

Abstract
CPT-11 (irinotecan), a DNA topoisomerase I inhibitor is one of the main treatments for colorectal cancer. The main dose limiting toxicities are neutropenia and late onset diarrhea. Though neutropenia is manageable, CPT-11 induced diarrhea is frequently severe, resulting in hospitalizations, dose reductions or omissions leading to ineffective treatment administration. Many potential agents have been tested in preclinical and clinical studies to prevent or ameliorate CPT-11 induced late onset diarrhea. It is predicted that prophylaxis of CPT-11 induced diarrhea will reduce sub-therapeutic dosing as well as hospitalizations and will eventually lead to dose escalations resulting in better response rates. This article reviews various experimental agents and strategies employed to prevent this debilitating toxicity. Covered topics include schedule/dose modification, intestinal alkalization, structural/chemical modification, genetic testing, anti-diarrheal therapies, transporter (ABCB1, ABCC2, BCRP2) inhibitors, enzyme (β-glucuronidase, UGT1A1, CYP3A4, carboxylesterase, COX-2) inducers and inhibitors, probiotics, antibiotics, adsorbing agents, cytokine and growth factor activators and inhibitors and other miscellaneous agents.
Keywords: Chemotherapy induced diarrhea, CPT-11 (irinotecan), diarrhea prevention and control, CPT-11 metabolism, toxicity, antidiarrheals/pharmacology, enzyme inhibitors/pharmacology
1. INTRODUCTION
Colorectal cancer (CRC), with an estimate of 51,690 deaths in 2012 is ranked second of top five causes of cancer related deaths [1]. One of the main treatments for this devastating disease is CPT-11 (irinotecan, 7-ethyl-10-[4-[1-piperidino]-1-piperidino] carbonyloxycamptothecin, Camptosar®, Camptothecin-11, Campto®) (Fig. 1), a semi synthetic camptothecin (Fig. 2) analog with topoisomerase-1 inhibitory action [2, 3]. This drug has been approved for treatment of metastatic CRC either alone or in combination with 5-FU and other relevant drugs (e.g., oxaliplatin, cape-citabine, bevacizumab, etc.) [4]. In addition, CPT-11 has shown activity in esophageal/gastric, lung, pancreatic, ovarian, leukemia, lymphoma, cervical, breast, head and neck and brain tumors [5, 6].
Fig. 1.
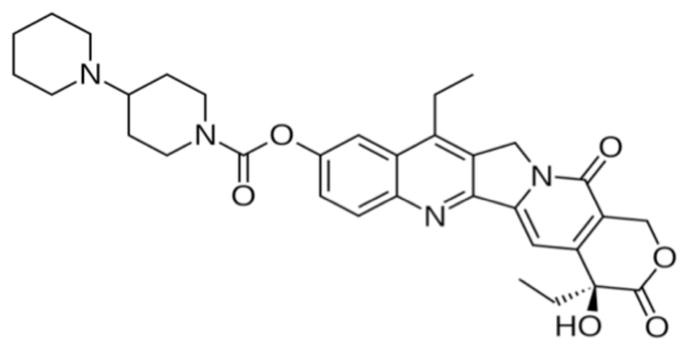
CPT-11.
Fig. 2.
Camptothecin.
The main dose-limiting side effects of CPT-11 include diarrhea and neutropenia [7]. Neutropenia is common, of short duration and there are treatments (e.g., use of growth factors such as G-CSF) that can ameliorate or prevent the condition [8]. Diarrhea, on the otherhand, continues to be a serious, debilitating and life threatening side effect particularly with high dose [9] and combination regimens (CPT-11 with bolus 5-FU) [10].
The overall incidence of clinically significant diarrhea with CPT-11 is up to 87%, with grade 3 or 4 diarrhea (National Cancer Institute Common Toxicity Criteria) ranging between 30–40% [9, 11–13]. Most patients require active treatment with anti-motility agents (e.g., loperamide, octreotide) and intravenous hydration. While most recover from these episodes, around 10% of the patients require prolonged hospitalization. Patients eventually require CPT-11 dose delays, omissions and dose reductions that impact the efficacy of the drug. Patients (~ 10%) with co-existent diarrhea, dehydration and neutropenia are at the highest risk of death (~3.5%) [10, 14].
In the past few years extensive research has been done on formulating novel strategies and agents to prophylax against this debilitating side effect. In this article we summarize the current state-of-art in prophylaxis against CPT-11 induced diarrhea [CID].
2. MATERIALS AND METHODS
We conducted an extensive search of the PUBMED database from 1985 to 2010 with keyword “CPT-11 and diarrhea”. All relevant articles were thoroughly reviewed and additional search was performed on referenced literature. Major focus was put on data related to the novel approaches to prevent CPT-11 induced late-onset diarrhea (CID).
3. METABOLISM
The metabolism of CPT-11 is extremely complex involving a number of enzymes and transporters. CPT-11 is a pro-drug and undergoes carboxylesterase (CES) mediated hydrolysis of carbamate bond between camptothecin moiety and dipiperidino side chain to form, 100–1000 times more cytotoxic metabolite SN-38 (Fig. 3). The conversion to SN-38 occurs primarily but not limited to, in the liver via three human carboxyesterases CES1A1, CES 2 and CES3 with different catalytic efficiency (CES2 > CES1A1 ≫ CES3) [3, 15]. CES-2 has been shown to be the major enzyme involved in CPT-11 hydrolysis and in addition to the liver; it is highly expressed in heart, skeletal muscle, colon, spleen and kidneys [15, 16].
Fig. 3.
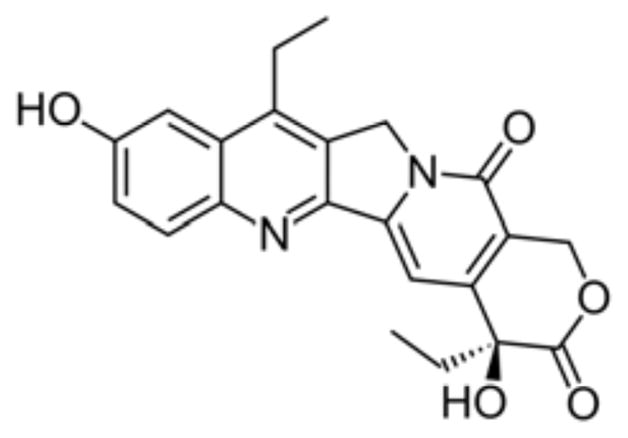
SN-38.
The second important metabolic pathway of CPT-11 is through cytochrome P450. CPT-11 preferentially undergoes CYP3A4 mediated oxidation of the terminal piperidine ring to form 7-ethyl-10[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxy-camptothecine (APC) and cleavage to form 7-ethyl-10-(4-amino-1-piperidino) carbonyloxy-camptothecine (NPC). CYP3A4 and CYP3A5 also generate several other clinically non significant minor metabolites [17–20]. Both APC and NPC are much less cytotoxic [17, 21]. However, CES and/or liver microsomes can hydrolyze NPC to SN-38 but not APC (or very poorly) [15, 17, 21].
Subsequently SN-38 is conjugated to inactive, non-toxic SN-38 glucuronide (SN-38G) by UGT1A1, UGT1A9 and extrahepatic UGT1A7 [22–24]. CPT-11 can be converted to SN-38 by intestinal CES [25]. Also SN-38G can be deglucuronidated to SN-38 by bacterial β-glucuronidase [26]. CPT-11 and SN-38 can both undergo enterohepatic circulation [27]. All these can potentiate SN-38 induced intestinal mucosal damage and toxicity.
The transportation of CPT-11 and its metabolites outside the cells include many transporters including ABCB1 [multidrug resistance (MDR1); P-glycoprotein (PgP)], ABCC1 [multi-drug resistance protein (MRP1)], ABCC2 [canalicular multispecific organic anion transporter (C-MOAT); MRP2] and ABCG2 [breast cancer resistance protein (BCRP)] [3]. They are known to influence the metabolism of CPT-11 and its metabolites by way of their involvement in intestinal absorption and excretion by biliary and renal mechanisms [3]. The predominant excretion route is feces (64%), followed by urine (32%). CPT-11 is the predominant circulating component in plasma (55%) and the chief excretion product in feces, urine and bile. The other most significant metabolites in feces are SN-38 and APC. However in urine and bile, the other most significant metabolites are APC and SN-38G. The mean plasma half lives of CPT-11, SN-38 and SN-38G are 14.6 h, 28.5 h and 35.5 h respectively [28].
4. CPT-11 ASSOCIATED DIARRHEA
CPT-11 is associated with two types of diarrhea. The early onset diarrhea (beginning within 24 hours) is mild, transient and part of a broader cholinergic syndrome which includes symptoms like gastrointestinal cramps, diaphoresis, salivation, visual disturbances, lacrimation, facial flushing, nausea, vomiting, miosis and rhinorrhea [11, 29]. It can be prevented by intravenous administration of 0.25–1 mg of atropine [11, 12, 29]. Premedication with diphenhydramine, ondansetron and scopolamine can also be used to decrease toxicity [30, 31].
CPT-11 induced late onset diarrhea (CID) begins more than 24 hours of infusion [11, 32]. CID is inconsistent, unpredictable, non-cumulative, schedule and dose dependent, with wide interpatient and intrapatient variability [33, 34]. It is often prolonged, leading to dehydration and electrolyte imbalance, making it a life-threatening condition. The underlying mechanism for CID is still not clearly defined but most likely involves direct intestinal mucosal damage due to SN-38 induced cytotoxicity [35]. CPT-11 has shown to injure tight junction proteins claudin-1 and occludin, damaging intestinal barrier and inducing bacterial translocation [36].
Intestinal mucosal changes as observed in animals after CPT-11 administration are described in Table 1.
Table 1.
Intestinal Mucosal Changes in Rats and Mice After CPT-11 Administration.
| S. No. | Target | Changes | References |
|---|---|---|---|
| 1 | Jejunum and ileum | Increased apoptosis, epithelial atrophy, infiltration of inflammatory cells in mucosal tissue, crypts hypoplasia, degenerative enterocytes within crypts, goblet cell alteration (or not), decreased Muc2 and Muc4 expression, reduction of villous height and crypts length in jejunum. Similar severe epithelial vacuolation and apoptosis in ileum. | [37–40] |
| 2 | Colon | Increased apoptosis, crypt hypoplasia, crypt dilation, presence of degenerative enterocytes within crypts, reduction in crypt length, goblet cell alteration (decrease or hyperplasia) and increased colonic mucosal/mucin secretion. caecal wall thickening, edema, hemorrhage and formation of a pseudomembrane like substance Increased PGE2, TXA2 and COX-2 expression. Decreased Muc2 expression. Increase in saturated fatty acids and decrease in mucosal diamine oxidase activity, total monounsaturated fatty acids, polyunsaturated fatty acids (eicosapentaenoic acid, docosahexaenoic acid) and eicosapentaenoic acid/arachidonic acid ratio. |
[37, 38, 40] [41] [40, 42–44] [45] |
SN-38 induced cytotoxicity is influenced by intestinal CES [25], bacterial β-glucuronidase [26] and enterohepatic recycling [27] all of which increase the intestinal concentration of the cytotoxic metabolite SN-38. Intestinal microflora also plays a major role in CPT-11 induced gastrointestinal toxicities as germ free mice have been found to be resistant to CID [46] and alteration to increased β-glucuronidase producing bacteria like E. coli after CPT-11 administration exacerbate CID [47]. CPT-11 administration is associated with increased PGE2, TXA2 and COX-2 expression in rat colon that play a key role in water, mucous and electrolyte imbalance in colon [42–44]. CPT-11 also increases nuclear factor-κb (NF-κb), cytokines (TNF-α, IL-1β, IL-6, and KC levels) and myeloperoxidase activity in intestinal tissue which may be involved in pathogenesis of CID [37, 48].
5. STRATEGIES TO BLOCK DELAYED DIARRHEA
Different prophylactic and curative strategies have been hypothesized and tested in animal models and clinical studies to block or treat CID. Prophylactic avoidance of CPT-11 induced diarrhea has been judged as the best strategy to prevent this serious drug complication [49]. Such strategies can improve the safety profile of the drug, cut down hospitalization costs, improve quality of life and can even lead to further dose escalations and improve efficacy with better tumor responses.
5.1. Schedule and Dose Modification
Schedule and dose modification has been one of the earliest strategies tested to alleviate CID. In rats same total dose of CPT-11 (240 mg/kg) showed more severe diarrhea with high dosage and alleviation with protracted schedule. The dose of 60 mg/kg once daily for 4 days induced severe diarrhea. However in comparison 30 mg/kg twice daily for 4 days with a 9 hour interval showed less diarrheal symptoms. Doses of 30 mg/kg for 8 days and 40 mg/kg for 6 days showed hardly any diarrhea [50]. Similarly with the same total dose, alleviation of incidence and severity of serious diarrhea was observed with the extension of infusion time in rats which correlated with decreased Cmax of CPT-11 (but not of SN-38) [41].
Therefore a decrease in Cmax of CPT-11, via low consecutive doses with an extension in infusion time will decrease incidence and severity of diarrhea. Although this approach will lower the magnitude of toxicities and improve quality of life, it might be less effective due to the decreased Cmax of CPT-11 theoretically leading to reduced antitumor activity. However, in rat xenograft models at the same total dose, CPT-11 has shown more significant antitumor activity with repeated administration [51]. Also due to S-phase inhibitory action, prolonged low dose CPT-11 infusion should have an appreciable efficacy as antitumor agent [52–54]. However in human trials even with low dose continuous intravenous infusion grade 3 adverse event of diarrhea have been observed [53–56].
A novel CPT-11 administration schedule was employed in treating 19 ovarian cancer patients. The initial administered dose was 70 mg/m2 followed by increasing doses up to 100 mg/m2 every 10 days for 9 cycles. A total response rate of 26% with no grade 2/3/4 diarrhea (p<0.001) was seen. However TJ-14 and G-CSF prophylaxis for CID and neutro-penia respectively were used in the trial [57]. In another pilot study the maximum tolerated dose (MTD) per month of CPT-11 was divided by 12 and administered three days a week as low-dose regimen (25 mg/m2 on days 1, 2 and 3 every week) to 21 metastatic colon and gastric cancer patients. No incidence of grade 3 or 4 diarrhea was recorded. Efficacy rate was 55.6% with 10 partial responses among the 18 evaluable patients with measurable disease [58].
In phase III trials on patients with 5-FU refractory CRC, CPT-11 in weekly schedule was found to be similar in efficacy but associated with a higher incidence of severe diarrhea as compared to once every 3 week schedule [59]. In a recent trial intermittent FOLFIRI treatment (2 months on and 2 months off) was found not inferior to standard chemotherapy in advanced CRC patients. However, no significant difference in diarrhea was observed between both arms [60].
Various studies have shown that dose escalation of standard regimens and high dose CPT-11 administration is feasible and may leads to greater benefit [61, 62]. However, delayed onset diarrhea still remains a predicament and it needs to be supplemented with an effective preventive or therapeutic approach.
5.2. Structural/Chemical Modification and Novel Drug Delivery Methods
Several CPT-11/SN-38 based agents and approaches are currently undergoing development with motive of decreasing toxicity and increasing activity. These novel approaches include pegylation (EZN 2208 [63], NKTR 102 [64]), micellar nanoparticles (NK012 [65]), conjugation with monoclonal antibody (labetuzumab-SN-38 immunoconjugates [66]), liposomalization (IHL-305 [67], LE-SN38 [68], CPX-1 [69], NL CPT-11 [70]), macromolecular carrier binding (hyaluronic acid + CPT-11 [71]) and multiple other formulations and approaches [72]. Enhancement of CPT-11 activity via adenovirus mediated expression of β-glucuronidase in tumors [73] and gene directed enzyme/prodrug therapy (CES/CPT-11) [74] may also indirectly help in alleviating CID. It has been hypothesized that intestinal damage will be less with targeted delivery and decreased concentrations of CPT-11 and SN-38 in bile due to liposomalization. But strategies to develop analogs of SN-38 or reformulate SN-38 have been in clinical development for nearly a decade and still their efficacy in comparison with CPT-11 is unknown. Indeed, these reformulation strategies are still not devoid of gut toxicity [63, 69, 71].
5.3. Genetic Testing
A large inter-patient variation in CPT-11 pharmacokinetics has been observed suggesting a major role of genetics in predicting CPT-11 toxicities [75, 76]. Most of the pharmacogenetic studies on CPT-11 have focused on UGT1A1 polymorphisms especially UGT1A1*28 relation with toxicities. UGT1A1*28 polymorphism is due to a change in the number of TA repeats in the TATA box of the UGT1A1 promoter region, from the wild-type 6 repeats to variant 7 repeats. UGT1A1*28 allele is associated with reduced levels of enzyme. Therefore, heterozygous (*1/*28) and homozygous (*28/*28) individuals are prone to higher levels of SN-38 and subsequent SN-38 mediated toxicities as compared to wild type (*1/*1) [77]. In a meta-analysis review of 20 published trials with 1760 pooled patients, dose dependent association between UGT1A1*28 polymorphism and CID was calculated. No association was found between UGT1A1*28 and severe diarrhea at low doses of CPT-11 (cut off <125 mg/m2). But a significant statistical association was found between risk of severe diarrhea at medium and high CPT-11 dose among patients with UGT1A1*28/*28 genotype as compared to those with a UGT1A1*1/*1 genotype (OR = 3.69, 95% CI = 2.00–6.83; P < 0.001). Also in patients with UGT1A1*1/*28 genotype, the risk of toxicity was higher than those with a *1/*1 genotype at medium and high doses (OR=1.92, 95% CI = 1.31–2.82; P = 0.001). No ethnic difference between UGT1A1*28 and diarrhea was observed [78]. However the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group (EWG) did not recommend routine UGT1A1 genotyping in metastatic CRC patients with the intent of targeted dosing to decrease adverse events [79]. Even if targeted dosing is found to be effective in reducing adverse events it is still unclear what will be the risks in terms of sub therapeutic doses and eventual anti-tumor activity.
5.4. Intestinal Alkalization
CPT-11, SN-38 and SN-38G all three have a labile α-hydroxy-δ-lactone ring that undergoes pH-dependent reversible hydrolysis. Direct effect of SN-38 on intestinal epithelium is believed to be the reason of late onset diarrhea. At physiological or higher pH, the less toxic and less potent carboxylate form is favored while at acidic pH, the more toxic and potent lactone form predominates. Thus changing the equilibrium towards carboxylate isomer will result in decreased CPT-11 toxicity. Also intestinal alkalization decreases reabsorption efficacy of CPT-11 and SN-38 by intestinal cells, with uptake decreasing by more than 65% above a pH of 6.8 [80]. In golden Syrian hamster model CPT-11 (20–50 mg/kg × 7 days) with oral sodium bicarbonate supplementation (5 mg/ml in drinking water) led to, 20% reduction in intestinal SN-38 lactone concentration and decrease in both small and large intestinal mucosal damage [81]. In Gunn rats, alkaline ionized water (administered 1 week before and continuing throughout study) along with CPT-11 (18 mg/kg) has shown to prevent an increase in diarrhea, reduced serum diamine oxidase levels and seemed to be more potent than glutamine as anti-diarrheal therapy [82]. A case-control study was done on 69 lung cancer patients receiving CPT-11 with cislatin to determine whether oral alkalinization decreased the incidence of CPT-11 induced diarrhea. Sodium bicarbonate, magnesium oxide, water and ursodeoxycholic acid were administered for 4 days after CPT-11. A total of 37 cases and 32 controls went into the study. Intestinal alkalization and control of defecation proved to be of significant benefit in controlling delayed diarrhea as well as nausea, vomiting and neutropenia [83]. In another study 24 patients with advanced gastrointestinal cancer on CPT-11 based regimens received 2 gm sodium bicarbonate per day with water. Observed incidence of grade 3 and 4 diarrhea was 16% as compared to expected 24% in large clinical phase III trials [84]. Alkalization did not appear to decrease blood levels of CPT-11 or its active metabolites [85] and did not affect antitumor response rates [83]. However in a case report intestinal alkalization significantly decreased plasma levels of SN-38 and CPT-11 [86]. Also the prophylaxis is extremely cumbersome requiring daily consumption of highly alkalized water in excess of 2–3L/day for the entire course of therapy [83].
5.5. Anti-Diarrheal Therapy
5.5.1. Loperamide
Loperamide is the first line therapy for treating CID [14]. Abigerges et al. have shown that with CPT-11 infusion every 3 weeks, diarrhea was the dose limiting toxicity at 350 mg/m2, but concomitant high-dose loperamide allowed administration of CPT-11 at doses up to 750 mg/m2 [32]. However concomitant loperamide has its own side effects.
5.5.2. Octreotide
Octreotide has been recommended as a second line agent to control CID after loperamide failure [14]. Though in a small case series report long acting octreotide has shown efficacy in secondary prophylaxis of refractory CID, quality studies in setting of CPT-11 induced diarrhea are lacking [87].
5.5.3. Acetorphan
Acetorphan (Racecadotril, Tiorfan®), an orally active enkephalinase inhibitor with antidiarrheal activity and antisecretory mechanism has been tried as an alternative to loperamide. In a small dose escalation study prophylactic treatment of acetorphan (200 mg, three times a day for 15 days, beginning 8 hours after CPT-11 infusion), effectively decreased incidence of diarrhea almost to zero without causing constipation. In further retrospective analysis to compare preventive acetorphan versus curative loperamide, acetorphan scored favorably in terms of less number of diarrheal episodes, cycles with episodes and also in doses of loperamide to control diarrhea [88]. In a randomized, open-label, multi-center, phase II study prophylactic acetorphan (300 mg/day from D0 to D15) was compared with no anti-diarrheal prophylaxis in 5-FU resistant advanced CRC patients receiving CPT-11 (350 mg/m2, once every 3 weeks). No difference between two arms in terms of CID incidence, severity and characteristic was observed. The study showed prophylactic acetorphan at 300 mg/day had no effect on CID [89].
5.5.4. Budesonide
Budesonide, is commonly used in inflammatory bowel disease. CID also exhibits inflammatory mucosal changes on colonoscopy. In a phase I trial budesonide (9 mg oral morning dose per day) was administered to 14 patients with CPT-11 induced grade 3/4 diarrhea with loperamide failure. Budesonide reduced diarrhea severity by at least 2 grades in 86% patients on CPT-11. All positive response patients were successfully treated with prophylactic budesonide [90]. However in a randomized, double blinded placebo controlled phase III trial oral budesonide failed to show significant benefit in prevention of CID in advanced CRC patients but results were in its favor. Oral budesonide (3 mg three times a day) prevented diarrhea in 58.3% patients as compared to 38.5% in placebo (p=0.257) arm. Patients on budesonide arm had fewer CID episodes (0.7 vs. 2.2), with shorter duration (1.8 vs. 4.2 days) and required lesser dose of loperamide (24.9 vs. 36.2 capsules) as compared to placebo. A higher response rate with placebo, uneven distribution of concomitant medications and diarrhea cut off as > 4 stools were points of discussion regarding the trial [91]. Further prospective studies are warranted to confirm these effects.
5.5.5. Miscellaneous
A study was conducted to assess the efficacy of combined prophylactic and curative anti-diarrheal medication in advanced CRC patients receiving CPT-11. Patients received sucralfate (4g/d) and nifuroxazide (600 mg/d) prophylactic treatment on days 0–7. If severe diarrhea was encountered, preventive treatment was replaced with loperamide (12 mg/d) and diosmectite (9 g/d). Of total 34 patients in trial, 29 received the preventive treatment at cycle 1 and grade 3 CID was observed in 14% patients and 3.7% of cycles with no grade 4 incidence. The preventive combined treatment seemed effective in reducing the incidence of CID, and it should be evaluated further [92].
5.6. Transporter Inhibition
As discussed above the transportation of CPT-11 include many transporters like ABCB, ABCC1, ABCC2 and ABCG2 [3]. Their inhibition by various drugs as mentioned below can influence CPT-11 pharmacokinetics, biliary SN-38 concentration and affect intestinal toxicities.
5.6.1. ABCB1 Inhibition
Diarrhea due to CPT-11 is largely due to direct intestinal cytotoxicity by SN-38. Both SN-38 and SN-38G are transported from systemic circulation into biliary tract by ABCB1 and ABCC2 and then they go eneterohepatic recycling [3]. Cyclosporine an ABCB1 and ABCC2 inhibitor has shown to decrease biliary excretion of CPT-11 metabolites in preclinical studies [93, 94].
In a phase I study of intravenous CPT-11 with cyclosporine (5 mg/kg bid for 3 days) on 37 patients, only one grade 3 diarrhea was reported. The MTD and recommended dose of CPT-11 for phase II studies was decided as 100 mg/m2 every 2 weeks [94]. Further treatment of 34 patients at recommended dose revealed a 3% rate of grade 3/4 diarrhea. However some patients withdrew from study due to cyclosporine toxicity [95]. In another phase II trial of CPT-11 (60 mg/m2/day, 90 min infusion, 3 hour after cyclosporine initiation) with cyclosporine (5 mg/kg/week, 6 hr infusion), 16 patients with advanced 5-FU refractory CRC were treated. Grade 3/4 diarrhea was 13% and 1 partial response was seen. But as noted by authors CPT-11 dose was too low in the study [96].
In Wistar rats phenobarbital pretreatment resulted in increase of AUC of SN-38G and decrease in AUCs of CPT-11 and SN-38 possibly due to induction of conjugation [97]. A phase I study was conducted on 92 patients with refractory tumors to modulate CPT-11 pharmacology first with cyclosporine alone and then by adding phenobarbital [98]. In this three step study, at the recommended phase II dose of 120 mg/m2, prevalence of grade 3 diarrhea was 18% with no grade 4 events as compared to 34% prevalence of grade 3 and 4 diarrhea in weekly regimens [98]. The study demonstrated that cyclosporine increased systemic SN-38 exposure and reduced CPT-11 clearance possibly by inhibition of ABCB1 and ABCC2 transporters. Phenobarbital increased CPT-11 clearance and reduced SN-38 AUC when compared with patients treated with cyclosporine alone by upregulating ABCC2 and ABCB1 gene expression and by inducing UGT1A1 mediated glucuronidation [98]. Although the combination did not permit a higher dose administration of CPT-11, it improved toxicity profile without altering anti-tumor activity as evident by 5 partial responses in the study. This may be due to increased availability of CPT-11 for intratumoral conversion to SN-38 and decreased SN-38 intestinal exposure [98]. Other ABCB1 inhibitors like verapamil and quercetin have also shown to increase bioavailability and decrease biliary excretion of CPT-11 and SN-38 in female Wistar rats [99, 100].
5.6.2. ABCC2 Inhibition
MRP2/ABCC2 mediates a majority of transport of SN-38 and SN-38G and to a much lesser extent of CPT-11 [76]. Probenecid (MRP2 inhibitor) co-administration, with CPT-11 in rats decreased biliary excretion of CPT-11, SN-38 and SN-38G and increased their plasma concentration. On decreasing the dose of CPT-11 to half with concomitant pro-benecid, the intestinal SN-38 concentration and CID decreased whereas the plasma concentration of SN-38 and bone marrow suppression was the same as that of control [101].
5.6.3. BCRP Inhibition
BCRP, one of the important CPT-11 transporting proteins is present in the bile canalicular membrane [3]. In CPT-11 treated male Sprague-Dawley rats co-administration of BCRP inhibitors (GF120918 and cyclosporine A) or excipients (Pluronic F68 and PEG 2000 stearate) ameliorated CID as compared to controls. GF120918 decreased biliary excretion of CPT-11 more than cyclosporine A. Co-administration of GF120918 and PEG 2000 stearate was shown to decrease CPT-11 induced intestinal mucosal damage [102].
5.7. Enzyme Induction and Inhibition
5.7.1. β-Glucuronidase Inhibition
As explained above, SN-38 undergoes glucuronidation in the liver to SN-38G by UDP glucuronyltransferases and is excreted via the biliary system into the GI tract [22, 23]. SN-38G is converted back into its active deglucuronidated form SN-38 by bacterial β-glucuronidase enzymes in the commensal microbiota resulting in increased intestinal exposure to the active metabolite which plays an essential role in the delayed onset diarrhea [26]. Various substances have been evaluated as potential inhibitors of bacterial β-glucuronidase to reduce the intestinal concentration of SN-38 with resulting decreased enterohepatic circulation and intestinal damage.
5.7.1.1. Kampo
Kampo (Japanese/Chinese herbal) medications have been shown to decrease enterohepatic circulation of SN-38. The four natural glucoronides baicalin, wogonoside, luteolin-3′-glucuronide and glycyrrhizin present in Kampo inhibit β-glucuronidase [103]. The 3 aglycons of these glucuronides; baicalein, luteolin and glycyrrhetic acid inhibit UDP-glucuronosyltransferase activity towards SN-38 as a substrate [104]. Baicalin (and sulfobromophthatlein) also inhibit a specific transporter which mediates SN-38 uptake across the apical membrane in Caco-2 cells [105]. Many Kampo medicines have shown to inhibit SN-38 glucuronidation by UGT1A1 [106].
Recently a Kampo medicine, Dai-kenchu-to has also shown to maintain the mucosal integrity and decrease apoptosis and CPT-11 induced inflammatory cytokines in intestinal mucosa of male Wistar rats. Probable mechanisms involved are increase in intestinal blood flow, decrease in gastrointestinal transit time and reduction in inflammation [107]. TJ-14 (Hange-Shashin-to), a Kampo medicine used to treat diarrhea symptoms is made from 7 medicinal herbs-Pinelliae tuber, Scutellariae radix, Glycyrrhizae radix, Zizy-phi fructus, Ginseng radix, Coptidis rhizoma and Zingiberis siccatum rhizoma. TJ-14 has shown to decrease colonic pros-taglandin E2 production [108], increase water absorption [108] and inhibit spontaneous contraction in distal colon through nitric oxide production in rats [109]. TJ-14 has also been shown to protect against CPT-11 induced weight loss, intestinal epithelial damage, anorexia and delayed onset of diarrhea in rats [110, 111]. In a study, 23 cancer patients were treated with various CPT-11 schedules in combination with prophylactic oral TJ-14 (7.5 g, three times a day, every day starting prior to CPT-11 infusion). No incidence of grade 2/3/4 diarrhea was seen. But constipation, a Kampo toxicity was seen in 10% of patients. Also compliance was not sufficient as 3 patients could not take TJ-14 due to odor/taste and 1 patient due to vomiting [112]. Later on, in a randomized controlled trial, TJ-14 was evaluated on chemo naïve non small cell lung cancer patients. Chemotherapy regimen consisted of CPT-11 with cisplatin. Experimental arm had 18 patients who received oral 7.5 g TJ-14 per day divided in 3 doses before each meal, started ≥3 days prior and continued minimum of 21 days after starting chemotherapy. As compared to control arm (23 patients), TJ-14 arm reported improvement in diarrhea grade (p=0.044) and decreased incidence of Garde3/4 diarrhea (1 vs 10; p=0.018). But no difference in frequency and duration of diarrhea was observed. Grade 1 constipation in 11% patients was the observed side effect of TJ-14 [113]. A study was conducted on male Wistar rat model to assess the most promising treatment in preventing CID from several available potential treatments mostly targeting β-glucuronidase. CPT-11, 60 mg/kg was given intravenously; from days 1 to 4. The investigative options were administered orally twice daily from days −1 to 4 and included streptomycin 20 mg/kg with penicillin 10 mg/kg (S/P), neomycin 20 mg/kg with bacitracin 10 mg/kg (N/B), TJ-14 1,000 mg/kg and activated charcoal 1000 mg/kg (AC). TJ-14, S/P and N/B improved weight loss and CID symptoms to similar extent while efficacy of AC was less but significant. In a separate experiment with the same regimen in rats with breast cancer (Walker 256-TC) TJ-14, N/B and AC, improved CPT-11 toxicity without decreasing antitumor activity. Also, unexpectedly twice daily oral doses of cyclosporine A (50 mg/kg) (a P-glycoprotein and cMOAT/MRP2 inhibitor) or valproic acid (200 mg/kg) (UDP-glucuronosyltranferase inhibitor), exacerbated the intestinal toxicity without modifying CPT-11’s antitumor activity [114].
5.7.1.2. D-saccharic Acid 1,4-Lactone
D-saccharic acid 1,4-lactone (SAL), a β-glucuronidase inhibitor has been studied in Wistar rats as a prospective CID blocking agent. Combination treatment with SAL significantly reduced CPT-11 induced mucosal damage in small intestine of rats [115].
5.7.1.3. Selective Bacterial β-Glucuronidase Inhibition
In a novel targeted approach bacterial β glucuronidase inhibition was tried without killing intestinal commensals or inhibiting mammalian β glucuronidase. Wallace et al. identified a specific 17 residue bacterial loop in the E. coli enzyme which is not found in the human ortholog, and found inhibitors specifically affecting this bacterial loop with lack of activity towards mammalian glucuronidase. Four lead compounds inhibiting bacterial β glucuronidase were tested in vitro on β glucuronidase positive and negative, aerobic and anaerobic bacteria and colon cancer cell lines (HCT116, Caco-2 and CMT93). The lead compounds didn’t adversely affect cell viability in any of them. A lead compound, Inhibitor 1 protected against CPT-11 induced intestinal epithelial damage in Balb/cJ mice [116]. Rasmussen et al. prepared a new compound, uronic-Noeurostegine, and demonstrated its potent, competitive E. coli β-glucuronidase inhibition, while mammalian β-glucuronidase inhibition from bovine liver was found to be less significant [117]. Recently nialamide, isocarboxazid, and amoxapine have been found to inhibit bacterial β-glucuronidase but with no significant activity against mammalian β-glucuronidase [118]. However, these novel compounds still need to be tested in clinical trials.
5.7.2. UGT1A1 Induction
UDP-glucuronosyltransferase 1-1 also known as UGT1A1 is an enzyme of the glucuronidation pathway which conjugates cytotoxic SN-38 to inactive, non-toxic SN-38 glucuronide (SN-38G) [3, 22, 23]. Chrysin has low oral bioavailability [119] and has shown to upregulate UGT1A1 [120]. Therefore it can selectively increase glucuronoconjugation of SN-38 to SN-38G in the gastrointestinal tract by UGT1A1 induction and reduce intestinal mucosal damage and delayed diarrhea without affecting systemic or intratumoral levels of SN-38. In a study on 20 previously treated advanced CRC patients, chrysin was administered twice daily for 1 week preceding and succeeding treatment with CPT-11 (single agent; 350 mg/m2 over 90 min every 3 weeks). Only 10% of patients experienced grade 3 diarrhea as compared to 19% in patients treated with the same schedule without chrysin in prior studies. No chrysin related toxicities were encountered, loperamide use was modest and mass ratio of plasma SN-38G/SN-38 was similar to historical controls. A randomized phase II placebo-controlled study of chrysin in combination with CPT-11 to examine the efficacy of chrysin in reducing delayed diarrhea is being done [121]. Other UGT1A1 inducers like phenobarbital (discussed above) and glucocorticoids have also been studied in clinical trials. Dexamethasone though a CYP3A4 and UGT1A1 inducer did not seem to alter AUC of SN-38 and CPT-11 [6, 98].
5.7.3. Carboxylesterases Inhibition and Activation
As discussed above one of the contributing factors of CID is local activation of bile excreted CPT-11 in the small intestine by human intestinal carboxylesterase (hiCE) [122]. Seven hiCE inhibitors, all sulfonamide derivatives, demonstrating greater than 200 times selectivity towards hiCE (as compared to liver) and non inhibitors of human acetylcholinesterase and butyrylcholinesterase have been developed by Wadkins et al. [123]. Further studies revealed that benzil and dimethylbenzil analog efficiently enter cells and inhibit hiCE intracellularly [124]. Yoon et al. developed 4 nitrophenol derivative lead compounds of which compound 3 has shown to preferentially inhibit hiCE (14% CES activity remaining) as compared to rabbit liver carboxylesterase (30% CES activity remaining) [125]. Recently a series of novel, highly potent, hiCE inhibitor fluorene analogs have been designed [126]. Results of their application in animal and human studies are awaited.
A novel strategy has been employed for tumor-specific activation of CPT-11. In this targeted approach human liver CES-2 was fused with anticarcinoembryonic antigen (CEA) side chain Fv fragment. This acted as a targeting molecule. The recombinant enzyme anchored CEA on the tumor cell, leading to extracellular CPT-11 metabolism. This fusion recombinant protein showed CPT-11 activation to SN-38, specific binding to CEA-expressing cells and exerted anti-proliferative effects on human cancer cells with CPT-11. This secreted tumor-targeted form of CES should also prevent enzyme leakage from the site of the tumor into the circulation. This tumor specific activation may lead to administration of less toxic CPT-11 dosing which might ultimately benefit CID [127].
5.7.4. CYP3A4 Inducers
Concomitant administration of CYP3A4 inducer anti epileptic drugs like phenytoin, carbamazepine and phenobarbital with or without dexamethasone have shown to increase the CPT-11 recommended dose to 750 mg/m2 every 3 weeks as compared to 350 mg/m2 every 3 weeks in patients not receiving them. As expected they increased CPT-11 clearance and decreased AUC of SN-38 [6]. Similar an increase in CPT-11 clearance and recommended dose (500 mg/m2 every 15 days on 28 day schedule) was found in another study with temozolomide in patients receiving enzyme inducing antiepileptic drugs [128].
5.7.5. COX-2 Inhibition
COX-2 is found to be overexpressed in metastatic CRC and promotes tumor growth by several mechanisms including PGE2 and TXA2 production [129]. CPT-11 administration is also associated with increased PGE2 and TXA2 [42–44] which contributes to the secretory diarrhea [44]. Thus it was suggested that COX-2 inhibitors might play a role in this regard. Trifan et al. hypothesized that CID could be partly secondary to COX-2 activation secondary to CPT-11 induced colonic mucosal damage. In two mouse models (HT-29 and colon-26 cells) celecoxib (selective COX-2 inhibitor) enhanced the antitumor effect of CPT-11. In Sprague Dawley male rat model celecoxib decreased the severity of CID and weight loss in a dose dependent manner. The effective COX-2 inhibitory, celecoxib dosage in the study ranged from 10–50 mg/kg/day which yielded a plasma concentration from 0.8 to 2.4 μg/ml [130]. In humans the same plasma concentration can be achieved by administering celecoxib 400 mg twice a day [130, 131]. Also celecoxib has shown anti-angiogenic and antitumor activities within the same range [130, 132, 133]. In another study on Ward CRC rat models; oral celecoxib (30 mg/kg daily in 2 divided doses) decreased CPT-11 toxicity and increased antitumor activity along with survival at otherwise lethal doses [134]. Multiple clinical studies have subsequently been conducted but they failed to show any improvements in CID or efficacy in CRC [135]. Diarrhea remained the major non hematological toxicity. The reason might be that COX-2 inhibition alone is not sufficient enough to ameliorate CID or the delivery of cele-coxib is not sufficient to the target tissues for protective effects [135]. A phase I/II study of CPT-11 with 5-FU and rofecoxib was conducted on metastatic CRC patients. In a cycle of 8 weeks CPT-11 (i.v.) was administered on days 1, 8, 15, and 22, rofecoxib orally 50 g/day, and 5-FU infusion at a fixed dose of 200 mg/m2 per day for 5 weeks followed by 3 weeks of therapy with rofecoxib alone. Recommended dose of CPT-11 for phase II study was 125 mg/m2. In the phase II part 33 patients were enrolled. Twenty-five patients (75.8%) experienced diarrhea. of which 12 patients (36.4%) had grade 3 diarrhea. However rofecoxib which was 10 times more potent than celecoxib has been withdrawn due to potential cardiac side effects by U. S. Food and Drug Administration [136].
5.8. Alteration of Intestinal Microflora
5.8.1. Prebiotics and Probiotics
In a nutritional modulation study on Ward colon tumor bearing rats treated with CPT-11 (125 mg/kg × 3 days), addition of prebiotic oligosaccharides to the diet (8%, w/w of diet; inulin and oligofructose in 1:1 w/w) starting 2 weeks prior to chemotherapy did not decrease the severity of diarrhea rather doubled the activity of β-glucuronidase in caecal contents [137]. VSL#3 is a commercially available novel probiotic compound comprising of strains of lactobacilli, bifidobacteria and streptococcus. In CPT-11 treated rats, VSL#3 has shown to increase crypt proliferation and decrease weight loss, diarrhea, intestinal apoptosis, mucin secretion and CPT-11 induced increase in goblet cells within jejunal crypts. But protective effects of VSL#3 are maximal only when given before and after chemotherapy [138]. In another study Lactobacillus casei strain Shirota (LcS) (1.64 × 10(11) cfu/0.5 g/3 mL saline) was administered orally in rats for 28 days followed by CPT-11 (14 days later, for 4 days; 100 mg/kg i. p.). LcS significantly inhibited CPT-11 induced weight loss, increased food intake and improved CID symptoms as compared to controls. Suggested possible reason was inhibitory β-glucuronidase activity of LcS [139]. Similarly in a study on rats, Saccharomyces boulardii administration (800 mg/kg, 3 days before and continuing throughout experiment) resulted in significant improvement in CID and mucositis. S. boulardii is also a probiotic, having multimodal effects ranging from effects on enteric pathogens to influencing cell signaling and cell growth and maturation [140]. Though probiotics seem to be moderately effective, they are cheap, non-toxic and convenient prophylactic agents who deserve to be investigated in humans.
5.8.2. Antibiotics
Oral antibiotics have been shown to be effective in reducing CPT-11 induced gastrointestinal toxicity in both preclinical and clinical studies. One of the proposed mechanisms is the reduced activity of bacterial β-glucuronidase by killing the intestinal micro flora resulting in decreased intestinal concentration of SN-38.
Preclinical Studies
In a study germ free mice were found to be more resistant to CPT-11 toxicities. Experiments showed that in holoxenic mice the lethal dose of CPT-11 was lower (60 to 80 mg vs. ≥150 mg) and they suffered significantly higher intestinal damage with 100 mg CPT-11 as compared to germ free mice. Even at 60 and 80 mg, CID incidence was 95% and 100% in holoxenic mice but only 0% and 5% respectively in germ-free mice [46]. This further suggests elimination of intestinal microflora via antibiotics can play a major role in CID alleviation.
In CPT-11 treated male Wistar rats addition of 1 mg penicillin and 2 mg streptomycin per ml of drinking water (beginning 5 days before and continuing throughout study) inhibited β-glucuronidase activity, markedly ameliorated diarrhea and reduced cecal damage. The segmental difference in the degree of intestinal damage after CPT-11 treatment was found to be correlated with the luminal β-glucuronidase activity [141]. Acute diarrhea was also inhibited probably due to inhibition of cholinergic transmission by streptomycin [141, 142]. Antibiotics didn’t show any effect on pharmacokinetics of CPT-11, SN-38 and SN-38G in blood, tissues or small intestine but decreased AUC1–24h of SN-38 by 85% in the large intestine without affecting that of CPT-11 [143]. Streptomycin has shown to decrease CID in Gunn rats via other mechanisms including inhibiting CPT-11 absorption from intestinal lumen, decreasing CES activity and increasing UGT activity in the intestinal epithelium [144]. In rats bearing Ward colon tumor, prophylactic ciprofloxacin nullified CPT-11 related mortality (45% in the control group). Though ciprofloxacin decreased CPT-11 related weight loss, it was unable to alter severity or course of CID. However it appeared to correct hyper-proliferative response of mesenteric lymph nodes, hypo-responsiveness of spleno-cytes, post-chemotherapy related immunological anergy and local pro-inflammatory response in intestinal epithelium, thereby limiting mucosal injury [145]. In a study on CPT-11 treated Sprague-Dawley rats, valproic acid and ceftriaxone, either alone or in combination had similar effects in preventing severe CID. However activated charcoal was surprisingly not found to be effective in the prevention of severe CID in the study [146].
Clinical Studies
In one study, seven 5-FU resistant CRC patients who experienced grade 2 diarrhea or more after CPT-11 (350 mg/m2; i.v. once every 3 weeks) therapy in the first cycle received the same dose with oral neomycin (1000 mg three times a day; beginning 2 days prior to 5 days after CPT-11 therapy) in the second course. Five patients did not experience any diarrhea in second course (p=0.0326), one experienced grade 1 diarrhea (3 days) and one experienced grade 3 diarrhea (5 days). There was no effect on the systemic exposure of CPT-11 or SN-38 (p≥0.22) and hematological toxicity. On phenolphthalein assay fecal β-glucuronidase activity was found to decrease from 7.03 ± 1.76 μg/h/mg to undetectable levels. There was also a decrease in fecal concentrations of SN-38 [26]. In another study CRC and small cell lung cancer patients on CPT-11 based regimen who experienced grade 3/4 diarrhea were administered neomycin (500 mg; twice daily) from subsequent cycles. No recurrence of severe diarrhea was reported after neomycin prophylaxis [147]. In another study 15 CRC patients experiencing diarrhea in first cycle while on Saltz regimen were administered 1000 mg Bimixin (25000 IU neomycin plus 2500 IU bacitracin) three times a day. Bimixin was administered on days 2 to 5 and 16 to 19 of each cycle during chemotherapy. Complete resolution of diarrhea was observed from second to fourth cycle. Two patients (all grade 1) in fifth cycle and five patients (2 grade 1, 3 grade 2) in last cycle had diarrhea [148].
In a double blinded, randomized, multicentre, placebo controlled trial, patients received CPT-11 (350 mg/m2 once every 3 weeks) with either neomycin (660 mg three times daily for three days, starting 2 days before chemotherapy) or placebo. Sixty two patients were evaluable for toxicity analysis. Though grade 3 diarrhea was less frequent in neomycin arm as compared to placebo (17.9% vs. 32.4%; p = 0.19), it was associated with a concomitant increase in grade 2 diarrhea. No significant difference in terms of overall incidence, severity and duration of CID was observed. The difference in results to diarrhea with neomycin as compared to earlier studies may be due to the different schedules of neomycin. Interestingly patients in neomycin arm were at significantly higher risk for grade 2 nausea than with those in placebo (39.3% vs. 8.8%; p< 0.01) possibly due to neomycin [149].
A more targeted approach towards aerobic β-glucuronidase producing bacteria was tried in a phase I study on 40 pediatric patients with refractory tumors. In this study cefexime (8 mg/kg/day beginning 5 days before CPT-11 and continuing throughout study) was co administered with 5 days daily oral CPT-11 (for 2 consecutive weeks; repeated every 21 days). The MTD without and with cefexime was 40 and 60 mg/m2/d respectively. At MTD, the median systemic SN-38 lactone exposure of oral 60 mg/m2/d CPT-11 with cefexime was significantly higher than without cefexime and was similar to protracted i.v. 20 mg/m2/d CPT-11 [150]. In a phase I trial, children with refractory solid tumors were treated with CPT-11, vincristine and temozolomide combination. Cefpodoxime was used for diarrhea prophylaxis. Grade 3 diarrhea was seen only in 5 of 111 courses (5%) with no C. difficile infection. Interestingly DLTs were pancreatitis and transaminitis [151]. In another phase I study in pediatric population with refractory solid tumors, cefpodoxime administration increased the MTD of CPT-11 from 20 to 30 mg/m2/dose. Diarrhea remained the dose limiting toxicity [152]. In a pooled analysis of 51 pediatric patients receiving protracted CPT-11 based therapy with cephalosporin prophylaxis overall incidence of grade 3/4 diarrhea was 7% only. This is important considering the fact that 72% of the courses were administered at CPT-11 doses higher than established MTDs without antibiotic support. But as a matter of concern, 3 cases of C. difficile enteritis cases were reported. Moreover the optimal timing for start of antibiotic prophylaxis is still not clear and non-compliance due to adverse events (e.g. nausea) can be a hindrance in administration [153]. However intravenous cephalosporin can be an option to reduce CID in patients unable to tolerate oral antibiotics [154].
In a phase II study on 51 metastatic CRC patients; CPT-11 (250 mg/m2, 4 applications every 2 weeks followed by 2 weeks break) was administered with levofloxacin (500 mg once a day; −1 to + 1 day) and cholestyramine (4 g three times a day, −1 to + 1 day). Only one patient experienced WHO grade 3 diarrhea and no grade 4 diarrhea was seen. The 2% incidence of severe diarrhea reported in the study was very low and the schedule appeared easy to be compliant. Combined treatment with levofloxacin and cho-lestyramine can thus eliminate intestinal microflora and also prevent enterohepatic recirculation of SN-38. Further studies are warranted to test dose escalation [155].
In a combination trial of gentamycin with sodium bicarbonate for prevention of CID, the experimental group (52 patients) received both gentamycin (80,000 U BID) and sodium bicarbonate (2 g TID) beginning one day prior to each CPT-11 dose and continuing for 4 days. The control group (46 patients) received only CPT-11. Incidence of diarrhea in the experimental group was only 13.7% as compared to 34.83% in the control group (p< 0.001). The experimental group also had decreased nausea, vomiting, myelosuppression, loperamide consumption and increased stool pH; all of which were statistically significant (p<0.001) [156]. In another study, 31 cancer patients on CPT-11 based chemotherapy were divided in interventional group (Chemotherapy+Bacillus licheniformis granules+Gentamycin) and control group (chemotherapy only) with cross-over self-controlled randomization. CID prophylaxis with Bacillus licheniformis granules and Gentamycin proved to be effective and a statistically significant decrease in CID incidence and severity was observed [157].
Despite the extensively published work on the use of antibiotics in CID, this approach has several drawbacks including side effects of antibiotics (nausea, vomiting), negative impact on intestinal metabolism, imbalance of intestinal micro flora, resulting in malabsorption and increased chances of antibiotic associated toxicities, diarrhea, C. difficile and fungal infection.
5.9. Prevention of Direct Intestinal Exposure
5.9.1. Activated Charcoal
Activated charcoal (AC) adsorbs free intestinal luminal SN-38 thereby decreasing the intestinal mucosal damage and reducing CID. It also acts by enhancing clearance via entero-capillary exsorption and enterohepatic circulation interruption [158]. In a study on male Sprague-Dawley rats, AC (Ultracarbon, orally 2.5 g/kg daily for 5 days) was given 10 min before CPT-11 (60 mg/kg i.v. daily for 5 days; total dose 300 mg/kg). Multiple oral doses of AC did not modulate CPT-11, SN-38 and SN-38G kinetics in rats. The control group had higher frequency of grade 3 diarrhea as compared to the experimental group (log OR: −1.06; 95% CI: −2.25, 0.13) but was not statistically significant (p=0.08) [159].
In a phase II trial, advanced CRC patients were treated with 125 mg/m2 CPT-11weekly infusion for 4 weeks every 6 weeks. In the first cycle AC (5ml aqueous Charcodote [1,000 mg AC] with 25 mL water) was given on the evening prior to chemotherapy and then three times a day for 48 hours after the dose. In cycle 2, no AC was given. In total 28 and 24 patients completed cycle 1 (with AC) and 2 (without AC) respectively. In cycle 1 grade 3/4 diarrhea was 7.1% and grade 0 diarrhea was 46.4%. Whereas in cycle 2 grade 3/4 diarrhea was 25% and grade 0 diarrhea was 20.8%. Also in cycle 1 as compared to cycle 2, a higher percentage of median planned doses were delivered (98% vs. 70%) and lesser patients took more than 10 loperamide tablets (25% vs. 54%). Compliance and tolerance of AC was found to be excellent [160]. In another study on pediatric patients on CPT-11 based chemotherapy, the interventional group received 250 mg AC three times a day during CPT-11 administration. In the interventional group (10 patients) a total of 13 events of diarrhea in 45 cycles were reported as compared to the control group (12 patients) in which 15 events occurred in 21 cycles (p=0.002). Grade 3 and 4 diarrhea was present only in 4.4% patients in AC group as compared to 52.3% in control group (p=0.010). Also chemotherapy was discontinued in fewer patients in the interventional group as compared to control group (6.6% vs 52.3%). Therefore AC increased CPT-11 compliance and reduced the frequency and severity of CID [161]. However AC has an incomplete effect, can absorb other orally administered comedications and is cumbersome due to intake three times per day.
5.9.2. AST-120
AST-120 (Kremezin) is another oral carbonaceous adsorbent tested as a potential agent to block delayed diarrhea. Kremezin has demonstrated significant adsorption capacity for CPT-11 in vitro and gastrointestinal tract of rats. In Kremezin treated rats the frequency of diarrhea was reduced by approximately 50% as compared to non treated rats along with a decrease in severity [162]. In a clinical trial 2 gram Kremezin three times a day, during and after CPT-11 treatment decreased CID along with a small effect on pharmacokinetics of CPT-11 and its metabolites [163].
5.10. Cytokine and Growth Factors Induction and Inhibition
5.10.1. Thalidomide
Thalidomide is an antiangiogenic, immunomodulatory and antineoplastic agent [164]. In animal studies thalidomide co-administration enhanced antitumor activity of CPT-11 and attenuated CPT-11 induced weight loss, myelosuppression, diarrhea, intestinal epithelial inflammation and apoptosis. The mechanisms involved TNF-α, IL-1β, IL-6 and IFN-γ inhibition. It also significantly increased the AUC of CPT-11 and reduced biliary excretion as well as cecal exposure of CPT-11, SN-38 and SN-38G. This might be due to the inhibitory effects on the hydrolysis of CPT-11 and enhanced effects in the intracellular accumulation of SN-38 by thalidomide and its major hydrolytic product phthaloyl glutamic acid via inhibition of PgP/MDR1, MRP1- and MRP2- mediated CPT-11 and SN-38 transport [165]. Thalidomide administration has shown to alleviate CPT-11 induced toxicities in rats, increase AUC of CPT-11 and significantly decrease AUC and t1/2 of SN-38. The hydrolytic products of thalidomide but not thalidomide itself significantly decrease the hepatic hydrolysis of CPT-11 and increase the intracellular accumulation of SN-38 (but not CPT-11) [166]. In another preclinical study, thalidomide significantly reduced mucosal lesions, MPO activity and TNF-α tissue levels in CPT-11 treated male Swiss mice but didn’t reduced severity of CID however pentoxifylline (cytokine production inhibitor) reduced all [48]. Apart from these mechanisms inhibition of TNF-α secretion by activation of nuclear transcription factor NF-κB by monocytes, TNF mediated downregulation of Na/K-ATPase, inhibition of COX, PG and TNF-α driven calcium and potassium secretion directly or through COX have been implicated in prevention of CID by thalidomide [33].
In combination studies of CPT-11 (300–350 mg/m2, q3 weeks) with thalidomide (400 mg at bed time) on metastatic CRC patients, gastrointestinal adverse events including diarrhea were minimal, response to CPT-11 was enhanced and quality of life during treatment was better [167, 168]. One study reported a significant decrease in CPT-11 metabolism to SN-38 with thalidomide [169] while others didn’t find any significant alteration of CPT-11 pharmacokinetics by thalidomide [170, 171]. In a randomized controlled trial of CPT-11 with fuorouracil and leucovorin on metastatic CRC with or without thalidomide (300 mg, oral on day 1–14, two weeks as a cycle) no significant difference between adverse events and response rate was observed [172].
Thalidomide with and without celecoxib has also been studied as potential modulators of CPT-11 toxicity. In the first step thalidomide, 200 mg daily (13 patients) and 400 mg daily (11 patients) were tested in combination with CPT-11 (125 mg/m2, 2 weeks out of 3). The dose of 400 mg thalidomide daily was associated with fewer adverse events. Addition of celecoxib (12 patients) to 400 mg thalidomide did not improve the safety profile [171].
5.10.2. Velafermin
In tumor bearing DA rats, velafermin (FGF-20) administered at 16 mg/kg prior to CPT-11 improved CPT-11 related gastrointestinal mucositis, diarrhea and mortality. Though rats that received velafermin prior to, or prior to and during CPT-11 treatment did develop severe or moderate diarrhea, however it occurred later, was of lesser severity and duration, was in fewer rats and was not associated with mortality as compared to 50% mortality in controls. There was no increase in tumor proliferation with velafermin. Other doses of velafermin were also assessed and were found to be less effective in reducing the severity of CID and mortality. Curiously some doses appeared to increase the diarrhea and mortality [173].
5.10.3. IL-15
IL-15 is a cytokine that has shown to significantly protect against CPT-11 induced intestinal toxicity and moderately potentiate its antitumor activity in advanced CRC bearing rat model. IL-15 protection was found to be dose and schedule dependent as 11 low doses of IL-15 at 100μg/kg/dose offered best protection in the experiment [174].
5.10.4. RDP58
RDP58 is a novel, oral, non–absorbable, anti-inflammatory, D-amino-acid decapeptide shown to inhibit TNF-α, IL-12 and IFN-γ production and up-regulate heme oxygenase 1 activity in vivo [175]. Oral administration of RDP58 significantly decreased incidence of diarrhea, body weight loss, intestinal mucosal inflammation and improved survival in CPT-11 and 5-FU treated C57BL/6 and BALB/c mice models. Also RDP58 administration allowed CPT-11 MTD to be doubled resulting in significantly enhanced tumor responses and prolongation of time to relapse without a concomitant increase in gastrointestinal toxicity [175].
5.10.5. JBT3002
JBT3002, an N-acylated derivative of ψ-amino-C1-C3 alkane-sulfonic acid, is a novel synthetic bacterial lipopeptide. In preclinical models it has shown to stimulate expression of IL-15 by macrophages in lamina propria and upregulate the expression of inducible nitric oxide synthase and metalloproteinases 2 and 9 in murine macrophages [176]. It has also shown to stimulate IL-1, IL-6 and TNF-α in human monocytes and activates murine macrophages and human monocytes along with IFN-γ to become tumorocidal [176]. In CT-26 colon cancer mice with liver metastasis, oral liposomal JBT3002 has shown to prevent CPT-11 induced dose limiting gastrointestinal toxicity, maintain mucosal immunity, lamina propria integrity and enhance tumoricidal properties in tissue macrophages resulting in increased therapeutic effect as well as dose intensification of CPT-11 [177].
5.10.6. Keratinocyte Growth Factor
Palifermin, a human recombinant keratinocyte growth factor has shown to reduce incidence, severity and duration of mucositis in phase III trials [178]. In tumor bearing DA rats’ single large dose of palifermin prior to CPT-11 administration was found to have delayed onset and reduced severity of diarrhea compared to all treatment groups. With this prophylaxis, only 5% of the animals experienced diarrhea as compared to 11% with multiple small palifermin dosage (p < 0.05) and 28% in CPT-11 alone control groups (p < 0.05). Single large dose palifermin also reduced mortality which was 2%, as compared to 11% in multiple small dose palifermin group and 28% in CPT-11 alone control group. It did not adversely affect tumor growth [179].
5.10.7. St. John’s Wort
St. John’s wort (SJW) is a commonly used herbal medicine with anti-inflammatory and gastro protective activity. It also induces cytochrome 450 isozymes and interacts with PgP transporter [180]. In a randomized crossover study on 5 patients, oral SJW decreased plasma concentration of SN-38 and myelosuppression [181]. This can lead to ineffective treatment dosing. In male Sprague Dawley rats pretreatment with SJW significantly reduced CID, intestinal lesions and inflammation. It was noted that combined long term treatment of SJW altered CPT-11 and SN-38 pharmacokinetics [182, 183]. The probable mechanism of SJW protective action against CID was through, lowering of SN-38 concentration and inhibition of pro-inflammatory cytokines and intestinal epithelial apoptosis [180, 182, 183].
5.11. Miscellaneous Medications and Agent
5.11.1. Oil Supplementation
In preclinical studies low level of dietary fish oil supplementation (3% or 6%) prior to and during CPT-11 treatment enhanced regression of MCF7 human breast cancer xenografts in nude mice. This increase in efficacy of CPT-11 was also associated with concomitant decrease in histopathological damage to intestine [184]. In Ward colon tumor bearing rats fed on standardized basal diet and treated with CPT-11 (125 mg/kg × 3 days), inclusion of n−3 fatty acids (5% w/w of total fat), did not affect severity or incidence of diarrhea. However it enhanced CPT-11 antitumor efficacy and suppressed tumor growth [137]. In a study on CPT-11 treated rats, perilla oil (10% of diet) decreased plasma PGE2 and increased ω-3 polyunsaturated fatty acids, α-linolenic acid, eicosapentaenoic acid and eicosapentaenoic acid/arachidonic acid ratio in colonic mucosa. But the amounts used were not sufficient to decrease CPT-11 induced intestinal damage [45].
5.11.2. GPI 15427
Wistar rats treated with CPT-11 (30 mg/kg/day × 3days) experienced gut mucosal damage and delayed diarrhea. However concomitant administration of oral GPI 15427 (40 mg/kg/q2 × 3d), a novel poly (ADP-ribose) polymerase (PARP) inhibitor protected against CPT-11 induced intestinal damage and reduced severity of CID [185]. GPI 15427 also reduced jejunal inflammation and severity of CID in animals receiving CPT-11 at higher doses than that used in combination with temozolomide. The observed protective effect was associated with inhibition of PARP in intestinal epithelium, indicating a role of PARP-1 over activation in CPT-11 induced gastrointestinal toxicity. Moreover GPI 15427 was shown to enhance the antitumor activity of CPT-11 and temozolomide combination in HT-29 and LoVo colon cancer xenograft models [185].
5.11.3. Glutamine
L-Glutamine is regarded as a conditionally essential amino acid due to body’s inability to synthesize it in adequate amounts during stress [186]. Glutamine is also an essential nutrient in regards of gut mucosal epithelial cell growth, differentiation, integrity and barrier function. It has shown to facilitate electrolyte absorption in animals with experimental diarrhea [186].
In female rats with Ward colon tumor, oral bolus glutamine (0.75 g/kg; 30 min prior to each CPT-11 dose) decreased both intensity and severity of CID (p<0.05). Glutamine was also associated with several potentially protective effects like increase in heat shock proteins 25, −70 and −90α in colon (p<0.05), suppression of colonic β glucuronidase activity (p<0.05), increased ratio of reduced to oxidized glutathione (p<0.05) and a proportionate increase in CD3+CD8+ lymphocytes and memory CD8+ subset in mesenteric lymph nodes. However there was no alteration in antitumor activity of CPT-11, amino acid concentrations, heat shock protein expression and ratio of reduced to oxidized glutathione in the tumor [187]. In Gunn rats daily oral administration of 0.4 g/kg of glutamine beginning 1 week before administration of 18 mg/kg i.v. CPT-11 prevented an increase in diarrhea score. Also glutamine treated rats showed less body weight loss and significantly reduced serum diamine oxidase activity compared to that of controls [82]. In Ward colon tumor bearing rats oral bolus glutamine (0.75 g/kg), prior to each CPT-11 chemotherapy (125 mg/kg × 3 days) reduced incidence of severe diarrhea by approximately 20% (p< 0.005) without affecting CPT-11’s antitumor efficacy. Identical results were obtained with intravenous bolus glutamine infusion [137]. Oral glutamine also partially prevents CPT-11 induced intestinal microflora alteration in rats [188].
In a small case series, oral glutamine supplementation prevented CID in 5 patients which facilitated reescalation of CPT-11 dose in 4 of the 5 patients in subsequent cycles [189]. However in a phase II trial of CPT-11, 5-FU and leucovorin combined with celecoxib and glutamine as first line therapy in colorectal patients, there was neither an increase in efficacy nor a decreased incidence of severe diarrhea [190].
5.11.4. PHY906
PHY906 formulation is derived from Huang Qin Tang, a multicomponent Chinese herbal supplement used for treatment of nausea, vomiting and diarrhea [191]. PHY906 is composed of 4 primary herbs Glycyrrhiza uralensis Fisch, Paeonia lactiflora Pall, Scutellaria baicalensis Georgi and Ziziphus jujuba Mill [192]. In preclinical studies PHY906 has shown to decrease CPT-11 induced weight loss and mortality while enhanced its antitumor activity [193]. On further studies in female BDF1 mice with murine colon 38 allografts, PHY906 did not protect against the initial CPT-11 induced intestinal DNA damage and apoptosis but restored the intestinal epithelium by 4 days after CPT-11 treatment. PHY906 seemed to act via promoting regeneration of intestinal progenitor or stem cells and several Wnt signaling components. It also exerted anti-inflammatory effects by decreasing the infiltration of neutrophils or macrophages, TNF–α expression in the intestine, proinflammatory cytokine concentrations in plasma and inhibition of nuclear factor κB, COX-2, and inducible nitric oxide synthase. [191]. The pro-apoptotic effects of PHY906 seemed to be tumor specific [191, 194].
PHY906 with bolus weekly regimen of irinotecan, 5-FU, and leucovrin (IFL) has been studied in a randomized, double blinded, placebo-controlled, cross over, phase I trial on advanced CRC patients. PHY906 also did not seem to alter pharmacokinetics of CPT-11. PHY906 seemed to reduce grade 3/4 diarrhea but the trial had relatively small number of patients [192]. Larger randomized studies are needed to evaluate the benefit in CID. Though high performance liquid chromatography and liquid/gas chromatography-mass spectroscopy has been used [192], economic and practical quality control can be an issue.
5.11.5. Amifostine
Amifostine is a cytoprotective adjuvant used with chemotherapy agents including alkylating agents (e.g. cyclophosphamide) and platinum containing agents (e.g. cisplatin) as well as radiotherapy [195]. However phase I and phase II clinical trials of amifostine (740 mg/m2 i.v.) with CPT-11 in metastatic CRC patients did not show a protective effect against CPT-11 induced toxicities [196, 197]. In a combination phase I study of cisplatin with CPT-11 in children with refractory cancer, addition of amifostine (825 mg/m2) was evaluated. This approach encountered amifostine related hypocalcemia and no apparent benefit in terms of diarrhea [198].
5.11.6. Tamoxifen
CPT-11 blocks intestinal epithelial cells in G2 phase which coincided with CID. Therefore by blocking intestinal epithelial cells in G0/G1 would protect them from CPT-11 toxicities [199]. Tamoxifen an antiestrogen, G0/G1 blocker [200] reduced CPT-11 induced weight loss and colonic damage in hamsters. Tamoxifen has also shown to potentiate SN-38 toxicity in Caco2 tumor cells [199].
5.11.7. Other Agents
Though selenium compounds reduced CPT-11 toxicities in mice [201], selenomethionine with CPT-11 in humans did not show any significant protection against CPT-11 induced toxicities [202, 203]. Co-administration of sulphobromophthalein (20 mg/body), a drug used for testing liver function with CPT-11 (500 mg/body) in Wistar rats partially inhibited the biliary excretion of SN-38G with a concomitant increase in its AUC. No significant alteration in biliary excretion and AUC of SN-38 was observed. These results suggest that sulphobromophthalein might decrease intraluminal SN-38 concentrations without altering its pharmacokinetics [204]. Intraperitoneal propolis preparation and related flavi-noids also seem to decrease CPT-11 induced toxicity effects in normal cells without affecting CPT-11 cytotoxicity in Ehrlich ascites tumor cells bearing mice [205]. Green tea poly-phenols have shown to partially ameliorated CPT-11 induced intestinal side effects, prevented increase of glutathione di-sulfide and MPO activity in ileum in mice [206]. JO-1, a recombinant adenovirus serotype 3–derived protein has shown to allow decrease in effective CPT-11 dosage and reduce CPT-11 mediated intestinal epithelial damage in pre-clinical models. However mild transient diarrhea is itself a side effect of this novel epithelial junction opener [207]. But most of these novel agents need to be tested in clinical trials.
CONCLUSIONS
CPT-11 is being used in the treatment of colorectal and various other cancers for almost two decades. However, CID continues to be a major problem, remains unpredictable and dose limiting. It is often severe, leads to dose reductions, omissions and admissions. This not only lowers efficacy of CPT-11 but also increases health costs and decreases quality of life. A lot of work has been done in past several years with a number of agents for prophylaxis of CID. But even after that results are not satisfactory with each agent having its own set of limitations (Table 2, Fig. 4). The reason for these failures can also be contributed to the complex metabolism of CPT-11, which encompasses a number of enzymes, metabolites, transporters and enterohepatic recycling. It is further compounded by different patient genotypes and clinical risk factors.
Table 2.
Experimental Strategies and Therapies for Prophylaxis of CPT-11 Induced Late Onset Diarrhea
| Section No. | Strategy or therapy | Possible Mechanisms of Action | Limitations and Comments | References |
|---|---|---|---|---|
| 5.1 | Schedule/dose modification | Decreased SN-38 intestinal exposure | May lead to administration of subtherapeutic dosage. Protracted dosage also associated with severe CID. May be beneficial with other strategies | [41, 50–62] |
| 5.2 | Structural and chemical modification- Novel analogs and drug delivery methods | Targeted delivery, less SN-38 intestinal exposure, higher efficacy | Though in development for a decade, but no encouraging results. CID is still a problem in some and efficacy is not established. | [63–74] |
| 5.3 | Genetic testing | Dose modification in susceptible patients based on genotyping | Clear association with CID unproved. Also no guidelines for dose reduction and even after that efficacy need to be tested. | [75–79] |
| 5.4 | Intestinal alkalization | Tilting equilibrium towards less gastrotoxic carboxylate isomer, decreasing intestinal reabsorption of CPT-11 and SN-38. | Daily consumption of highly alkalized 2–3 L water per day is cumbersome. Effective with defecation control but compliance will be a problem. | [80–83] |
| 5.5.1 | Loperamide | Retardation of small intestinal transit, stimulation of anal sphincter pressure and fecal continence, opiate agonsim, calcium channel blockage, calcium-calmodulin antagonist, selective inhibition of CPT-11 biliary excretion in rats. | Aggressive treatment with loperamide beginning with first sign of diarrhea is the most effective strategy. Once CID is fulminant rarely successful. Even with loperamide 1/3 patients develop severe diarrhea. Decreased gut motility can lead to extended SN-38 exposure of intestinal mucosa which may augment mucosal damage. | [14, 32, 208, 209] |
| 5.5.2 | Octreotide | Antisecretory, prolongs intestinal transit time, improves absorption. | Recommended second line strategy for treatment. Decreased intestinal transit can cause more intestinal damage. Quality studies for prophylaxis lacking. | [14, 87] |
| 5.5.3 | Acetorphan | Enkephalinase inhibitor, antidiarrheal activity and antisecretory mechanism | Failed to show activity as prophylactic agent. | [88, 89] |
| 5.5.4 | Budesonide | Anti inflammatory | Can be recommended. However unknown safety data in clinical setting. Can cause possible toxicities dry mouth, stomach upset, bad taste etc. | [90, 91] |
| 5.6.1 | Cyclosporine A | Decrease in CPT-11 biliary excretion by ABCB1, ABCC2 and ABCG2 inhibition. | Cyclosporine toxicity. Prophylaxis not complete. | [93–96, 98] |
| 5.6.2 | Probenecid | ABCC2 inhibition | Untested in clinical setting. Can itself cause gastrointestinal toxicities. | [76, 101] |
| 5.6.3 | GF120918, PEG 2000 stearate | ABCG2 inhibitors | Untested in clinical setting. Due to multiple receptors may not be totally effective in prophylaxis. | [102] |
| 5.7.1.1 | Kampo, TJ-14 | β-glucuronidase inhibitors, UGT1A1 inhibition | Compliance is poor due to odor, taste and constipation. Quality control is a matter of concern. Not fully effective clinically. | [103–114] |
| 5.7.2 | Chrysin | UGT1A1 upregulator | CID still 10%. Results of phase II awaited. | [119–121] |
| 5.7.3 | Carboxylesterases inhibition | Intestinal carboxylesterase inhibition | Results awaited in preclinical models. Still a long time to know clinical benefit. | [122–126] |
| 5.7.4 | Anti-epileptic drugs | CYP3A4 inducers | Increased CPT-11 MTD twofold. However anti epileptic drugs have narrow therapeutic index. So caution should be taken. | [6, 128] |
| 5.7.5 | Celecoxib | COX-2and PGE2 inhibition | Encouraging activity shown in preclinical models absent in clinical trials. | [130–135] |
| 5.8.1 | Probiotics | Inhibitory β-glucuronidase activity, epithelial proliferation, decreased intestinal apoptosis and prevents increase in goblet cell number and mucin secretion | Though only moderately effective, are cheap, non-toxic and convenient prophylactic agents. | [138–140] |
| 5.8.2 | Antibiotics | Elimination of β glucuronidase producing bacteria. Streptomycin also decrease intestinal absorption of CPT-11 and CES activity and increase UGT activity in the intestinal epithelium | Feasible but additional antibiotic toxicities are a problem. They include C. difficile, enterohemorrhagic E. coli, fungal infection, due to removal of commensal bacteria and toxicities due to antibiotics. | [46, 141–157] |
| 5.9.1 | Activated charcoal | Decreased CPT-11 absorption | Incomplete effect, can absorb other orally administered comedications and is cumbersome due to delivery three times per day. | [158–161] |
| 5.9.2 | AST-120 | Decreased CPT-11 absorption | Incomplete effect, can absorb other orally administered comedications and is cumbersome due to delivery three times per day. | [162, 163] |
| 5.10.1 | Thalidomide | TNF-α, IL-1β, IL-6 and IFN-γ inhibition, inhibition of ABCB1, ABCC2. | Showed encouraging results in preventing CID but further studies are warranted in this regard. | [33, 48, 164–172] |
| 5.10.2 | Velafermin | Epithelial repair | Some doses appeared to increase the diarrhea and mortality. Also FGF has a role in tumor growth. Not tested in clinical trials. | [173] |
| 5.10.3 | IL-15 | Apoptosis inhibition | Not tested in clinical trials. Also MTD increase didn’t result in enhanced response in preclinical studies.. | [174] |
| 5.10.4 | RDP58 | TNF-α, IL-12 and IFN-γ inhibition, anti-inflammatory | Not tested in clinical trials. | [175] |
| 5.10.5 | JBT-3002 | IL-1α, IL-1β, IL-6, IL-15, TNF-α and nitric oxide activation | Not tested in clinical trials. | [176, 177] |
| 5.10.6 | Palifermin | Antimucotoxic | Did not preserve intestinal morphometry and gastro intestinal apoptosis nor was totally effective in preventing diarrhea in rats. | [178, 179] |
| 5.10.7 | St. John’s wort | Anti-inflammatory | Altered pharmacokinetics of CPT-11 due to CYP450 and PgP induction | [180–183] |
| 5.11.1 | Oil supplementation | Mucosal protection | In preclinical models enhanced CPT-11 efficacy but didn’t prevent intestinal toxicities. Not tested in clinical trials. | [45, 137, 184] |
| 5.11.2 | GPI 15427 | inhibition of poly (ADP-ribose) polymerase | Not tested in clinical trials. | [185] |
| 5.11.3 | Glutamine | Increase in heat shock proteins 25, −70 and −90α, prevention of β glucuronidase upregulation, increased ratio of reduced glutathione to oxidized glutathione | Cumbersome as glutamine needs to be taken three or four times a day. No benefit in phase II trials. | [186–190] |
| 5.11.4 | PHY906 | Promote expression of intestinal progenitor or stem cell markers and increase proliferative cells in the intestine | Potentiated antitumor activity and proliferated cells in the intestine after CPT-11 treatment. Quality control is a matter of concern. Not fully effective clinically. Needs larger phase I trials. | [191–194] |
| 5.11.5 | Amifostine | Cytoprotectant | Potential for dose escalation but additional amifostine toxicities | [198] |
| 5.11.6 | Tamoxifen | G0/G1 blocker | Not tested in clinical trials. | [199] |
| 5.11.7 | Selenomethionine | increased phosphorylation of kinase chk2, down-regulation of cdc6 expression, increased poly(ADP-ribose) polymerase and activation of caspase-3 | Benefit evident in preclinical models was not seen in clinical trials. | [201–203] |
Fig. 4.

Strategies to Target CPT-11 induced Diarrhea.
It has been postulated that with complete prophylaxis of CID there can be further dose escalations of CPT-11 with enhanced tumor response. It will also definitely lead to better quality of life and drastically cut down health costs. Therefore, further research is warranted in mechanism of CID and targeted strategies like bacterial β-glucuronidase inhibition, individualized therapy based on genotyping and intratumoral drug activation. Also careful management of polypharmacy [210] and proper use of CPT-11 in only indicated situations [211] can help in bringing down incidence of CID.
Acknowledgments
The work was supported in part by NIH (CA 127231) and the Damon Runyon Clinical Investigator Award.
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Tanizawa A, Fujimori A, Fujimori Y, Pommier Y. Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J Natl Cancer Inst. 1994;86(11):836–42. doi: 10.1093/jnci/86.11.836. [DOI] [PubMed] [Google Scholar]
- 3.Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In vitro. 2006;20(2):163–75. doi: 10.1016/j.tiv.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 4.NCCN. [accessed Nov 16, 2012];National Comprehensive Cancer Network Guidelines. http://www.nccn.org/professionals/physician_gls/pdf/colon.pdf.
- 5.Rosen LS. Irinotecan in lymphoma, leukemia, and breast, pancreatic, ovarian, and small-cell lung cancers. Oncology (Williston Park) 1998;12(8 Suppl 6):103–9. [PubMed] [Google Scholar]
- 6.Prados MD, Yung WK, Jaeckle KA, et al. Phase 1 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium study. Neuro Oncol. 2004;6(1):44–54. doi: 10.1215/S1152851703000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armand JP. CPT-11: clinical experience in phase I studies. Semin Oncol. 1996;23(1 Suppl 3):27–33. [PubMed] [Google Scholar]
- 8.Hecht JR, Pillai M, Gollard R, et al. A randomized, placebo-controlled phase ii study evaluating the reduction of neutropenia and febrile neutropenia in patients with colorectal cancer receiving pegfilgrastim with every-2-week chemotherapy. Clin Colorectal Cancer. 2010;9(2):95–101. doi: 10.3816/CCC.2010.n.013. [DOI] [PubMed] [Google Scholar]
- 9.Merrouche Y, Extra JM, Abigerges D, et al. High dose-intensity of irinotecan administered every 3 weeks in advanced cancer patients: a feasibility study. J Clin Oncol. 1997;15(3):1080–6. doi: 10.1200/JCO.1997.15.3.1080. [DOI] [PubMed] [Google Scholar]
- 10.Rothenberg ML, Meropol NJ, Poplin EA, Van Cutsem E, Wadler S. Mortality associated with irinotecan plus bolus fluorouracil/leucovorin: summary findings of an independent panel. J Clin Oncol. 2001;19(18):3801–7. doi: 10.1200/JCO.2001.19.18.3801. [DOI] [PubMed] [Google Scholar]
- 11.Rougier P, Bugat R, Douillard JY, et al. Phase II study of irinotecan in the treatment of advanced colorectal cancer in chemotherapy-naive patients and patients pretreated with fluorouracil-based chemotherapy. J Clin Oncol. 1997;15(1):251–60. doi: 10.1200/JCO.1997.15.1.251. [DOI] [PubMed] [Google Scholar]
- 12.Bleiberg H, Cvitkovic E. Characterisation and clinical management of CPT-11 (irinotecan)-induced adverse events: the European perspective. Eur J Cancer. 1996;32A (Suppl 3):S18–23. doi: 10.1016/0959-8049(96)00293-6. [DOI] [PubMed] [Google Scholar]
- 13.Cunningham D, Pyrhonen S, James RD, et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet. 1998;352(9138):1413–8. doi: 10.1016/S0140-6736(98)02309-5. [DOI] [PubMed] [Google Scholar]
- 14.Benson AB, 3rd, Ajani JA, Catalano RB, et al. Recommended guidelines for the treatment of cancer treatment-induced diarrhea. J Clin Oncol. 2004;22(14):2918–26. doi: 10.1200/JCO.2004.04.132. [DOI] [PubMed] [Google Scholar]
- 15.Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, Bosron WF. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos. 2004;32(5):505–11. doi: 10.1124/dmd.32.5.505. [DOI] [PubMed] [Google Scholar]
- 16.Wu MH, Chen P, Remo BF, Cook EH, Jr, Das S, Dolan ME. Characterization of multiple promoters in the human carboxylesterase 2 gene. Pharmacogenetics. 2003;13(7):425–35. doi: 10.1097/00008571-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Dodds HM, Haaz MC, Riou JF, Robert J, Rivory LP. Identification of a new metabolite of CPT-11 (irinotecan): pharmacological properties and activation to SN-38. J Pharmacol Exp Ther. 1998;286(1):578–83. [PubMed] [Google Scholar]
- 18.Haaz MC, Rivory L, Riche C, Vernillet L, Robert J. Metabolism of irinotecan (CPT-11) by human hepatic microsomes: participation of cytochrome P-450 3A and drug interactions. Cancer Res. 1998;58(3):468–72. [PubMed] [Google Scholar]
- 19.Santos A, Zanetta S, Cresteil T, et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res. 2000;6(5):2012–20. [PubMed] [Google Scholar]
- 20.Lokiec F, du Sorbier BM, Sanderink GJ. Irinotecan (CPT-11) metabolites in human bile and urine. Clin Cancer Res. 1996;2(12):1943–9. [PubMed] [Google Scholar]
- 21.Rivory LP, Riou JF, Haaz MC, et al. Identification and properties of a major plasma metabolite of irinotecan (CPT-11) isolated from the plasma of patients. Cancer Res. 1996;56(16):3689–94. [PubMed] [Google Scholar]
- 22.Ciotti M, Basu N, Brangi M, Owens IS. Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38) by the human UDP-glucuronosyltransferases encoded at the UGT1 locus. Biochem Biophys Res Commun. 1999;260(1):199–202. doi: 10.1006/bbrc.1999.0453. [DOI] [PubMed] [Google Scholar]
- 23.Gagne JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) Mol Pharmacol. 2002;62(3):608–17. doi: 10.1124/mol.62.3.608. [DOI] [PubMed] [Google Scholar]
- 24.Iyer L, King CD, Whitington PF, et al. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest. 1998;101(4):847–54. doi: 10.1172/JCI915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed F, Vyas V, Cornfield A, et al. In vitro activation of irinotecan to SN-38 by human liver and intestine. Anticancer Res. 1999;19(3A):2067–71. [PubMed] [Google Scholar]
- 26.Kehrer DF, Sparreboom A, Verweij J, et al. Modulation of irinotecan-induced diarrhea by cotreatment with neomycin in cancer patients. Clin Cancer Res. 2001;7(5):1136–41. [PubMed] [Google Scholar]
- 27.Catimel G, Chabot GG, Guastalla JP, et al. Phase I and pharmacokinetic study of irinotecan (CPT-11) administered daily for three consecutive days every three weeks in patients with advanced solid tumors. Ann Oncol. 1995;6(2):133–40. doi: 10.1093/oxfordjournals.annonc.a059108. [DOI] [PubMed] [Google Scholar]
- 28.Slatter JG, Schaaf LJ, Sams JP, et al. Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos. 2000;28(4):423–33. [PubMed] [Google Scholar]
- 29.Gandia D, Abigerges D, Armand JP, et al. CPT-11-induced cholinergic effects in cancer patients. J Clin Oncol. 1993;11(1):196–7. doi: 10.1200/JCO.1993.11.1.196. [DOI] [PubMed] [Google Scholar]
- 30.Rowinsky EK, Grochow LB, Ettinger DS, et al. Phase I and pharmacological study of the novel topoisomerase I inhibitor 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) administered as a ninety-minute infusion every 3 weeks. Cancer Res. 1994;54(2):427–36. [PubMed] [Google Scholar]
- 31.Zampa G, Magnolfi E, Borgomastro A. Premedication for irinotecan. J Clin Oncol. 2000;18(1):237. [PubMed] [Google Scholar]
- 32.Abigerges D, Chabot GG, Armand JP, Herait P, Gouyette A, Gandia D. Phase I and pharmacologic studies of the camptothecin analog irinotecan administered every 3 weeks in cancer patients. J Clin Oncol. 1995;13(1):210–21. doi: 10.1200/JCO.1995.13.1.210. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Hu Z, Chan SY, et al. Novel agents that potentially inhibit irinotecan-induced diarrhea. Curr Med Chem. 2005;12(11):1343–58. doi: 10.2174/0929867054020972. [DOI] [PubMed] [Google Scholar]
- 34.Lokiec F, Canal P, Gay C, et al. Pharmacokinetics of irinotecan and its metabolites in human blood, bile, and urine. Cancer Chemother Pharmacol. 1995;36(1):79–82. doi: 10.1007/BF00685737. [DOI] [PubMed] [Google Scholar]
- 35.Hecht JR. Gastrointestinal toxicity or irinotecan. Oncology (Williston Park) 1998;12(8 Suppl 6):72–8. [PubMed] [Google Scholar]
- 36.Nakao T, Kurita N, Komatsu M, et al. Irinotecan Injures Tight Junction and Causes Bacterial Translocation in Rat. J Surg Res. 2012;173(2):341–7. doi: 10.1016/j.jss.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Logan RM, Gibson RJ, Bowen JM, Stringer AM, Sonis ST, Keefe DM. Characterisation of mucosal changes in the alimentary tract following administration of irinotecan: implications for the pathobiology of mucositis. Cancer Chemother Pharmacol. 2008;62(1):33–41. doi: 10.1007/s00280-007-0570-0. [DOI] [PubMed] [Google Scholar]
- 38.Gibson RJ, Bowen JM, Inglis MR, Cummins AG, Keefe DM. Irinotecan causes severe small intestinal damage, as well as colonic damage, in the rat with implanted breast cancer. J Gastroenterol Hepatol. 2003;18(9):1095–100. doi: 10.1046/j.1440-1746.2003.03136.x. [DOI] [PubMed] [Google Scholar]
- 39.Ikuno N, Soda H, Watanabe M, Oka M. Irinotecan (CPT-11) and characteristic mucosal changes in the mouse ileum and cecum. J Natl Cancer Inst. 1995;87(24):1876–83. doi: 10.1093/jnci/87.24.1876. [DOI] [PubMed] [Google Scholar]
- 40.Stringer AM, Gibson RJ, Logan RM, et al. Irinotecan-induced mucositis is associated with changes in intestinal mucins. Cancer Chemother Pharmacol. 2009;64(1):123–32. doi: 10.1007/s00280-008-0855-y. [DOI] [PubMed] [Google Scholar]
- 41.Kurita A, Kado S, Kaneda N, Onoue M, Hashimoto S, Yokokura T. Alleviation of side effects induced by irinotecan hydrochloride (CPT-11) in rats by intravenous infusion. Cancer Chemother Pharmacol. 2003;52(5):349–60. doi: 10.1007/s00280-003-0682-0. [DOI] [PubMed] [Google Scholar]
- 42.Kase Y, Hayakawa T, Togashi Y, Kamataki T. Relevance of irinotecan hydrochloride-induced diarrhea to the level of prostaglandin E2 and water absorption of large intestine in rats. Jpn J Pharmacol. 1997;75(4):399–405. doi: 10.1254/jjp.75.399. [DOI] [PubMed] [Google Scholar]
- 43.Sakai H, Sato T, Hamada N, et al. Thromboxane A2, released by the anti-tumour drug irinotecan, is a novel stimulator of Cl- secretion in isolated rat colon. J Physiol. 1997;505 (Pt 1):133–44. doi: 10.1111/j.1469-7793.1997.133bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakai H, Diener M, Gartmann V, Takeguchi N. Eicosanoid-mediated Cl- secretion induced by the antitumor drug, irinotecan (CPT-11), in the rat colon. Naunyn Schmiedebergs Arch Pharmacol. 1995;351(3):309–14. doi: 10.1007/BF00233252. [DOI] [PubMed] [Google Scholar]
- 45.Usami M, Ohata A, Kishimoto K, et al. Phospholipid fatty acid composition and diamine oxidase activity of intestinal mucosa from rats treated with irinotecan hydrochloride (CPT-11) under vegetable oil-enriched diets: comparison between perilla oil and corn oil. JPEN J Parenter Enteral Nutr. 2006;30(2):124–32. doi: 10.1177/0148607106030002124. [DOI] [PubMed] [Google Scholar]
- 46.Brandi G, Dabard J, Raibaud P, et al. Intestinal microflora and digestive toxicity of irinotecan in mice. Clin Cancer Res. 2006;12(4):1299–307. doi: 10.1158/1078-0432.CCR-05-0750. [DOI] [PubMed] [Google Scholar]
- 47.Stringer AM, Gibson RJ, Bowen JM, et al. Irinotecan-induced mucositis manifesting as diarrhoea corresponds with an amended intestinal flora and mucin profile. Int J Exp Pathol. 2009;90(5):489–99. doi: 10.1111/j.1365-2613.2009.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melo ML, Brito GA, Soares RC, et al. Role of cytokines (TNF-alpha, IL-1beta and KC) in the pathogenesis of CPT-11-induced intestinal mucositis in mice: effect of pentoxifylline and thalidomide. Cancer Chemother Pharmacol. 2008;61(5):775–84. doi: 10.1007/s00280-007-0534-4. [DOI] [PubMed] [Google Scholar]
- 49.Alimonti A, Gelibter A, Pavese I, et al. New approaches to prevent intestinal toxicity of irinotecan-based regimens. Cancer Treat Rev. 2004;30(6):555–62. doi: 10.1016/j.ctrv.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Kurita A, Kado S, Kaneda N, Onoue M, Hashimoto S, Yokokura T. Modified irinotecan hydrochloride (CPT-11) administration schedule improves induction of delayed-onset diarrhea in rats. Cancer Chemother Pharmacol. 2000;46(3):211–20. doi: 10.1007/s002800000151. [DOI] [PubMed] [Google Scholar]
- 51.Furuta T, Yokokura T. Effect of administration schedules on the antitumor activity of CPT-11, a camptothecin derivative. Gan To Kagaku Ryoho. 1990;17(1):121–30. [PubMed] [Google Scholar]
- 52.Gerrits CJ, de Jonge MJ, Schellens JH, Stoter G, Verweij J. Topoisomerase I inhibitors: the relevance of prolonged exposure for present clinical development. Br J Cancer. 1997;76(7):952–62. doi: 10.1038/bjc.1997.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takimoto CH, Morrison G, Harold N, et al. Phase I and pharmacologic study of irinotecan administered as a 96-hour infusion weekly to adult cancer patients. J Clin Oncol. 2000;18(3):659–67. doi: 10.1200/JCO.2000.18.3.659. [DOI] [PubMed] [Google Scholar]
- 54.Herben VM, Schellens JH, Swart M, et al. Phase I and pharmacokinetic study of irinotecan administered as a low-dose, continuous intravenous infusion over 14 days in patients with malignant solid tumors. J Clin Oncol. 1999;17(6):1897–905. doi: 10.1200/JCO.1999.17.6.1897. [DOI] [PubMed] [Google Scholar]
- 55.Ma MK, Zamboni WC, Radomski KM, et al. Pharmacokinetics of irinotecan and its metabolites SN-38 and APC in children with recurrent solid tumors after protracted low-dose irinotecan. Clin Cancer Res. 2000;6(3):813–9. [PubMed] [Google Scholar]
- 56.Ohe Y, Sasaki Y, Shinkai T, et al. Phase I study and pharmacokinetics of CPT-11 with 5-day continuous infusion. J Natl Cancer Inst. 1992;84(12):972–4. doi: 10.1093/jnci/84.12.972. [DOI] [PubMed] [Google Scholar]
- 57.Fujii H, Koshiyama M, Konishi M, Yoshida M, Tauchi K. Intermittent, repetitive administrations of irinotecan (CPT-11) reduces its side-effects. Cancer Detect Prev. 2002;26(3):210–2. doi: 10.1016/s0361-090x(02)00060-0. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi Y, Kitakata H, Yamashita K, et al. Pilot study of low-dose, divided maximum tolerated dose of CPT-11 in 21 consecutive patients with metastatic colorectal or gastric cancer. Surg Today. 2004;34(3):246–50. doi: 10.1007/s00595-003-2688-y. [DOI] [PubMed] [Google Scholar]
- 59.Fuchs CS, Moore MR, Harker G, Villa L, Rinaldi D, Hecht JR. Phase III comparison of two irinotecan dosing regimens in second-line therapy of metastatic colorectal cancer. J Clin Oncol. 2003;21(5):807–14. doi: 10.1200/JCO.2003.08.058. [DOI] [PubMed] [Google Scholar]
- 60.Labianca R, Sobrero A, Isa L, et al. Intermittent versus continuous chemotherapy in advanced colorectal cancer: a randomised ‘GISCAD’ trial. Ann Oncol. 2011;22(5):1236–42. doi: 10.1093/annonc/mdq580. [DOI] [PubMed] [Google Scholar]
- 61.Hebbar M, Ychou M, Ducreux M. Current place of high-dose irinotecan chemotherapy in patients with metastatic colorectal cancer. J Cancer Res Clin Oncol. 2009;135(6):749–52. doi: 10.1007/s00432-009-0580-x. [DOI] [PubMed] [Google Scholar]
- 62.Mineur L, Sabatier R, Kirscher S, Plat F, Goubely Y, Molinari N. Are we turning to more than a first line treatment of metastatic colorectal cancer with high dose irinotecan?: A monocentric institution safety analysis of 46 patients. Clin Res Hepatol Gastroenterol. 2011;35(2):125–31. doi: 10.1016/j.gcb.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 63.Kurzrock R, Goel S, Wheler J, et al. Safety, pharmacokinetics, and activity of EZN-2208, a novel conjugate of polyethylene glycol and SN38, in patients with advanced malignancies. Cancer. 2012;118(24):6144–51. doi: 10.1002/cncr.27647. [DOI] [PubMed] [Google Scholar]
- 64.Azim HA, Jr, Awada A. Clinical development of new formulations of cytotoxics in solid tumors. Curr Opin Oncol. 2012;24(3):325–31. doi: 10.1097/CCO.0b013e328351fb29. [DOI] [PubMed] [Google Scholar]
- 65.Hamaguchi T, Doi T, Eguchi-Nakajima T, et al. Phase I study of NK012, a novel SN-38-incorporating micellar nanoparticle, in adult patients with solid tumors. Clin Cancer Res. 2010;16(20):5058–66. doi: 10.1158/1078-0432.CCR-10-0387. [DOI] [PubMed] [Google Scholar]
- 66.Govindan SV, Cardillo TM, Moon SJ, Hansen HJ, Goldenberg DM. CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clin Cancer Res. 2009;15(19):6052–61. doi: 10.1158/1078-0432.CCR-09-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Infante JR, Keedy VL, Jones SF, et al. Phase I and pharmacokinetic study of IHL-305 (PEGylated liposomal irinotecan) in patients with advanced solid tumors. Cancer Chemotherapy Pharmacol. 2012;70(5):699–705. doi: 10.1007/s00280-012-1960-5. [DOI] [PubMed] [Google Scholar]
- 68.Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270(1–2):93–107. doi: 10.1016/j.ijpharm.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 69.Batist G, Gelmon KA, Chi KN, et al. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin Cancer Res. 2009;15(2):692–700. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]
- 70.Noble CO, Krauze MT, Drummond DC, et al. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006;66(5):2801–6. doi: 10.1158/0008-5472.CAN-05-3535. [DOI] [PubMed] [Google Scholar]
- 71.Gibbs P, Clingan PR, Ganju V, et al. Hyaluronan-Irinotecan improves progression-free survival in 5-fluorouracil refractory patients with metastatic colorectal cancer: a randomized phase II trial. Cancer Chemother Pharmacol. 2011;67(1):153–63. doi: 10.1007/s00280-010-1303-3. [DOI] [PubMed] [Google Scholar]
- 72.Venditto VJ, Simanek EE. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm. 2010;7(2):307–49. doi: 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang PT, Chen KC, Prijovich ZM, Cheng TL, Leu YL, Roffler SR. Enhancement of CPT-11 antitumor activity by adenovirus-mediated expression of beta-glucuronidase in tumors. Cancer Gene Ther. 2011;18(6):381–9. doi: 10.1038/cgt.2011.3. [DOI] [PubMed] [Google Scholar]
- 74.Kojima A, Hackett NR, Ohwada A, Crystal RG. In vivo human carboxylesterase cDNA gene transfer to activate the prodrug CPT-11 for local treatment of solid tumors. J Clin Invest. 1998;101(8):1789–96. doi: 10.1172/JCI119888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Canal P, Gay C, Dezeuze A, et al. Pharmacokinetics and pharmacodynamics of irinotecan during a phase II clinical trial in colorectal cancer. Pharmacology and Molecular Mechanisms Group of the European Organization for Research and Treatment of Cancer. J Clin Oncol. 1996;14(10):2688–95. doi: 10.1200/JCO.1996.14.10.2688. [DOI] [PubMed] [Google Scholar]
- 76.Mathijssen RH, van Alphen RJ, Verweij J, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clin Cancer Res. 2001;7(8):2182–94. [PubMed] [Google Scholar]
- 77.Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22(8):1382–8. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 78.Hu ZY, Yu Q, Zhao YS. Dose-dependent association between UGT1A1*28 polymorphism and irinotecan-induced diarrhoea: a meta-analysis. Eur J Cancer. 2010;46(10):1856–65. doi: 10.1016/j.ejca.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 79.Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med. 2009;11(1):15–20. doi: 10.1097/GIM.0b013e31818efd9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kobayashi K, Bouscarel B, Matsuzaki Y, Ceryak S, Kudoh S, Fromm H. pH-dependent uptake of irinotecan and its active metabolite, SN-38, by intestinal cells. Int J Cancer. 1999;83(4):491–6. doi: 10.1002/(sici)1097-0215(19991112)83:4<491::aid-ijc10>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 81.Ikegami T, Ha L, Arimori K, et al. Intestinal alkalization as a possible preventive mechanism in irinotecan (CPT-11)-induced diarrhea. Cancer Res. 2002;62(1):179–87. [PubMed] [Google Scholar]
- 82.Anami S, Saegusa K, Nishikata M, et al. Effect of glutamine or alkaline ionized water on late diarrhea induced by irinotecan hydrochloride in Gunn rats. Asian Journal of Pharmaceutical Sciences. 2009;4(2):96–105. [Google Scholar]
- 83.Takeda Y, Kobayashi K, Akiyama Y, et al. Prevention of irinotecan (CPT-11)-induced diarrhea by oral alkalization combined with control of defecation in cancer patients. Int J Cancer. 2001;92(2):269–75. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1179>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 84.Valenti Moreno V, Brunet Vidal J, Manzano Alemany H, et al. Prevention of irinotecan associated diarrhea by intestinal alkalization. A pilot study in gastrointestinal cancer patients. Clin Transl Oncol. 2006;8(3):208–12. doi: 10.1007/s12094-006-0012-1. [DOI] [PubMed] [Google Scholar]
- 85.Tamura T, Yasutake K, Nishisaki H, et al. Prevention of irinotecan-induced diarrhea by oral sodium bicarbonate and influence on pharmacokinetics. Oncology. 2004;67(5–6):327–37. doi: 10.1159/000082915. [DOI] [PubMed] [Google Scholar]
- 86.Hamada A, Aoki A, Terazaki H, et al. Pharmacokinetic changes of irinotecan by intestinal alkalinization in an advanced colorectal cancer patient. Ther Drug Monit. 2005;27(4):536–8. doi: 10.1097/01.ftd.0000165505.01298.00. [DOI] [PubMed] [Google Scholar]
- 87.Rosenoff SH. Octreotide LAR resolves severe chemotherapy-induced diarrhoea (CID) and allows continuation of full-dose therapy. Eur J Cancer Care (Engl) 2004;13(4):380–3. doi: 10.1111/j.1365-2354.2004.00511.x. [DOI] [PubMed] [Google Scholar]
- 88.Goncalves E, de Costa L, Abigerges D, Armand JP. A new enkephalinase inhibitor as an alternative to loperamide in the prevention of diarrhea induced by CPT-11. J Clin Oncol. 1995;13(8):2144–6. doi: 10.1200/JCO.1995.13.8.2144. [DOI] [PubMed] [Google Scholar]
- 89.Ychou M, Douillard JY, Rougier P, et al. Randomized comparison of prophylactic antidiarrheal treatment versus no prophylactic antidiarrheal treatment in patients receiving CPT-11 (irinotecan) for advanced 5-FU-resistant colorectal cancer: an open-label multicenter phase II study. Am J Clin Oncol. 2000;23(2):143–8. doi: 10.1097/00000421-200004000-00008. [DOI] [PubMed] [Google Scholar]
- 90.Lenfers BH, Loeffler TM, Droege CM, Hausamen TU. Substantial activity of budesonide in patients with irinotecan (CPT-11) and 5-fluorouracil induced diarrhea and failure of loperamide treatment. Ann Oncol. 1999;10(10):1251–3. doi: 10.1023/a:1008390308416. [DOI] [PubMed] [Google Scholar]
- 91.Karthaus M, Ballo H, Abenhardt W, et al. Prospective, double-blind, placebo-controlled, multicenter, randomized phase III study with orally administered budesonide for prevention of irinotecan (CPT-11)-induced diarrhea in patients with advanced colorectal cancer. Oncology. 2005;68(4–6):326–32. doi: 10.1159/000086971. [DOI] [PubMed] [Google Scholar]
- 92.Duffour J, Gourgou S, Seitz JF, et al. Efficacy of prophylactic anti-diarrhoeal treatment in patients receiving Campto for advanced colorectal cancer. Anticancer Res. 2002;22(6B):3727–31. [PubMed] [Google Scholar]
- 93.Gupta E, Safa AR, Wang X, Ratain MJ. Pharmacokinetic modulation of irinotecan and metabolites by cyclosporin A. Cancer Res. 1996;56(6):1309–14. [PubMed] [Google Scholar]
- 94.Chester JD, Joel SP, Cheeseman SL, et al. Phase I and pharmacokinetic study of intravenous irinotecan plus oral ciclosporin in patients with fuorouracil-refractory metastatic colon cancer. J Clin Oncol. 2003;21(6):1125–32. doi: 10.1200/JCO.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 95.Vasudev NS, Jagdev S, Anthoney DA, Seymour MT. Intravenous irinotecan plus oral ciclosporin. Clin Oncol (R Coll Radiol) 2005;17(8):646–9. doi: 10.1016/j.clon.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 96.Desai AA, Kindler HL, Taber D, et al. Modulation of irinotecan with cyclosporine: a phase II trial in advanced colorectal cancer. Cancer Chemother Pharmacol. 2005;56(4):421–6. doi: 10.1007/s00280-005-1020-5. [DOI] [PubMed] [Google Scholar]
- 97.Gupta E, Wang X, Ramirez J, Ratain MJ. Modulation of glucuronidation of SN-38, the active metabolite of irinotecan, by valproic acid and phenobarbital. Cancer Chemother Pharmacol. 1997;39(5):440–4. doi: 10.1007/s002800050595. [DOI] [PubMed] [Google Scholar]
- 98.Innocenti F, Undevia SD, Ramirez J, et al. A phase I trial of pharmacologic modulation of irinotecan with cyclosporine and phenobarbital. Clin Pharmacol Ther. 2004;76(5):490–502. doi: 10.1016/j.clpt.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 99.Bansal T, Mishra G, Jaggi M, Khar RK, Talegaonkar S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur J Pharm Sci. 2009;36(4–5):580–90. doi: 10.1016/j.ejps.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 100.Bansal T, Awasthi A, Jaggi M, Khar RK, Talegaonkar S. Preclinical evidence for altered absorption and biliary excretion of irinotecan (CPT-11) in combination with quercetin: possible contribution of P-glycoprotein. Life Sci. 2008;83(7–8):250–9. doi: 10.1016/j.lfs.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 101.Horikawa M, Kato Y, Sugiyama Y. Reduced gastrointestinal toxicity following inhibition of the biliary excretion of irinotecan and its metabolites by probenecid in rats. Pharm Res. 2002;19(9):1345–53. doi: 10.1023/a:1020358910490. [DOI] [PubMed] [Google Scholar]
- 102.Zhang XX, Pan WS, Gan L, Zhu CL, Gan Y. Effects of breast cancer resistance protein inhibitors and pharmaceutical excipients on decreasing gastrointestinal toxicity of camptothecin analogs. Acta Pharmacol Sin. 2008;29(11):1391–8. doi: 10.1111/j.1745-7254.2008.00883.x. [DOI] [PubMed] [Google Scholar]
- 103.Narita M, Nagai E, Hagiwara H, Aburada M, Yokoi T, Kamataki T. Inhibition of beta-glucuronidase by natural glucuronides of kampo medicines using glucuronide of SN-38 (7-ethyl-10-hydroxycamptothecin) as a substrate. Xenobiotica. 1993;23(1):5–10. doi: 10.3109/00498259309059356. [DOI] [PubMed] [Google Scholar]
- 104.Yokoi T, Narita M, Nagai E, Hagiwara H, Aburada M, Kamataki T. Inhibition of UDP-glucuronosyltransferase by aglycons of natural glucuronides in kampo medicines using SN-38 as a substrate. Jpn J Cancer Res. 1995;86(10):985–9. doi: 10.1111/j.1349-7006.1995.tb03011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Itagaki S. Intestinal absorption and secretion mechanism of carboxylate drugs. Yakugaku Zasshi. 2009;129(11):1341–9. doi: 10.1248/yakushi.129.1341. [DOI] [PubMed] [Google Scholar]
- 106.Katoh M, Yoshioka Y, Nakagawa N, Yokoi T. Effects of Japanese herbal medicine, Kampo, on human UGT1A1 activity. Drug Metab Pharmacokinet. 2009;24(3):226–34. doi: 10.2133/dmpk.24.226. [DOI] [PubMed] [Google Scholar]
- 107.Chikakiyo M, Shimada M, Nakao T, et al. Kampo medicine “Dai-kenchu-to” prevents CPT-11-induced small-intestinal injury in rats. Surgery Today. 2012;42(1):60–7. doi: 10.1007/s00595-011-0014-7. [DOI] [PubMed] [Google Scholar]
- 108.Kase Y, Hayakawa T, Ishige A, Aburada M, Komatsu Y. The effects of Hange-shashin-to on the content of prostaglandin E2 and water absorption in the large intestine of rats. Biol Pharm Bull. 1997;20(9):954–7. doi: 10.1248/bpb.20.954. [DOI] [PubMed] [Google Scholar]
- 109.Kito Y, Teramoto N. Effects of Hange-shashin-to (TJ-14) and Keishi-ka-shakuyaku-to (TJ-60) on contractile activity of circular smooth muscle of the rat distal colon. Am J Physiol Gastrointest Liver Physiol. 2012;303(9):G1059–66. doi: 10.1152/ajpgi.00219.2012. [DOI] [PubMed] [Google Scholar]
- 110.Takasuna K, Kasai Y, Kitano Y, et al. Protective effects of kampo medicines and baicalin against intestinal toxicity of a new anticancer camptothecin derivative, irinotecan hydrochloride (CPT-11), in rats. Jpn J Cancer Res. 1995;86(10):978–84. doi: 10.1111/j.1349-7006.1995.tb03010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kase Y, Hayakawa T, Aburada M, Komatsu Y, Kamataki T. Preventive effects of Hange-shashin-to on irinotecan hydrochloride-caused diarrhea and its relevance to the colonic prostaglandin E2 and water absorption in the rat. Jpn J Pharmacol. 1997;75(4):407–13. doi: 10.1254/jjp.75.407. [DOI] [PubMed] [Google Scholar]
- 112.Sakata Y, Suzuki H, Kamataki T. Preventive effect of TJ-14, a kampo (Chinese herb) medicine, on diarrhea induced by irinotecan hydrochloride (CPT-11) Gan To Kagaku Ryoho. 1994;21(8):1241–4. [PubMed] [Google Scholar]
- 113.Mori K, Kondo T, Kamiyama Y, Kano Y, Tominaga K. Preventive effect of Kampo medicine (Hangeshashin-to) against irinotecan-induced diarrhea in advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2003;51(5):403–6. doi: 10.1007/s00280-003-0585-0. [DOI] [PubMed] [Google Scholar]
- 114.Takasuna K, Hagiwara T, Watanabe K, et al. Optimal antidiarrhea treatment for antitumor agent irinotecan hydrochloride (CPT-11)-induced delayed diarrhea. Cancer Chemother Pharmacol. 2006;58(4):494–503. doi: 10.1007/s00280-006-0187-8. [DOI] [PubMed] [Google Scholar]
- 115.Fittkau M, Voigt W, Holzhausen HJ, Schmoll HJ. Saccharic acid 1. 4-lactone protects against CPT-11-induced mucosa damage in rats. J Cancer Res Clin Oncol. 2004;130(7):388–94. doi: 10.1007/s00432-004-0557-8. [DOI] [PubMed] [Google Scholar]
- 116.Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330(6005):831–5. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rasmussen TS, Koldso H, Nakagawa S, Kato A, Schiott B, Jensen HH. Synthesis of uronic-noeurostegine--a potent bacterial beta-glucuronidase inhibitor. Org Biomol Chem. 2011;9(22):7807–13. doi: 10.1039/c1ob06038d. [DOI] [PubMed] [Google Scholar]
- 118.Ahmad S, Hughes MA, Yeh LA, Scott JE. Potential repurposing of known drugs as potent bacterial beta-glucuronidase inhibitors. J Biomol Screen. 2012;17(7):957–65. doi: 10.1177/1087057112444927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Walle T, Otake Y, Brubaker JA, Walle UK, Halushka PV. Disposition and metabolism of the flavonoid chrysin in normal volunteers. Br J Clin Pharmacol. 2001;51(2):143–6. doi: 10.1111/j.1365-2125.2001.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Walle T, Otake Y, Galijatovic A, Ritter JK, Walle UK. Induction of UDP-glucuronosyltransferase UGT1A1 by the flavonoid chrysin in the human hepatoma cell line hep G2. Drug Metab Dispos. 2000;28(9):1077–82. [PubMed] [Google Scholar]
- 121.Tobin PJ, Beale P, Noney L, Liddell S, Rivory LP, Clarke S. A pilot study on the safety of combining chrysin, a non-absorbable inducer of UGT1A1, and irinotecan (CPT-11) to treat metastatic colorectal cancer. Cancer Chemother Pharmacol. 2006;57(3):309–16. doi: 10.1007/s00280-005-0053-0. [DOI] [PubMed] [Google Scholar]
- 122.Khanna R, Morton CL, Danks MK, Potter PM. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000;60(17):4725–8. [PubMed] [Google Scholar]
- 123.Wadkins RM, Hyatt JL, Yoon KJ, et al. Discovery of novel selective inhibitors of human intestinal carboxylesterase for the amelioration of irinotecan-induced diarrhea: synthesis, quantitative structure-activity relationship analysis, and biological activity. Mol Pharmacol. 2004;65(6):1336–43. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 124.Hyatt JL, Tsurkan L, Wierdl M, Edwards CC, Danks MK, Potter PM. Intracellular inhibition of carboxylesterases by benzil: modulation of CPT-11 cytotoxicity. Mol Cancer Ther. 2006;5(9):2281–8. doi: 10.1158/1535-7163.MCT-06-0160. [DOI] [PubMed] [Google Scholar]
- 125.Yoon KJ, Hyatt JL, Morton CL, Lee RE, Potter PM, Danks MK. Characterization of inhibitors of specific carboxylesterases: development of carboxylesterase inhibitors for translational application. Mol Cancer Ther. 2004;3(8):903–9. [PubMed] [Google Scholar]
- 126.Hicks LD, Hyatt JL, Stoddard S, et al. Improved, selective, human intestinal carboxylesterase inhibitors designed to modulate 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (Irinotecan; CPT-11) toxicity. J Med Chem. 2009;52(12):3742–52. doi: 10.1021/jm9001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Uchino J, Takayama K, Harada A, et al. Tumor targeting carboxylesterase fused with anti-CEA scFv improve the anticancer effect with a less toxic dose of irinotecan. Cancer Gene Ther. 2008;15(2):94–100. doi: 10.1038/sj.cgt.7701100. [DOI] [PubMed] [Google Scholar]
- 128.Loghin ME, Prados MD, Wen P, et al. Phase I study of temozolomide and irinotecan for recurrent malignant gliomas in patients receiving enzyme-inducing antiepileptic drugs: a north american brain tumor consortium study. Clin Cancer Res. 2007;13(23):7133–8. doi: 10.1158/1078-0432.CCR-07-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Voutsadakis IA. Pathogenesis of colorectal carcinoma and therapeutic implications: the roles of the ubiquitin-proteasome system and Cox-2. J Cell Mol Med. 2007;11(2):252–85. doi: 10.1111/j.1582-4934.2007.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Trifan OC, Durham WF, Salazar VS, et al. Cyclooxygenase-2 inhibition with celecoxib enhances antitumor efficacy and reduces diarrhea side effect of CPT-11. Cancer Res. 2002;62(20):5778–84. [PubMed] [Google Scholar]
- 131.Karim A, Boynton J, Wallemark C, Kent J, Frazier-O’Bannon L. Effects of celecoxib (cyp2c9 substrate) and omeprazole (cyp2c9 substrate/inhibitor) on systemic exposure to each other. AAPS Pharm Sci. 2001;3:T3669. [Google Scholar]
- 132.Masferrer JL, Leahy KM, Koki AT, et al. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000;60(5):1306–11. [PubMed] [Google Scholar]
- 133.Leahy KM, Ornberg RL, Wang Y, Zweifel BS, Koki AT, Masferrer JL. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res. 2002;62(3):625–31. [PubMed] [Google Scholar]
- 134.Javle MM, Cao S, Durrani FA, et al. Celecoxib and mucosal protection: translation from an animal model to a phase I clinical trial of celecoxib, irinotecan, and 5-fluorouracil. Clin Cancer Res. 2007;13(3):965–71. doi: 10.1158/1078-0432.CCR-06-0551. [DOI] [PubMed] [Google Scholar]
- 135.Fakih MG, Rustum YM. Does celecoxib have a role in the treatment of patients with colorectal cancer? Clin Colorectal Cancer. 2009;8(1):11–4. doi: 10.3816/CCC.2009.n.002. [DOI] [PubMed] [Google Scholar]
- 136.Gasparini G, Gattuso D, Morabito A, et al. Combined therapy with weekly irinotecan, infusional 5-fluorouracil and the selective COX-2 inhibitor rofecoxib is a safe and effective second-line treatment in metastatic colorectal cancer. Oncologist. 2005;10(9):710–7. doi: 10.1634/theoncologist.10-9-710. [DOI] [PubMed] [Google Scholar]
- 137.Xue H, Sawyer MB, Field CJ, Dieleman LA, Baracos VE. Nutritional modulation of antitumor efficacy and diarrhea toxicity related to irinotecan chemotherapy in rats bearing the ward colon tumor. Clin Cancer Res. 2007;13(23):7146–54. doi: 10.1158/1078-0432.CCR-07-0823. [DOI] [PubMed] [Google Scholar]
- 138.Bowen JM, Stringer AM, Gibson RJ, Yeoh AS, Hannam S, Keefe DM. VSL#3 probiotic treatment reduces chemotherapy-induced diarrhea and weight loss. Cancer Biol Ther. 2007;6(9):1449–54. doi: 10.4161/cbt.6.9.4622. [DOI] [PubMed] [Google Scholar]
- 139.Ooi K, Miya T, Sasaki H, Morimoto Y. Prevention of irinotecan hydrochloride-induced diarrhea by oral administration of Lactobacillus casei strain Shirota in rats. Gan To Kagaku Ryoho. 2008;35(6):951–4. [PubMed] [Google Scholar]
- 140.Sezer A, Usta U, Cicin I. The effect of Saccharomyces boulardii on reducing irinotecan-induced intestinal mucositis and diarrhea. Med Oncol. 2009;26(3):350–7. doi: 10.1007/s12032-008-9128-1. [DOI] [PubMed] [Google Scholar]
- 141.Takasuna K, Hagiwara T, Hirohashi M, et al. Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 1996;56(16):3752–7. [PubMed] [Google Scholar]
- 142.Said AA, Matsuki N, Kasuya Y. Effects of aminoglycoside antibiotics on cholinergic autonomic nervous transmission. Pharmacol Toxicol. 1995;76(2):128–32. doi: 10.1111/j.1600-0773.1995.tb00117.x. [DOI] [PubMed] [Google Scholar]
- 143.Takasuna K, Hagiwara T, Hirohashi M, et al. Inhibition of intestinal microflora beta-glucuronidase modifies the distribution of the active metabolite of the antitumor agent, irinotecan hydrochloride (CPT-11) in rats. Cancer Chemother Pharmacol. 1998;42(4):280–6. doi: 10.1007/s002800050818. [DOI] [PubMed] [Google Scholar]
- 144.Kurita A, Kado S, Matsumoto T, et al. Streptomycin alleviates irinotecan-induced delayed-onset diarrhea in rats by a mechanism other than inhibition of beta-glucuronidase activity in intestinal lumen. Cancer Chemother Pharmacol. 2011;67(1):201–13. doi: 10.1007/s00280-010-1310-4. [DOI] [PubMed] [Google Scholar]
- 145.Xue H, Field CJ, Sawyer MB, Dieleman LA, Baracos VE. Prophylactic ciprofloxacin treatment prevented high mortality, and modified systemic and intestinal immune function in tumour-bearing rats receiving dose-intensive CPT-11 chemotherapy. Br J Cancer. 2009;100(10):1581–8. doi: 10.1038/sj.bjc.6605051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chowbay B, Sharma A, Zhou QY, Cheung YB, Lee EJ. The modulation of irinotecan-induced diarrhoea and pharmacokinetics by three different classes of pharmacologic agents. Oncol Rep. 2003;10(3):745–51. [PubMed] [Google Scholar]
- 147.Schmittel A, Jahnke K, Thiel E, Keilholz U. Neomycin as secondary prophylaxis for irinotecan-induced diarrhea. Ann Oncol. 2004;15(8):1296. doi: 10.1093/annonc/mdh310. [DOI] [PubMed] [Google Scholar]
- 148.Alimonti A, Satta F, Pavese I, Burattini E, Zoffoli V, Vecchione A. Prevention of irinotecan plus 5-fluorouracil/leucovorin-induced diarrhoea by oral administration of neomycin plus bacitracin in first-line treatment of advanced colorectal cancer. Ann Oncol. 2003;14(5):805–6. doi: 10.1093/annonc/mdg193. [DOI] [PubMed] [Google Scholar]
- 149.de Jong FA, Kehrer DF, Mathijssen RH, et al. Prophylaxis of irinotecan-induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: a double-blind, randomized, placebo-controlled study. Oncologist. 2006;11(8):944–54. doi: 10.1634/theoncologist.11-8-944. [DOI] [PubMed] [Google Scholar]
- 150.Furman WL, Crews KR, Billups C, et al. Cefixime allows greater dose escalation of oral irinotecan: a phase I study in pediatric patients with refractory solid tumors. J Clin Oncol. 2006;24(4):563–70. doi: 10.1200/JCO.2005.03.2847. [DOI] [PubMed] [Google Scholar]
- 151.McNall-Knapp RY, Williams CN, Reeves EN, Heideman RL, Meyer WH. Extended phase I evaluation of vincristine, irinotecan, temozolomide, and antibiotic in children with refractory solid tumors. Pediatr Blood Cancer. 2010;54(7):909–15. doi: 10.1002/pbc.22460. [DOI] [PubMed] [Google Scholar]
- 152.McGregor LM, Stewart CF, Crews KR, et al. Dose escalation of intravenous irinotecan using oral cefpodoxime: a phase I study in pediatric patients with refractory solid tumors. Pediatr Blood Cancer. 2012;58(3):372–9. doi: 10.1002/pbc.23075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wagner LM, Crews KR, Stewart CF, et al. Reducing irinotecan-associated diarrhea in children. Pediatr Blood Cancer. 2008;50(2):201–7. doi: 10.1002/pbc.21280. [DOI] [PubMed] [Google Scholar]
- 154.Nariai H, Shitara T, Oto Y, Natsumeda T, Taniuchi M, Sato Y. How can we ameliorate irinotecan-associated diarrhea in children with poor compliance for oral cephalosporin? Pediatr Blood Cancer. 2009;52(1):145. doi: 10.1002/pbc.21739. [DOI] [PubMed] [Google Scholar]
- 155.Flieger D, Klassert C, Hainke S, Keller R, Kleinschmidt R, Fischbach W. Phase II clinical trial for prevention of delayed diarrhea with cholestyramine/levofloxacin in the second-line treatment with irinotecan biweekly in patients with metastatic colorectal carcinoma. Oncology. 2007;72(1–2):10–6. doi: 10.1159/000111083. [DOI] [PubMed] [Google Scholar]
- 156.Mei Q, Cao Z, Xiong H, Chen Y. Preliminary results of gentamycin combined with sodium bicarbonate for prevention of irinotecan-induced diarrhea. Front Med China. 2009;3(4):470–474. [Google Scholar]
- 157.Zhao-Xia D, Lei S, Bing W, Yan W, Jun C, Yang Z. Bacillus licheniformis granules combined with Gentamycin for prevention of irinotecan-induced diarrhea. Zhongguo Weishengtaxixue Zazhi/Chinese Journal of Microecology. 2009;21(6):543–544. 546. [Google Scholar]
- 158.Chyka PA. Multiple-dose activated charcoal and enhancement of systemic drug clearance: summary of studies in animals and human volunteers. J Toxicol Clin Toxicol. 1995;33(5):399–405. doi: 10.3109/15563659509013748. [DOI] [PubMed] [Google Scholar]
- 159.Balram C, Zhou QY, Cheung YB, Lee EJ. Influence of multiple dose activated charcoal on the disposition kinetics of irinotecan in rats. Drug Metabol Drug Interact. 2002;19(2):137–48. doi: 10.1515/dmdi.2002.19.2.137. [DOI] [PubMed] [Google Scholar]
- 160.Michael M, Brittain M, Nagai J, et al. Phase II study of activated charcoal to prevent irinotecan-induced diarrhea. J Clin Oncol. 2004;22(21):4410–7. doi: 10.1200/JCO.2004.11.125. [DOI] [PubMed] [Google Scholar]
- 161.Sergio GC, Felix GM, Luis JV. Activated charcoal to prevent irinotecan-induced diarrhea in children. Pediatr Blood Cancer. 2008;51(1):49–52. doi: 10.1002/pbc.21491. [DOI] [PubMed] [Google Scholar]
- 162.Hidaka M, Yamasaki K, Okumura M, et al. Adsorption of irinotecan onto oral adsorbent AST-120 (Kremezin) for preventing delayed diarrhea. Cancer Chemother Pharmacol. 2007;59(3):321–8. doi: 10.1007/s00280-006-0273-y. [DOI] [PubMed] [Google Scholar]
- 163.Maeda Y, Ohune T, Nakamura M, Yamasaki M, Kiribayashi Y, Murakami T. Prevention of irinotecan-induced diarrhoea by oral carbonaceous adsorbent (Kremezin) in cancer patients. Oncol Rep. 2004;12(3):581–5. [PubMed] [Google Scholar]
- 164.Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–71. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 165.Yang XX, Hu ZP, Xu AL, et al. A mechanistic study on reduced toxicity of irinotecan by coadministered thalidomide, a tumor necrosis factor-alpha inhibitor. J Pharmacol Exp Ther. 2006;319(1):82–104. doi: 10.1124/jpet.106.103606. [DOI] [PubMed] [Google Scholar]
- 166.Yang XX, Hu ZP, Chan SY, et al. Pharmacokinetic mechanisms for reduced toxicity of irinotecan by coadministered thalidomide. Curr Drug Metab. 2006;7(4):431–55. doi: 10.2174/138920006776873517. [DOI] [PubMed] [Google Scholar]
- 167.Govindarajan R. Irinotecan and thalidomide in metastatic colorectal cancer. Oncology (Williston Park) 2000;14(12 Suppl 13):29–32. [PubMed] [Google Scholar]
- 168.Govindarajan R, Safar AM, Maddox A-M, Hutchins LF. Irinotecan and thalidomide prolong disease free and overall survival in 5FU refractory metastatic colorectal cancer. Proc Am Soc Clin Oncol. 2003;22:abstract no. 997. [Google Scholar]
- 169.Allegrini G, Di Paolo A, Cerri E, et al. Irinotecan in combination with thalidomide in patients with advanced solid tumors: a clinical study with pharmacodynamic and pharmacokinetic evaluation. Cancer Chemother Pharmacol. 2006;58(5):585–93. doi: 10.1007/s00280-006-0205-x. [DOI] [PubMed] [Google Scholar]
- 170.Ramirez J, Wu K, Janisch L, et al. The effect of thalidomide on the pharmacokinetics of irinotecan and metabolites in advanced solid tumor patients. Cancer Chemother Pharmacol. 2011;68(6):1629–32. doi: 10.1007/s00280-011-1727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Villalona-Calero M, Schaaf L, Phillips G, et al. Thalidomide and celecoxib as potential modulators of irinotecan’s activity in cancer patients. Cancer Chemother Pharmacol. 2007;59(1):23–33. doi: 10.1007/s00280-006-0249-y. [DOI] [PubMed] [Google Scholar]
- 172.Zhang HG, Li J, Qin SK, Zhang YJ, Song SP, Chu DT. A randomized trial of irinotecan plus fuorouracil and leucovorin with thalidomide versus without thalidomide in the treatment for advanced colorectal cancer. Zhonghua Zhong Liu Za Zhi. 2007;29(3):228–31. [PubMed] [Google Scholar]
- 173.Gibson RJ, Stringer AM, Bowen JM, et al. Velafermin improves gastrointestinal mucositis following irinotecan treatment in tumor-bearing DA rats. Cancer Biol Ther. 2007;6(4):541–7. doi: 10.4161/cbt.6.4.3848. [DOI] [PubMed] [Google Scholar]
- 174.Cao S, Black JD, Troutt AB, Rustum YM. Interleukin 15 offers selective protection from irinotecan-induced intestinal toxicity in a preclinical animal model. Cancer Res. 1998;58(15):3270–4. [PubMed] [Google Scholar]
- 175.Zhao J, Huang L, Belmar N, Buelow R, Fong T. Oral RDP58 allows CPT-11 dose intensification for enhanced tumor response by decreasing gastrointestinal toxicity. Clin Cancer Res. 2004;10(8):2851–9. doi: 10.1158/1078-0432.ccr-03-0496. [DOI] [PubMed] [Google Scholar]
- 176.Zhang F, Zhao G, Dong Z. Phosphatidylcholine-specific phospholipase C regulates activation of RAW264. 7 macrophage-like cells by lipopeptide JBT3002. J Leukoc Biol. 2001;69(6):1060–6. [PubMed] [Google Scholar]
- 177.Shinohara H, Killion JJ, Kuniyasu H, Kumar R, Fidler IJ. Prevention of intestinal toxic effects and intensification of irinotecan’s therapeutic efficacy against murine colon cancer liver metastases by oral administration of the lipopeptide JBT 3002. Clin Cancer Res. 1998;4(9):2053–63. [PubMed] [Google Scholar]
- 178.Athar U, Gentile TC. Keratinocyte growth factor. Expert Opin Biol Ther. 2009;9(6):779–87. doi: 10.1517/14712590902976837. [DOI] [PubMed] [Google Scholar]
- 179.Gibson RJ, Bowen JM, Keefe DM. Palifermin reduces diarrhea and increases survival following irinotecan treatment in tumor-bearing DA rats. Int J Cancer. 2005;116(3):464–70. doi: 10.1002/ijc.21082. [DOI] [PubMed] [Google Scholar]
- 180.Rahimi R, Abdollahi M. An update on the ability of St. John’s wort to affect the metabolism of other drugs. Expert Opin Drug Metab Toxicol. 2012;8(6):691–708. doi: 10.1517/17425255.2012.680886. [DOI] [PubMed] [Google Scholar]
- 181.Mathijssen RH, Verweij J, de Bruijn P, Loos WJ, Sparreboom A. Effects of St. John’s wort on irinotecan metabolism. J Natl Cancer Inst. 2002;94(16):1247–9. doi: 10.1093/jnci/94.16.1247. [DOI] [PubMed] [Google Scholar]
- 182.Hu Z, Yang X, Ho PC, et al. St. John’s Wort modulates the toxicities and pharmacokinetics of CPT-11 (irinotecan) in rats. Pharm Res. 2005;22(6):902–14. doi: 10.1007/s11095-005-4585-0. [DOI] [PubMed] [Google Scholar]
- 183.Hu ZP, Yang XX, Chan SY, et al. St. John’s wort attenuates irinotecan-induced diarrhea via down-regulation of intestinal pro-inflammatory cytokines and inhibition of intestinal epithelial apoptosis. Toxicol Appl Pharmacol. 2006;216(2):225–37. doi: 10.1016/j.taap.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 184.Hardman WE, Moyer MP, Cameron IL. Fish oil supplementation enhanced CPT-11 (irinotecan) efficacy against MCF7 breast carcinoma xenografts and ameliorated intestinal side-effects. Br J Cancer. 1999;81(3):440–8. doi: 10.1038/sj.bjc.6690713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tentori L, Leonetti C, Scarsella M, et al. Inhibition of poly(ADP-ribose) polymerase prevents irinotecan-induced intestinal damage and enhances irinotecan/temozolomide efficacy against colon carcinoma. FASEB J. 2006;20(10):1709–11. doi: 10.1096/fj.06-5916fje. [DOI] [PubMed] [Google Scholar]
- 186.Rao RK, Samak G. Role of glutamine in protection of intestinal epithelial tight junctions. J Epithelial Biol Pharmacol. 2012;5(Suppl11-M7):47–54. doi: 10.2174/1875044301205010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xue H, Sawyer MB, Field CJ, Dieleman LA, Murray D, Baracos VE. Bolus oral glutamine protects rats against CPT-11-induced diarrhea and differentially activates cytoprotective mechanisms in host intestine but not tumor. J Nutr. 2008;138(4):740–6. doi: 10.1093/jn/138.4.740. [DOI] [PubMed] [Google Scholar]
- 188.Lin XB, Dieleman LA, Ketabi A, et al. Irinotecan (CPT-11) chemotherapy alters intestinal microbiota in tumour bearing rats. PLoS One. 2012;7(7):e39764. doi: 10.1371/journal.pone.0039764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Savarese D, Al-Zoubi A, Boucher J. Glutamine for irinotecan diarrhea. J Clin Oncol. 2000;18(2):450–1. doi: 10.1200/JCO.2000.18.2.450. [DOI] [PubMed] [Google Scholar]
- 190.Pan CX, Loehrer P, Seitz D, et al. A phase II trial of irinotecan, 5-fluorouracil and leucovorin combined with celecoxib and glutamine as first-line therapy for advanced colorectal cancer. Oncology. 2005;69(1):63–70. doi: 10.1159/000087302. [DOI] [PubMed] [Google Scholar]
- 191.Lam W, Bussom S, Guan F, et al. The four-herb Chinese medicine PHY906 reduces chemotherapy-induced gastrointestinal toxicity. Sci Transl Med. 2010;2(45):45ra59. doi: 10.1126/scitranslmed.3001270. [DOI] [PubMed] [Google Scholar]
- 192.Kummar S, Copur MS, Rose M, et al. A phase I study of the chinese herbal medicine PHY906 as a modulator of irinotecan-based chemotherapy in patients with advanced colorectal cancer. Clinical colorectal cancer. 2011;10(2):85–96. doi: 10.1016/j.clcc.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 193.Liu S-H, Jiang Z, Su T-M, et al. Developing PHY906 as a broad-spectrum modulator of chemotherapeutic agents in cancer therapy. Proc Amer Assoc Cancer Res. 2004;45:abstract no. 557. [Google Scholar]
- 194.Wang E, Bussom S, Chen J, et al. Interaction of a traditional Chinese Medicine (PHY906) and CPT-11 on the inflammatory process in the tumor microenvironment. BMC Med Genomics. 2011;4:38. doi: 10.1186/1755-8794-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Santini V. Amifostine: chemotherapeutic and radiotherapeutic protective effects. Expert Opin Pharmacother. 2001;2(3):479–89. doi: 10.1517/14656566.2.3.479. [DOI] [PubMed] [Google Scholar]
- 196.Wakelee H, Fisher GA. A phase I trial of irinotecan (CPT-11) with amifostine in patients with metastatic colorectal cancer. Invest New Drugs. 2005;23(3):241–2. doi: 10.1007/s10637-005-6732-1. [DOI] [PubMed] [Google Scholar]
- 197.Delioukina ML, Prager D, Parson M, Hecht JR, Rosen P, Rosen LS. Phase II trial of irinotecan in combination with amifostine in patients with advanced colorectal carcinoma. Cancer. 2002;94(8):2174–9. doi: 10.1002/cncr.10432. [DOI] [PubMed] [Google Scholar]
- 198.Souid AK, Dubowy RL, Blaney SM, et al. Phase I clinical and pharmacologic study of weekly cisplatin and irinotecan combined with amifostine for refractory solid tumors. Clin Cancer Res. 2003;9(2):703–10. [PubMed] [Google Scholar]
- 199.Ulrich RG. Pharmacia & Upjohn Company; Kalamazoo, MI: United States Patent 5955466. Tamoxifen as a therapy to reduce irinotecan hydrochloride-induced diarrhea. 1999 Sep 21; (Gurnee, IL)
- 200.Otto AM, Paddenberg R, Schubert S, Mannherz HG. Cell-cycle arrest, micronucleus formation, and cell death in growth inhibition of MCF-7 breast cancer cells by tamoxifen and cisplatin. J Cancer Res Clin Oncol. 1996;122(10):603–12. doi: 10.1007/BF01221192. [DOI] [PubMed] [Google Scholar]
- 201.Cao S, Durrani FA, Rustum YM. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clin Cancer Res. 2004;10(7):2561–9. doi: 10.1158/1078-0432.ccr-03-0268. [DOI] [PubMed] [Google Scholar]
- 202.Fakih MG, Pendyala L, Brady W, et al. A Phase I and pharmacokinetic study of selenomethionine in combination with a fixed dose of irinotecan in solid tumors. Cancer Chemother Pharmacol. 2008;62(3):499–508. doi: 10.1007/s00280-007-0631-4. [DOI] [PubMed] [Google Scholar]
- 203.Fakih MG, Pendyala L, Smith PF, et al. A phase I and pharmacokinetic study of fixed-dose selenomethionine and irinotecan in solid tumors. Clin Cancer Res. 2006;12(4):1237–44. doi: 10.1158/1078-0432.CCR-05-2004. [DOI] [PubMed] [Google Scholar]
- 204.Itoh T, Itagaki S, Sasaki K, Hirano T, Takemoto I, Iseki K. Pharmacokinetic modulation of irinotecan metabolites by sulphobromophthalein in rats. J Pharm Pharmacol. 2004;56(6):809–12. doi: 10.1211/0022357023420. [DOI] [PubMed] [Google Scholar]
- 205.Orsolic N, Benkovic V, Lisicic D, Dikic D, Erhardt J, Horvat Knezevic A. Protective effects of propolis and related polyphenolic/flavonoid compounds against toxicity induced by irinotecan. Med Oncol. 2010;27(4):1346–58. doi: 10.1007/s12032-009-9387-5. [DOI] [PubMed] [Google Scholar]
- 206.Wessner B, Strasser EM, Koitz N, Schmuckenschlager C, Unger-Manhart N, Roth E. Green tea polyphenol administration partly ameliorates chemotherapy-induced side effects in the small intestine of mice. J Nutr. 2007;137(3):634–40. doi: 10.1093/jn/137.3.634. [DOI] [PubMed] [Google Scholar]
- 207.Beyer I, Cao H, Persson J, et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin Cancer Res. 2012;18(12):3340–51. doi: 10.1158/1078-0432.CCR-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tobin PJ, Hong Y, Seale JP, Rivory LP, McLachlan AJ. Loperamide inhibits the biliary excretion of irinotecan (CPT-11) in the rat isolated perfused liver. J Pharm Pharmacol. 2005;57(1):39–45. doi: 10.1211/0022357055100. [DOI] [PubMed] [Google Scholar]
- 209.Ruppin H. Review: loperamide--a potent antidiarrhoeal drug with actions along the alimentary tract. Aliment Pharmacol Ther. 1987;1(3):179–90. doi: 10.1111/j.1365-2036.1987.tb00617.x. [DOI] [PubMed] [Google Scholar]
- 210.Sasaki T, Fujita KI, Sunakawa Y, et al. Concomitant polypharmacy is associated with irinotecan-related adverse drug reactions in patients with cancer. Int J Clin Oncol. 2012 doi: 10.1007/s10147-012-0425-5. [DOI] [PubMed] [Google Scholar]
- 211.Tadokoro J, Kakihata K, Shimazaki M, et al. Post-marketing surveillance (PMS) of all patients treated with irinotecan in Japan: clinical experience and ADR profile of 13,935 patients. Jpn J Clin Oncol. 2011;41(9):1101–11. doi: 10.1093/jjco/hyr105. [DOI] [PubMed] [Google Scholar]