1 hour and were destained in methanol, acetic acid, water (1 : 0.75 : 8.25) until the zones of proteolysis had cleared. The addition of molecular-weight markers and conditioned media to the gels facilitated identification of the enzymes and allowed comparisons between gels. The relative quantities of proteolytic enzymes were analysed by scanning densitometry (Umax Colour Scanner, GMbH, Willich, Germany) and NIH image software (National Institutes of Health, Bethesda, MD, USA) [23] and expressed as absorbance units per milligram wet weight of explant.
Proteoglycan release
Proteoglycan release into the medium of cartilage explant cultures was measured by the dimethylmethylene blue (DMMB) assay for sulfated glycosaminoglycan (sGAG), using chondroitin sulfate C from shark cartilage as a standard, as described previously [18]. After treatment, explants were digested in 300 μg/ml of papain in 20 mM sodium phosphate (pH 6.8), 1 mM EDTA, 2 mM dithiothreitol at 60°C for 1 hour and sGAG was determined as above. Differences in the levels of sGAG associated with culture treatment were expressed as micrograms of GAG per milligram wet weight of cartilage.
Cytotoxicity assay
Cell death was measured using the CytoTox 96® assay (Promega, Southampton, UK). This assay quantitatively measures lactate dehydrogenase present in the culture medium that has been released upon natural lysis of cells during the culture period [24]. Differences in the release of lactate dehydrogenase associated with culture treatment were expressed as absorbance units per milligram wet weight of cartilage.
Statistical analysis
Data are presented as mean ± SEM (n ≥ 4 cartilage explants) of a representative experiment and analysed, where appropriate, by Student's unpaired two-sample t-test or Mann–Whitney test (MMP-9 data). Differences were considered significant at P values less than 0.05. Where significance was not obtained, but the P value was considered informative, this is also shown. All experiments were performed three times with similar results.
Results
C2-ceramide and TNF-α increase both synthesis and activation of MMP-2 in cartilage explants
Articular cartilage explants were cultured in the presence of C2-ceramide or TNF-α with and without the addition of the PKR inhibitor 2-AP for 24 hours and media were analysed for MMP-2 and MMP-9 activity by gelatin substrate zymography. The presence of MMP-2 (Fig. 1a) and of MMP-9 (Fig. 1b) was confirmed by comparisons with standards known to contain both enzymes. An additional proteolytic band was detected that appeared to be increased by TNF-α treatment and migrated at a molecular weight previously reported to be a rat/murine-specific glycosylated form of MMP-2 [25].
Figure 1.
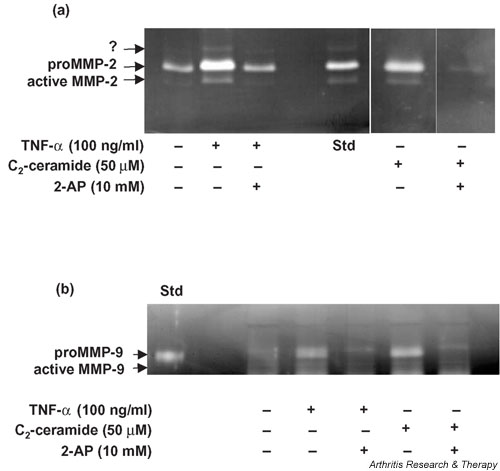
Detection of (a) MMP-2 and (b) MMP-9 activity in media collected from bovine articular cartilage explants treated with TNF-α or C2-ceramide for 24 hours. Medium was collected 24 hours after treatment of explants with 100 ng/ml TNF-α or 50 μM C2-ceramide in the presence or absence of the PKR inhibitor 2-AP (10 mM) and analysed by gelatin substrate zymography. A standard known to contain either MMP-2 or MMP-9 was included (Std). An unidentified enzyme is indicated by '?'. 2-AP, 2-aminopurine; MMP, matrix metalloproteinase; TNF, tumour necrosis factor.
The relative levels of MMP-2 were determined by scanning densitometry of the zymograms (Fig. 2a). C2-ceramide (50 μM) treatment of cartilage explants resulted in a significant release of both pro- (15.8 ± 1.8; P = 0.004) and active (3.4 ± 1.0; P = 0.036) MMP-2 into the culture medium over 24 hours in comparison with basal levels produced by unstimulated control explants (8.2 ± 1.0 and 1.4 ± 0.3 respectively). TNF-α (100 ng/ml) treatment resulted in a small, but not significant, increase in levels of proMMP-2 and active MMP-2 in the media.
Figure 2.
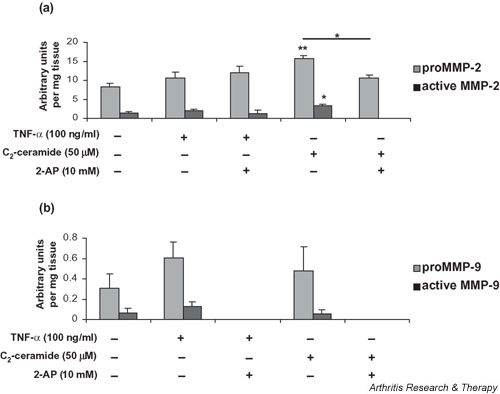
Quantitative analysis of (a) MMP-2 and (b)MMP-9 synthesis by bovine articular cartilage. Medium was collected 24 hours after treatment of explants with 100 ng/ml TNF-α or 50 μM C2-ceramide in the presence or absence of 2-AP (10 mM) and analysed by gelatin substrate zymography. The area (absorbance units) of substrate gel cleared by proMMP-9 and active MMP-9 was measured by scanning densitometry. Each area obtained was related to the gel clearance obtained with the fibroblast-conditioned-medium standard in order to facilitate comparisons between gels. Data shown are arbitrary units per milligram of wet weight of tissue and are expressed as means ± SEM. *Significantly different from control explants at P < 0.05; **P < 0.01. 2-AP, 2-aminopurine; MMP, matrix metalloproteinase; TNF, tumour necrosis factor.
C2-ceramide-induced regulation of MMP-2 is abrogated by inhibition of PKR
To determine a role for PKR in ceramide-induced regulation of MMP-2, we treated explant cultures with C2-ceramide in the presence of the PKR inhibitor 2-AP. With the addition of 10 mM 2-AP, the C2-ceramide-induced increase in proMMP-2 (15.8 ± 1.8) was significantly reduced (10.6 ± 0.9; P = 0.029) to near basal levels (8.2 ± 1.0) and activation of MMP-2 was completely abolished (Fig. 2a). A reduction in the activation of MMP-2 was also observed when 2-AP was added to explants treated with TNF-α, although this was not statistically significant (P = 0.109). Addition of 2-AP (1, 5, or 10 mM) to control cultures alone had no significant effect on MMP-2 (data not shown).
De novo synthesis and activation of MMP-9 is regulated by PKR
The relative levels of MMP-9 were determined by scanning densitometry (Fig. 2b). A two-fold increase in the production (P = 0.196) and activation (P = 0.123) of MMP-9 was observed in cartilage explants stimulated with TNF-α (100 ng/ml) in comparison with levels produced by unstimulated control cultures. C2-ceramide (50 μM) had no effect on the levels of MMP-9 produced by cartilage explants. However, in contrast, the addition of 2-AP (10 mM) to explant cultures completely abolished the production and activation of MMP-9 irrespective of ceramide or TNF treatment. Addition of 2-AP (1, 5, or 10 mM) to control cultures alone had no significant effect on MMP-9 (data not shown).
Treatment with TNF-α or C2-ceramide increases the release of proteoglycan from bovine articular cartilage
An increase in sGAG release from cartilage explants was observed following 24 hours of treatment with 50 ng/ml TNF-α (1.78 ± 0.5; P = 0.06), which was highly significant at 100 ng/ml TNF-α (2.44 ± 0.5; P < 0.001) in comparison with release from control cultures (0.73 ± 0.07) (Fig. 3a).
Figure 3.
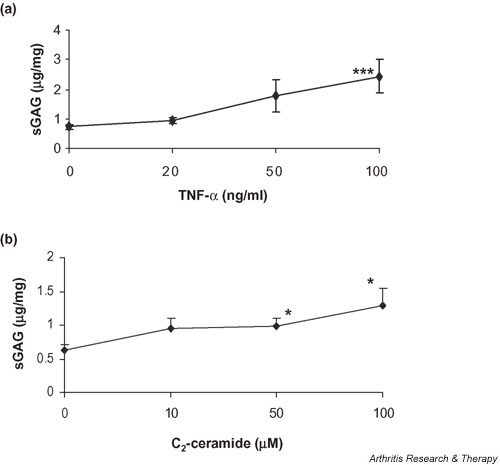
Treatment with TNF-α or ceramide induces proteoglycan release from articular cartilage. Cartilage explants were cultured for 24 hours in the presence of (a) TNF-α (0–100 ng/ml) or (b) C2-ceramide (0–100 μM) and media were analysed for release of sulfated GAGs by dimethylmethylene blue assay. Differences in the release of sGAG associated with culture treatment are expressed as micrograms GAG per milligram wet weight of cartilage. *Significantly different from untreated, control explants at P < 0.05; **P < 0.001. sGAG, sulfated glycosaminoglycan; TNF, tumour necrosis factor.
The release of sGAG into the medium increased 1.5-fold after treatment with C2-ceramide at 10 μM (P = 0.092) and 50 μM (P = 0.05) and two-fold with C2-ceramide at 100 μM (P = 0.029) (Fig. 3b).
2-aminopurine inhibits proteoglycan release from bovine articular cartilage
A role for PKR in C2-ceramide- and TNF-α-induced proteoglycan release was examined using the PKR inhibitor 2-AP. Control cultures were treated with 2-AP alone to determine the effects of the inhibitor on basal sGAG release (Fig. 4a). No effect was seen at 1 and 5 mM 2-AP. However, a two-fold inhibition of GAG release occurred when control cultures were treated with 10 mM 2-AP (P = 0.01).
Figure 4.
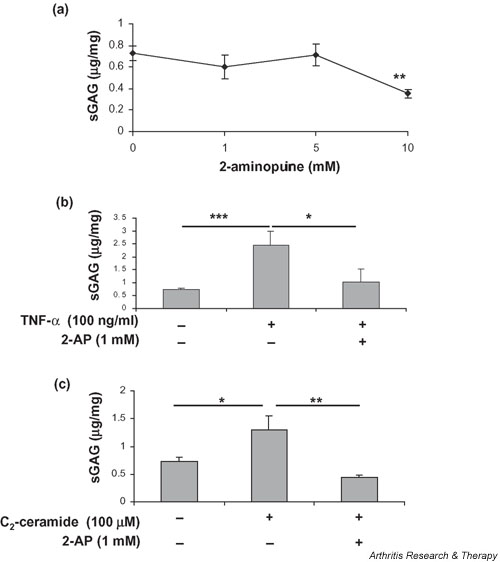
The PKR inhibitor 2-AP blocks both basal and TNF-α and C2-ceramide-induced proteoglycan release from articular cartilage. Bovine articular cartilage explants were cultured for 24 hours in the presence of (a) various concentrations of 2-AP (0–10 mM) alone, (b) TNF-α (100 ng/ml) with or without 2-AP (1 mM) or (c) C2-ceramide (100 μM) with or without 2-AP (1 mM). Medium was analysed for release of sGAGs by dimethylmethylene blue assay, expressed as micrograms of glycosaminoglycan released per milligram wet weight of cartilage. *Significantly different from control explants at P < 0.05; **P < 0.01; ***P < 0.001. 2-AP, 2-aminopurine; PKR, protein kinase R; sGAG, sulfated glycosaminoglycan; TNF, tumour necrosis factor.
A concentration of 1 mM 2-AP was therefore subsequently used to determine whether inhibition of PKR can abrogate the effects of ceramide or TNF-α on GAG release. The addition of 2-AP (1 mM) to explants 1 hour before treatment with TNF-α (100 ng/ml) significantly inhibited sGAG release into the medium (2-AP 1.03 ± 0.5 vs TNF-α 2.44 ± 0.5; P = 0.027) (Fig. 4b). A significant inhibition of sGAG release was also observed when explants were incubated with 2-AP (1 mM) before the addition of 100 μM C2-ceramide (2-AP 0.65 ± 0.14 vs ceramide 0.82 ± 0.1; P = 0.005) (Fig. 4c). These concentrations of TNF-α and C2-ceramide were chosen because they stimulated the largest proteoglycan release (Fig. 3).
TNF-α and C2-ceramide-induced increases in cartilage proteoglycan are PKR dependent
The amount of sGAG remaining within the cartilage explant after 24 hours of treatment with TNF-α and C2-ceramide in the presence and absence of 2-AP was determined by papain digestion and DMMB assay (Fig. 5). Interestingly, over this period, both TNF-α and C2-ceramide treatment resulted in a small (1.3-fold, P = 0.02, and 1.8-fold, P = 0.003, respectively) but significant increase in the sGAG content of the cartilage explants. Treatment with 2-AP alone (1 mM) had no effect on the amount of sGAG left within the explant and so this concentration was used to determine whether inhibition of PKR can abrogate the effect of TNF-α or C2-ceramide. The addition of 2-AP (1 mM) to explants 1 hour before the addition of TNF-α or C2-ceramide blocked their effect on sGAG synthesis within the cartilage.
Figure 5.
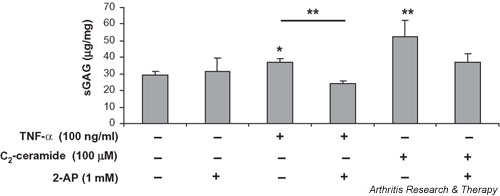
The PKR inhibitor 2-AP blocks TNF-α and C2-ceramide-induced proteoglycan synthesis. Cartilage explants were cultured for 24 hours in the presence of 2-AP (1 mM) alone, TNF-α (100 ng/ml) with or without 2-AP (1 mM) or C2-ceramide (100 μM) with or without 2-AP (1 mM). Explants were digested by papain (300 μg/ml) as described in Materials and methods and the amount of sGAG present determined as described above and expressed as micrograms glycosaminoglycan per milligram wet weight of cartilage. *Significantly different from control explants at P < 0.05; ** P < 0.01. 2-AP, 2-aminopurine; PKR, protein kinase R; sGAG, sulfated glycosaminoglycan; TNF, tumour necrosis factor.
TNF-α and C2-ceramide induce chondrocyte cell death via a mechanism involving PKR
The viability of articular cartilage explants after treatments was assessed by quantitatively measuring lactate dehydrogenase released into the medium, reflecting cell lysis during the culture period. Treatment of cartilage explants with 100 ng/ml TNF-α resulted in a 4.5-fold increase (P = 0.058) in cell death in comparison with control explants (Fig. 6a). The addition of 2-AP (10 mM) 1 hour before the treatment of cartilage with TNF-α (100 ng/ml) significantly inhibited TNF-α-induced cell death (5.6-fold; P = 0.002) and appeared to promote cell survival in comparison with levels of cell death in untreated controls (P = 0.032).
Figure 6.
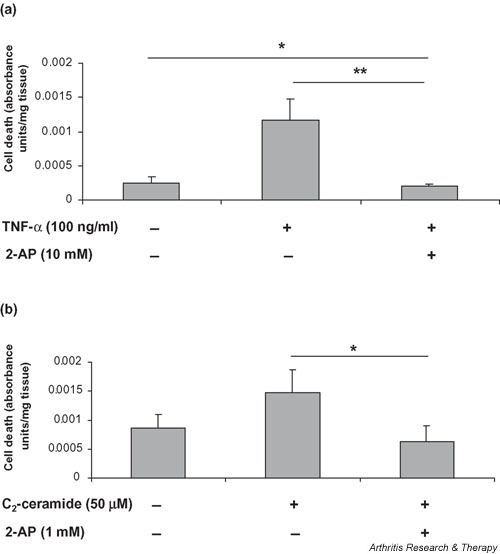
TNF-α and C2-ceramide induce cell death in articular cartilage via a mechanism involving PKR. Bovine articular cartilage explants were cultured for 24 hours in the presence of (a)TNF-α (100 ng/ml) or (b) C2-ceramide (50 μM). 2-AP (1 or 10 mM) was added 1 hour before the addition of treatments. Viability of explant tissue was determined using the CytoTox 96® assay, which quantitatively measures lactate dehydrogenase released into culture media upon cell death during the culture period. Data shown are absorbance units per milligrams of starting tissue and are expressed as means ± SEM. *Significantly different from control explants at P < 0.05; **P < 0.01. 2-AP, 2-aminopurine; PKR, protein kinase R; TNF, tumour necrosis factor.
C2-ceramide induced a 1.7-fold loss of cell viability at 50 μM (P = 0.141) (Fig. 6b). This loss became significant at 100 μM of C2-ceramide (P = 0.014) (data not shown). The addition of 2-AP (1 mM) before the addition of C2-ceramide abrogated the effect on cell death induced by C2-ceramide (2.3-fold; P = 0.05) and again appeared to promote cell survival.
Discussion
PKR is a serine/threonine protein kinase that can phosphorylate a limited number of cellular proteins, including the eukaryotic initiation factor 2α (eIF2α), resulting in a block on translation. Present at low levels in all cells, PKR is activated by double-stranded RNA, other polyanionic molecules such as heparin, and the protein activator PACT, and has been shown to be important in transcriptional pathways activated by specific cytokines [21,26], growth factors [27], and extracellular stresses [28]. PKR is involved in a number of cellular responses, including signal transduction, differentiation, and apoptosis [29-31], that may be involved in cartilage degradation. In the current study, we used the nucleoside analogue 2-AP as an inhibitor of PKR to investigate the role of PKR in the TNF-α- and ceramide-signalling pathways in cartilage degradation. The use of 2-AP as an inhibitor of PKR has been widely reported and there remains little doubt as to the importance of this compound in identifying PKR-dependent pathways in many cell types [17,19-21,32].
Since PKR has been shown to mediate TNF-α signalling in other cell types, and we had detected an up-regulation of PACT at the onset of osteoarthritis [15], we investigated whether PKR mediates TNF-α-induced degradative pathways in chondrocytes. Our data demonstrate that TNF-α treatment of bovine cartilage explants resulted in a small increase in the expression and activation of MMP-2 and -9. The low level of expression of MMP-9 within our control explants may represent activation of stress response pathways in chondrocytes that have been disrupted at the explant edge, since MMP-9 is not constitutively expressed in chondrocytes and its presence usually represents a degradative or diseased state [1]. The complete loss of expression of both pro- and active MMP-9 in the presence of 2-AP is an interesting finding and suggests that PKR may be a critical regulator of TNF/ceramide induced MMP-9 expression, although this will require further investigation. Inactivation of PKR signalling, therefore, due to TNF/ceramide is likely to abrogate MMP-9 expression below control levels. This regulation of MMP-9 may be through the nuclear factor κB (NFκB) response element in the promoter region of its gene, since NFκB is known to be a transcriptional activator of MMP expression in chondrocytes [33,34] and PKR is known to mediate TNF-α activation of NFκB in a number of cell types [21,26].
TNF-α signalling has been linked to the ceramide pathway in other cell types [9] and ceramide has been shown to increase MMP expression and activation in rabbit cartilage [12,13]. Importantly, binding of TNF-α to its cell-surface receptor (TNF-R55) activates neutral sphingomyelinase, which in turn releases ceramide as a second messenger [10]. TNF-R55 is known to be increased in arthritic disease [5]. We therefore tested whether the catabolic effects of C2-ceramide are also mediated through PKR in cartilage. Our studies show that C2-ceramide increased pro- and active MMP-2 and -9 in bovine cartilage explants and that this effect was significantly diminished (MMP-2) or completely abolished (MMP-9) by treatment with the PKR inhibitor 2-AP. The mechanism of MMP-2 activation observed in our study remains to be elucidated, but involvement of membrane type 1 MMP (MT1-MMP) seems likely, given that previous studies in hepatic myofibroblasts show that ceramide induces apoptosis and MMP-2 activation through increased MT1-MMP expression [35]. In addition, studies within our own laboratory suggest that C2-ceramide treatment of chondrocytes increases levels of MT1-MMP (data not shown). The increased expression and activation of MMP-2 and -9, induced by C2-ceramide treatment of chondrocytes, is a novel finding and the significant inhibitory effects of 2-AP provide compelling evidence that PKR is a critical mediator of this response in chondrocytes. Studies are being carried out to determine whether the effects of inhibiting PKR on MMPs are mediated by decreased levels of MT1-MMP or increased levels of TIMPs.
Previous work has shown that treatment of cartilage explants with TNF-α [36] or ceramide [12,13] results in proteoglycan release. Our data confirmed this in bovine cartilage explants, in which either TNF-α or ceramide treatment for 24 hours significantly enhanced proteoglycan release. Somewhat surprisingly, exposure to TNF/ceramide over 24 hours resulted in an increase in the amount of sGAG left within the cartilage, a finding that is suggestive of an anabolic response. Previous studies have shown that exposure to TNF-α or C2-ceramide over periods of 3 days or longer results in a net loss of proteoglycan from the cartilage, which is reflected in the increased levels found within the culture medium [13,18]. The short-term period of culture used in this study may reflect events that occur in the early stages of osteoarthritis where, as a response to damaged matrix, chondrocytes show enhanced production of collagen and proteoglycans [37].
Both TNF-α- and C2-ceramide-induced proteoglycan release from, and synthesis within, the explants was significantly reduced by treatment with the PKR inhibitor 2-AP at a concentration (1 mM) that does not affect constitutive proteoglycan release/synthesis. This suggests a novel role for PKR in proteoglycan metabolism in chondrocytes. Treatment of cartilage explants with a higher concentration of 2-AP (10 mM), in the absence of other treatments, blocked basal sGAG release without affecting cell viability. Since 10 mM 2-AP did not affect basal levels of MMP production, sGAG release must be due to the activity of alternative enzymes such as the aggrecanases ADAMTS4 and 5. Here we have shown a novel mechanism for proteoglycan catabolism involving PKR that may be important in cartilage degradation. Others have shown that ceramide stimulates aggrecanase-mediated degradation of proteoglycans in articular cartilage, but the mechanism of action remains unknown [13]. Our future studies will therefore investigate whether this occurs via the PKR signalling pathway.
In the current study, we aimed to determine whether TNF-α and C2-ceramide can induce cell death in our in vitro model of cartilage degradation and whether any such effect is mediated by PKR in chondrocytes. Changes in chondrocyte proliferation and viability are thought to be important in arthritic disease (for a review see [38]) and in animal models of osteoarthritis [39], although the role of apoptosis in arthritis remains controversial. In other cell types, PKR activation has been reported to mediate TNF-α- and ceramide-induced apoptosis [17,40-42]. We show that both TNF-α and C2-ceramide increase chondrocyte death and that this death can be significantly reduced by the addition of 2-AP, confirming a role for PKR in this event. Previous studies have shown that TNF-α treatment of primary chondrocytes and chondrocyte cell lines results in increased apoptosis and caspase activity [43,44]. Since we have previously shown that TNF-α increases PACT protein expression and phosphorylation of PKR and eIF2α in chondrocytes [16] and that this is known to trigger the apoptotic pathway in other cell types [40,41,45], it is tempting to speculate that the cell death observed in this current study is due to apoptosis. This will be confirmed in future studies.
Conclusion
Our data have shown that PKR potentially is an important mediator of degradative and death pathways in chondrocytes. We hypothesise that the TNF-α signalling pathway depicted in Fig. 7 that has been elucidated for other cell types also operates in chondrocytes. Since both the expression/activation of degradative enzymes and the number of chondrocytes committed to cell death are important determinants of cartilage integrity, our results suggest a pivotal role for the PKR pathway in arthritic disease onset and progression that requires further investigation.
Figure 7.
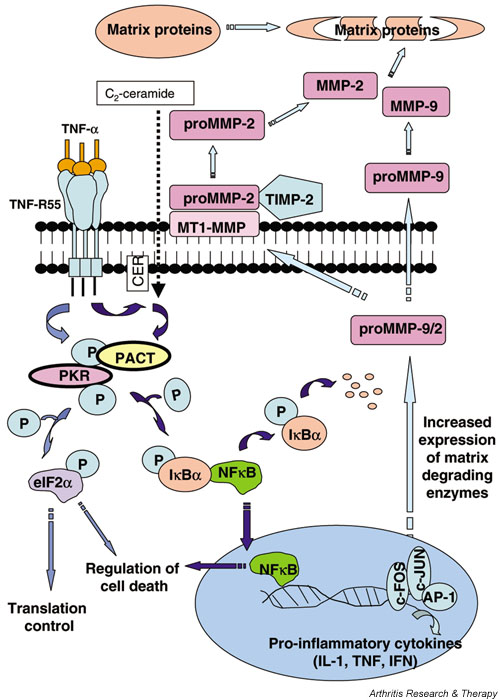
The potential role of PKR in cartilage degradation. Results from the current study have led us to hypothesise that TNF-α-induced degradative pathways in cartilage may be mediated via activation of PKR. TNF-α binding to its receptor (TNF-R55) may activate PKR either directly or through ceramide (CER) to increase transcription and activation of MMPs via induction of NFκB and early response genes (c-fos and c-jun). Activation of PKR and subsequent phosphorylation of eIF2α would lead to an inhibition of protein synthesis and increased apoptosis, which may also affect cartilage integrity. Increased expression and activation of MMPs, in the absence of a corresponding increase in their inhibitors, would shift the balance of homeostasis towards matrix catabolism. AP-1, activator protein-1; eIF2α, eukaryotic initiation factor 2α ; IκBα, inhibitor kappa B alpha; MMP, matrix metalloproteinase; NFκB, nuclear factor κB; P, phosphorylated; PACT, PKR-activating protein; PKR, protein kinase R; TIMP, tissue inhibitors of MMP; TNF, tumour necrosis factor; TNF-R55, tumour necrosis factor receptor-55.
Competing interests
None declared.
Abbreviations
2-AP = 2-aminopurine; DMEM = Dulbecco's Modified Eagle's Medium; DMMB = dimethylmethylene blue; eIF2α = eukaryotic initiation factor 2α; FCS = fetal calf serum; MMP = matrix metalloproteinase; MT1-MMP = membrane type 1 MMP; NFκB = nuclear factor κB; PACT = PKR-activating protein; PKR = protein kinase R; SDS = sodium dodecyl sulfate; sGAG = sulfated glycosaminoglycan; TIMPs = tissue inhibitors of MMPs; TNF-α = tumour necrosis factor α; TNF-R55 = tumour necrosis factor receptor-55.
Acknowledgments
Acknowledgements
The authors would like to thank the Arthritis Research Campaign for funding this work (Grant number M0650) and Dr Emma Blain for her expertise.
References
- Dean DD, Martel-Pelletier J, Pelletier JP, Howell DS, Woessner JF., Jr Evidence for metalloproteinase and metalloproteinase inhibitor imbalance in human osteoarthritic cartilage. J Clin Invest. 1989;84:678–685. doi: 10.1172/JCI114215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner T, Zien A, Gehrsitz A, Gebhard PM, McKenna L. Anabolic and catabolic gene expression pattern analysis in normal versus osteoarthritic cartilage using complementary DNA-array technology. Arthritis Rheum. 2001;44:2777–2789. doi: 10.1002/1529-0131(200112)44:12<2777::AID-ART465>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Yoshihara Y, Nakamura H, Obata K, Yamada H, Hayakawa T, Fujikawa K, Okada Y. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis. Ann Rheum Dis. 2000;59:455–461. doi: 10.1136/ard.59.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring MB. Osteoarthritis and cartilage: The role of cytokines. Curr Rheumatol Rep. 2000;2:459–465. doi: 10.1007/s11926-000-0021-y. [DOI] [PubMed] [Google Scholar]
- Westacott CI, Barakat AF, Wood L, Perry MJ, Neison P, Bisbinds I, Armstrong L, Millar AB, Elson CJ. Tumour necrosis factor-alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthritis Cartilage. 2000;8:213–221. doi: 10.1053/joca.1999.0292. [DOI] [PubMed] [Google Scholar]
- Westacott CI, Whicher JT, Barnes IC, Thompson D, Swan AJ, Dieppe PA. Synovial fluid concentration of five different cytokines in rheumatic diseases. Ann Rheum Dis. 1990;49:676–681. doi: 10.1136/ard.49.9.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. The role of cytokines in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 1999;Suppl 2:3–7. [PubMed] [Google Scholar]
- Serhan CN, Haeggstrom JZ, Leslie CC. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J. 1996;10:1147–1158. doi: 10.1096/fasebj.10.10.8751717. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol Res. 2003;47:383–392. doi: 10.1016/S1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Reunanen N, Westermarck J, Hakkinen L, Holmstrom TH, Elo I, Eriksson JE, Kahari VM. Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J Biol Chem. 1998;273:5137–5145. doi: 10.1074/jbc.273.9.5137. [DOI] [PubMed] [Google Scholar]
- Buisson-Legendre N, Bernard P, Bobichon H, Emonard H, Schneider C, Maquart FX, Haye B, Hornebeck W. Involvement of the 92-kDa gelatinase (matrix metalloproteinase-9) in the ceramide-mediated inhibition of human keratinocyte growth. Biochem Biophys Res Commun. 1999;260:634–640. doi: 10.1006/bbrc.1999.0565. [DOI] [PubMed] [Google Scholar]
- Sabatini M, Rolland G, Leonce S, Thomas M, Lesur C, Perez V, de Nanteuil G, Bonnet J. Effects of ceramide on apoptosis, proteoglycan degradation, and matrix metalloproteinase expression in rabbit articular cartilage. Biochem Biophys Res Commun. 2000;267:438–444. doi: 10.1006/bbrc.1999.1983. [DOI] [PubMed] [Google Scholar]
- Sabatini M, Thomas M, Deschamps C, Lesur C, Rolland G, de Nanteuil G, Bonnet J. Effects of ceramide on aggrecanase activity in rabbit articular cartilage. Biochem Biophys Res Commun. 2001;283:1105–1110. doi: 10.1006/bbrc.2001.4920. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SJ, Duance VC, Mason DJ. Identification of changes in chondrocyte gene expression in an in vivo animal model of osteoarthritis [abstract] Osteoarthritis Cartilage. 2000;Suppl B:S33. [Google Scholar]
- Gilbert SJ, Duance VC, Mason DJ. Tumour necrosis factor alpha up-regulates protein kinase R (PKR)-activating protein (PACT) and increases phosphorylation of PKR and eukaryotic initiation factor 2-alpha in articular chondrocytes. Biochem Soc Trans. 2002;30:886–889. doi: 10.1042/bst0300886. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Gao FG, Blalock WL, Deng X, Stratford May W. Ceramide regulates protein synthesis by a novel mechanism involving the cellular PKR activator RAX. J Biol Chem. 2001;276:11754–11758. doi: 10.1074/jbc.M011400200. [DOI] [PubMed] [Google Scholar]
- Little CB, Flannery CR, Hughes CE, Mort JS, Roughly PJ, Dent C, Caterson B. Aggrecanase versus matrix metalloproteinases in the catabolism of the interglobular domain of aggrecan in vitro. Biochem J. 1999;344:61–68. doi: 10.1042/0264-6021:3440061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pataer A, Vorburger SA, Barber GN, Chada S, Mhashilkar AM, Zou-Yang H, Stewart AL, Balachandran S, Roth JA, Hunt KK, Swisher SG. Adenoviral transfer of the melanoma differentiation-associated gene 7 (mda7) induces apoptosis of lung cancer cells via up-regulation of the double-stranded RNA-dependent protein kinase (PKR) Cancer Res. 2002;62:2239–2243. [PubMed] [Google Scholar]
- Osman F, Jarrous N, Ben-Asouli Y, Kaempfer R. A cis-acting element in the 3'-untranslated region of human TNF-alpha mRNA renders splicing dependent on the activation of protein kinase PKR. Genes Dev. 1999;1324:3280–3293. doi: 10.1101/gad.13.24.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshire JL, Williams BRG, Baldwin AS., Jr Involvement of double-stranded RNA-activated protein kinase in the synergistic activation of nuclear factor-kB by tumour necrosis factor-α and γ-interferon in preneuronal cells. J Biol Chem. 1999;274:4801–4806. doi: 10.1074/jbc.274.8.4801. [DOI] [PubMed] [Google Scholar]
- Blain EJ, Gilbert SJ, Wardale RJ, Capper SJ, Mason DJ, Duance VC. Up-regulation of matrix metalloproteinase expression and activation following cyclical compressive loading of articular cartilage in vitro. Arch Biochem Biophys. 2001;396:49–55. doi: 10.1006/abbi.2001.2575. [DOI] [PubMed] [Google Scholar]
- Vaughan-Thomas A, Gilbert SJ, Duance VC. Elevated levels of proteolytic enzymes in the aging human vitreous. Invest Ophthalmol Vis Sci. 2000;41:3299–3304. [PubMed] [Google Scholar]
- Milner JM, Rowan AD, Elliott SF, Cawston TE. Inhibition of furin-like enzymes blocks interleukin-1alpha/oncostatin M-stimulated cartilage degradation. Arthritis Rheum. 2003;48:1057–1066. doi: 10.1002/art.10873. [DOI] [PubMed] [Google Scholar]
- Lalu MM, Csonka C, Giricz Z, Csont T, Schulz R, Ferdinandy P. Preconditioning decreases ischemia/reperfusion-induced release and activation of matrix metalloproteinase-2. Biochem Biophys Res Commun. 2002;296:937–941. doi: 10.1016/S0006-291X(02)02019-3. [DOI] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Mogensen TH, DIDonato JA, Williams BRG. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol Cell Biol. 2000;20:1278–1290. doi: 10.1128/MCB.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundschau LJ, Faller DV. Platelet-derived growth factor signal transduction through the interferon-inducible kinase PKR. J Biol Chem. 1995;270:3100–3106. doi: 10.1074/jbc.270.7.3100. [DOI] [PubMed] [Google Scholar]
- Ito T, Jagus R, May WS. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependant protein kinase. Proc Natl Acad Sci USA. 1994;91:7455–7459. doi: 10.1073/pnas.91.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpick BW, Graziano V, Schneider D, Maitra RK, Lee X, Williams BRG. Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- Chu W-M, Ostertag D, Li Z-W, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M. JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity. 1999;11:721–731. doi: 10.1016/s1074-7613(00)80146-6. [DOI] [PubMed] [Google Scholar]
- Clemens MJ, Elia A. The double-stranded RNA–dependant protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–524. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- Williams BR. Signal integration via PKR. Sci STKE. 2001;89:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- Borden P, Heller RA. Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit Rev Eukaryot Gene Expr. 1997;7:159–178. doi: 10.1615/critreveukargeneexpr.v7.i1-2.90. [DOI] [PubMed] [Google Scholar]
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Preaux AM, D'ortho MP, Bralet MP, Laperche Y, Mavier P. Apoptosis of human hepatic myofibroblasts promotes activation of matrix metalloproteinase-2. Hepatology. 2002;363:615–622. doi: 10.1053/jhep.2002.35279. [DOI] [PubMed] [Google Scholar]
- Curtis CL, Hughes CE, Flannery CR, Little CB, Harwood JL, Caterson B. n-3 fatty acids specifically modulate catabolic factors involved in articular cartilage degradation. J Biol Chem. 2000;275:721–724. doi: 10.1074/jbc.275.2.721. [DOI] [PubMed] [Google Scholar]
- van der Kraan PM, van den Berg WB. Anabolic and destructive mediators in osteoarthritis. Curr Opin Clin Nutr Metab Care. 2000;3:205–211. doi: 10.1097/00075197-200005000-00007. [DOI] [PubMed] [Google Scholar]
- Aigner T, Kim HA. Apoptosis and cellular vitality. Issues in osteoarthritic cartilage degeneration. Arthritis Rheum. 2002;46:1986–1996. doi: 10.1002/art.10554. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Takahashi K, Amiel D, Coutts RD, Lotz M. Chondrocyte apoptosis and nitric oxide production during experimentally induced osteoarthritis. Arthritis Rheum. 1998;41:1266–1274. doi: 10.1002/1529-0131(199807)41:7<1266::AID-ART18>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Der SD, Yang YL, Weissmann C, Williams BR. A double-stranded RNA-activated protein kinase-dependant pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:8841–8845. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- Yeung MC, Liu J, Lau AS. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc Natl Acad Sci USA. 1996;93:12451–12455. doi: 10.1073/pnas.93.22.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttal ME, Nadeau DP, Fisher PW, Wang F, Keller PM, DeWolf WE, Goldring MB, Badger AM, Lee D, Levy MA, Gowen M, Lark MW. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J Orthop Res. 2000;18:356–363. doi: 10.1002/jor.1100180306. [DOI] [PubMed] [Google Scholar]
- Aizawa T, Kon T, Einhorn TA, Gerstenfeld LC. Induction of apoptosis in chondrocytes by tumour necrosis factor-alpha. J Orthop Res. 2001;19:785–796. doi: 10.1016/S0736-0266(00)00078-4. [DOI] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced, double-stranded RNA activated protein kinase, PKR. J Biol Chem. 2000;275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]