

Abstract
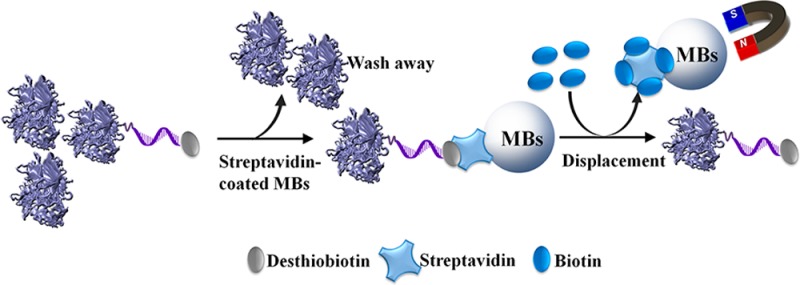
DNA–protein conjugates are very useful in analytical chemistry for target recognition and signal amplification. While a number of methods for conjugating DNA with proteins are known, methods for purification of DNA–protein conjugates from reaction mixture containing unreacted proteins are much less investigated. In this work, a simple and efficient approach to purify DNA–invertase conjugates from reaction mixture via a biotin displacement strategy to release desthiobiotinylated DNA–invertase conjugates from streptavidin-coated magnetic beads was developed. The conjugates purified by this approach were utilized for quantitative detection of cocaine and DNA using a personal glucose meter through structure-switching DNA aptamer sensors and competitive DNA hybridization assays, respectively. In both cases, the purified DNA–invertase conjugates showed better performance compared to the same assays using unpurified conjugates. The approach demonstrated here can be further expanded to other DNA and proteins to generate purified DNA–protein conjugates for analytical and other applications.
DNA molecules have been widely used in analytical chemistry as an excellent class of recognition moiety for selective detection of many target substances.1 The targets include not only complementary DNA or RNA through nucleic acid hybridization,2 but also metal ions, organic molecules, proteins and even cells, through functional DNAs that are capable of either catalyzing reactions (DNAzymes),3−8 binding target molecules (DNA aptamers),9−16 or both (DNA aptazymes).17−19 Functional DNAs are obtained via a combinatorial technique known as in vitro selection or systematic evolution of ligands by exponential enrichment (SELEX),3,20,21 and have been found to recognize a variety of analytes with high specificity and affinity.22−24 By using DNAs labeled with suitable signal reporters as sensors, a series of analytical techniques, such as fluorescence,15,25−31 colorimetry,32−37 electrochemistry,14,38−42 flow cytometry,43−45 magnetic resonance,46,47 and surface enhanced Raman scattering48−50 have been successfully applied for the detection with high sensitivity and selectivity.
Given the high selectivity of these DNA molecules, it is desirable to combine them with the functional versatility of proteins.51,52 Such DNA–protein conjugates would find a broad range of applications,53−61 including more sensitive detection via signal amplification and more diverse signal output via various enzymatic reactions.62 For example, DNA was conjugated with lipase for nucleic acid hybridization assays by forming sandwich complex with surface-immobilized DNA sensors,63 where each target nucleic acid molecule induced the binding of one DNA–enzyme conjugate on the surface to produce many product molecules for signal amplification. Two split cocaine aptamer fragments were also conjugated with a pair of cascade enzymes,55 respectively, to enable target-induced structure changes of the DNA–enzyme conjugates, which then brought the two enzymes closer and enhanced the cascade reactions for sensitive detection.
A functional DNA–invertase conjugation approach also played a key role in converting the widely available personal glucose meters (PGM) into a more versatile device to detect a broad range of nonglucose targets.64−71 PGM is currently the most successful and widely used personal diagnosis device,72−74 but its usage was limited in blood glucose monitoring for diabetes until the invertase-based approach was established.64−71 The advantages of using PGM for detecting targets beyond glucose include simplicity, low cost, portability and wide availability. The mechanism of the detection was based on the target-induced release or binding of DNA–invertase conjugates from the surface of magnetic beads for target recognition, and the invertase-catalyzed hydrolysis of sucrose into glucose for PGM measurement.64−67,70,71 The approach enabled PGMs to detect many targets such as toxins and disease biomarkers for environmental monitoring and medical diagnosis by the general public. Besides analytical applications, DNA–protein conjugates have also been widely utilized to construct nanoassemblies with various functions. The covalent attachment of DNA to streptavidin with four native binding sites for biotin allowed them to serve as selective connectors in the DNA-directed self-assembly of proteins.75 Photoresponsible DNA-HRP/GOx conjugates were also constructed and characterized by means of colorimetric signal change.58 To realize the full potentials of these important applications, efficient methods to prepare the DNA–protein conjugates are required.
To prepare DNA–protein conjugates, both covalent approach, such as chemical reaction through functional groups on the biomolecules,76 and noncovalent approach, such as through biotin–streptavidin and metal–histidine tag interactions77 have been developed. For most approaches, the conjugation between a large DNA and a large protein molecule often results in a moderate yield. As a result, considerable amounts of unconjugated DNAs and proteins are still present in the reaction mixture containing the DNA–protein conjugate products.51 To avoid interference from the starting materials, the conjugate products are required to be purified from the reaction mixtures using methods such as chromatography and electrophoresis.61,77 However, it is usually time-consuming to use chromatography to achieve fine separation of conjugation products in high yield, and electrophoresis under nondenaturing condition to preserve the native structure and activity of the proteins is also laborious and the recovery of conjugation products from electrophoresis gels often resulted in low yields. In this work, we report a simple and efficient method to separate conjugation product from the starting materials, by developing an on-bead biotin displacement method based on the much stronger affinity of biotin to streptavidin than a biotin analog called d-desthiobiotin. The purified conjugates using this method were shown to display enhanced analytical performance over the unpurified conjugates in sensing applications.
Experimental Section
Materials
ACCU-CHEK Aviva glucose meter was used for the tests. Streptavidin-coated magnetic beads (mean diameter 1.5 μm) and Amicon centrifugal filters (10 kDa and 100 kDa molecular weight cutoff) were purchased from Bangs Laboratories Inc. (Fishers, IN) and Millipore Inc. (Billerica, MA), respectively. Grade VII invertase from baker’s yeast (Saccharomyces cerevisiae), desthiobiotin, biotin, sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS), and other chemicals for buffers were purchased from Sigma-Aldrich Inc. (St. Louis, MO). All solutions were prepared using Milli-Q water with electrical resistance over 18 MΩ·cm. The oligonucleotides used in this work were synthesized from Integrated DNA Technologies Inc. (Coralville, IA) and the sequences and modifications are as follows (5′ to 3′): target DNA, GATCGACAATGAGTCTCCCGAGATAACCGACCATAA; single-mismatch DNA (S-mis), GATCGACAATGAGTCTCCAGAGATAACCGACCATAA; double mismatch DNA (D-mis), GATCGACAATGAGTCTCAAGAGATAACCGACCATAA; random DNA, GATCGACAATGAACTCAGGACGCCAACCGACCATAA; biotin-modified DNA1 (biotin-DNA1), TCACAGATGAGTAAAAAAAAAAAA-biotin; biotin-modified DNA2 (biotin-DNA2), biotin-AAAAAAATCTCGGGAGAC; 5-thiol-3-amine-modified DNA, HS-AAAAAAAAAAAAGTCTCCCGAGATAAAAAAAAAAAA-NH2; cocaine aptamer (coc-Apt), TTTTTTACTCATCTGTGAATCTCGGGAGACAAGGATAAATCCTTCAATGAAGTGGGTCTCCC. The buffer used in this work (buffer A) was composed of 0.1 M NaCl, 0.1 M sodium phosphate buffer, pH 7.3, 0.05% Tween-20 (Tween-20 was added as a surfactant to reduce the nonspecific binding of biomolecules to magnetic beads, and also to prevent the magnetic beads from sticking to the inner wall of microtubes).
Desthiobiotin–DNA Conjugation
A mixture of 3.8 mg of desthiobiotin, 7.6 mg of EDC, and 7.6 mg of sulfo-NHS was added to 100 μL of DMF. This mixture was kept at room temperature for 2 h. Then 80 μL of 500 μM 5-thiol-3-amine-modified DNA containing a 5′ thiol (originally as a disulfide modification as shown in Figure 1a and then reduced by TCEP to generate active thiol for later conjugation) and a 3′ amine, 20 μL of 1 M NaHCO3–Na2CO3 buffer at pH 8.7 were added to the DMF solution. The resulting solution was kept at room temperature for overnight. Then purified by Amicon-10 K using Millipore water by 8 times.
Figure 1.

(a) Conjugation of desthiobiotin–DNA and desthiobiotin–DNA–invertase, where “RS–S” indicates a disulfide modification that can be reduced to thiol by TCEP. (b) Purification of desthiobiotinylated DNA–invertase conjugates from reaction mixture.
Desthiobiotinylated DNA–Invertase Conjugation
The procedure was according to the method reported in our previous work with some minor modifications.64 Briefly, 30 μL of 1 mM desthiobiotin–DNA, 2 μL of 1 M sodium phosphate buffer at pH 5.5, and 2 μL of 30 mM TCEP were mixed, the solution was placed on a shaker for 1 h at room temperature. Then purified by Amicon-10 K using buffer A without Tween-20 by 8 times. In addition, 1 mg of sulfo-SMCC was mixed with 400 μL of 20 mg/mL invertase in buffer A without Tween-20. After vortexing, the solution was kept at room temperature for 1 h. Then the mixture was centrifuged to remove the insoluble excess sulfo-SMCC. The supernatant was purified for 8 times by Amicon-100 K using buffer A without Tween-20. The above solution of desthiobiotinylated DNA-SH was mixed with sulfo-SMCC activated invertase. The resulting solution was kept at room temperature for 48 h. Then it was purified by Amicon-100 K using buffer A without Tween-20 by 8 times.
Procedures for Cocaine Detection Using PGM
Sensor Preparation
One milliliter of 1 mg/mL streptavidin-coated magnetic beads was first washed using buffer A twice by a magnetic rack and then dispersed in buffer A. Twelve microliters of 0.5 mM biotin-DNA1 and 12 μL of 0.5 mM cocaine aptamer were added to the MBs solution and mixed for 30 min at room temperature. After that, it was washed three times using buffer A to remove excess biotin-DNA1 and cocaine aptamer. Purified DNA–invertase conjugates (about 20 mg/mL) was added to the MBs solution and well mixed for 30 min at room temperature. After that, the MBs were separated from the solution by the magnetic rack, and excess purified DNA–invertase conjugates were washed off by buffer A three times. Then separated as each portion of 60 μL 1 mg/mL MBs in buffer A.
Cocaine Detection via the Structure−Switching Assay
Twenty microliters of various concentration of target cocaine in buffer A was added to each portion of the above MBs and well mixed for 15 min. Then the solution was separated using a magnetic rack. Ten microliters of the clear solution was mixed with 10 μL of 1 M sucrose in buffer A. After it stood at room temperature for 15 min, 5 μL of the final solution was tested by a commercially available PGM.
Procedures for Competitive DNA Detection Using PGM
Sensor Preparation
One milliliter of 1 mg/mL streptavidin-coated magnetic beads was buffer exchanged to buffer A twice by a magnetic rack and finally resuspended in buffer A. Fifty microliters of 100 μM biotin-DNA2 was added to the solution, and the mixture was placed on a roller for 30 min at room temperature. After that, the MBs were washed three times using buffer A containing 1 mM biotin to remove unbound biotin-DNA2 and block nonspecific binding sites by biotin. Then, they were separated into portions containing 20 μL of 1 mg/mL MBs in buffer A.
DNA Detection via the Competitive Assay
Each portion of the above MBs was separated by a magnetic rack, and the residual MBs were used as the sensor for tests. Ten microliters of the DNA sample of various concentrations in buffer A was mixed with 10 μL of 40 nM purified DNA–invertase conjugates. This was then added to the MBs, and the mixture was mixed on a roller for 2 h at room temperature. After magnetic separation, a 10 μL of 1 M sucrose in buffer A was added to the supernatant. Finally, 5 μL of the solution was tested by a PGM after 1 h.
Results and Discussion
Design and Demonstration of Biotin Displacement Method for Purification of DNA–Protein Conjugates
The method developed in this study relies on the different dissociation constants between biotin–streptavidin (Kd = 1.3 × 10−15 M at pH 5.0) and desthiobiotin–streptavidin (Kd = 9 × 10−13 M for d-desthiobiotin and Kd = 6 × 10−11 M for l-desthiobiotin at pH 4.0).78 The much stronger binding affinity of streptavidin toward biotin over desthiobiotin suggests desthiobiotin-labeled conjugates can be efficiently displaced by biotin and released from streptavidin, as shown in Figure 1. To a DNA strand modified with an amine at 3′ end and a thiol (as disulfide) at 5′ end, desthiobiotin was covalently attached to the 3′ amine of the DNA through the EDC/sulfo-NHS condensation (Figure 1a). The production of desthiobiotinylated DNA with almost 100% modification yield was confirmed by the result from matrix-assisted laser desorption ionization-time-of-flight mass spectrum (MALDI-TOF MS, see Figure S1 in Supporting Information). Subsequently, the 5′ thiol of the desthiobiotinylated DNA was activated using TECP, and invertase was functionalized through its surface amines by a commercial cross-linker, Sulfo-SMCC. Finally, the desthiobiotinylated DNA containing reactive thiols and Sulfo-SMCC-activated invertase were conjugated via the maleimide–thiol reaction to yield the desthiobiotinylated DNA–invertase conjugates (Figure 1a). After removing unconjugated desthiobiotinylated DNA (<12 kDa) by a centrifuge filter membrane with 100 kDa molecular weight cutoff, the resulting solution containing desthiobiotinylated DNA–invertase conjugates and unreacted invertase (both >100 kDa) was treated with streptavidin-coated magnetic beads (MBs) to immobilize desthiobiotinylated DNA–invertase conjugates through desthiobiotin–streptavidin binding (Figure 1b). The unreacted invertase in solution phase was washed away after magnetic separation. Then, 1 mM biotin was used to release desthiobiotinylated DNA–invertase conjugates from MBs, because biotin could displace desthiobiotin from streptavidin as a result of its much higher binding affinity (Figure 1b). After multiple times of biotin displacement and magnetic separation, the solutions containing purified DNA–invertase conjugates were combined and condensed to a desired concentration using a centrifuge filter membrane with 100 kDa cutoff, through which biotin was also removed.
In the above method using biotin displacement and magnetic separation to purify the DNA–protein conjugates, the separation was only determined by whether the protein was conjugated by DNA (containing desthiobiotin) or not. Other properties of the protein, such as molecular weight, hydrophobicity and isoelectric point did not play any role in the separation, suggesting the method can be generally applicable for all the proteins using almost the same protocol as long as surface amines are available on the proteins for conjugation. In contrast, considerations such as molecular weight, hydrophobicity or isoelectric point of proteins may need to be taken into account to identify the desired peaks for DNA–protein conjugates in chromatography and electrophoresis techniques, usually making case-by-case optimizations required for different proteins.
The activities of the released DNA–invertase conjugate solutions after each time of biotin displacement were tested by measuring the amount of glucose production from sucrose by the conjugates (Supporting Information Figure S2), suggesting DNA conjugation on invertase did not disrupt the enzyme activity and almost all the conjugates were washed off by 5 times of biotin displacement. The efficiency of this displacement method was calculated by adding up the amount of DNA–invertase conjugates during each time of displacement using UV–vis spectra. As shown in Table S1 (see Supporting Information for more details), the average yield of recovery for the biotin displacement approach was estimated as 91.8%, suggesting the high efficiency of the method.
To demonstrate the purified solution contained only DNA–invertase conjugates and no unreacted DNA or invertase, native PAGE (10%) and SDS PAGE (4–20% gradient gel) were carried out to analyze its composite. After stain of DNA components using ethidium bromide, the native PAGE image (Figure 2a) suggested unreacted DNA (lane 5) was efficiently removed from the product mixture (lane 4) by centrifuge filter membranes (lane 3). The band stuck on the wells are purified DNA–invertase conjugates (lane 2), because their molecular weights were too large (>100 kDa) for them to migrate on 10% PAGE. On the other hand, the SDS PAGE image after stain of invertase components using coomassie brilliant blue (Figure 2b) indicated that unreacted invertase (lane 3) was also washed away from the mixture (lane 2) to yield purified DNA–invertase conjugates (lane 1) by the biotin displacement method. The faint top bands in lane 2 were probably some high molecular weight aggregates formed because of high invertase and DNA concentrations used for the conjugation reaction. As shown in lane 1, these aggregates were removed during the biotin displacement method and not present in the purified DNA–invertase conjugates.
Figure 2.
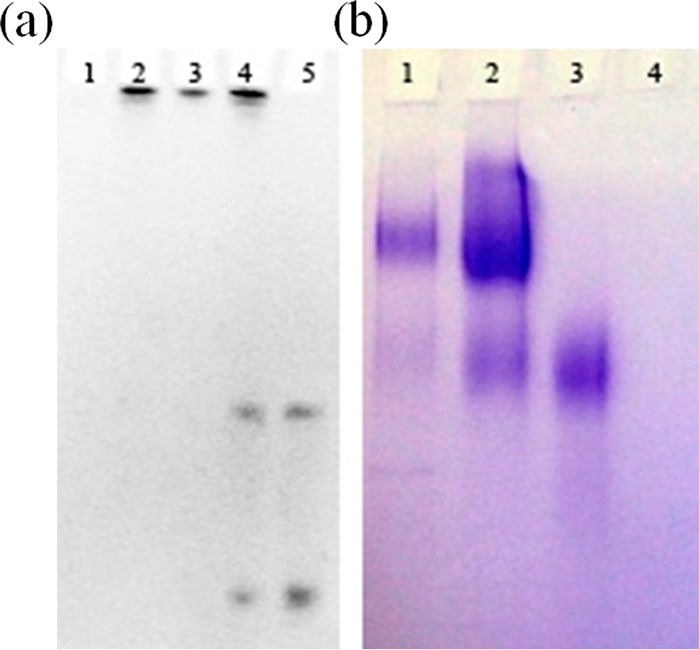
PAGE gels for the conjugation products. (a) Fluorescence imaging of ethidium bromide stained 10% native PAGE: (1) invertase, (2) purified DNA–invertase conjugates; (3) reaction mixture after amicon-100 kDa to remove free DNA (containing 1 and 2), (4) reaction mixture (containing 1, 2, and free DNA), and (5) desthiobiotinylated DNA. The two bands of DNA are because of the thiol–DNA and its disulfide-containing dimer form. (b) Coomassie brilliant blue stained 4–20% gradient SDS PAGE: (1) purified DNA–invertase conjugates, (2) reaction mixture (containing 1, 3, and 4), (3) invertase, and (4) desthiobiotinylated DNA.
Since the purified DNA–invertase conjugates were free of unreacted DNA and invertase, we could then use them to estimate the average number of DNA strands on each invertase in the conjugates, which was a question not fully addressed in our previous studies.64−66,70 In UV–vis absorption spectra (Figure 3), the maximum absorption of the purified conjugates (blue line) is at 260 nm, which was similar as that of DNA (black line) but did have a mild shoulder around 280 nm originated from invertase (red line) in the conjugates. The conjugates exhibited a much stronger UV absorbance at 260 nm (DNA component) compared with 280 nm (invertase component), because of the much higher molecular extinction coefficient (ε) of DNA than invertase. On the basis of the absorbance ratio (260 vs 280 nm) of DNA, invertase and DNA–invertase conjugates, the molecular ratio of DNA versus invertase in the purified conjugates was calculated to be ∼5 DNA per invertase (see Supporting Information for details of calculation).
Figure 3.
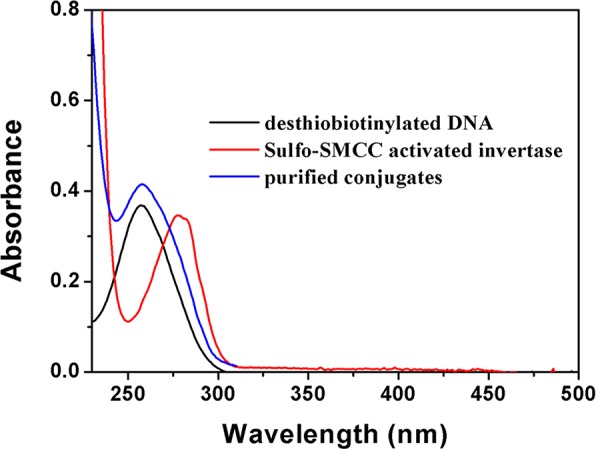
Absorption spectra of the conjugation products.
Application in Cocaine Detection by the Structure-Switching Aptamer
To examine the ability of the purified DNA–invertase conjugates to enhance the performance of DNA aptamer sensors, we chose the structure-switching aptamer for cocaine detection as an example.11 Cocaine is one of the most used recreational drugs in the United States and its detection is important in the fight against drug abuse and trafficking. As shown in Figure 4, a biotin-DNA1 containing 18 complementary nucleotides to the cocaine aptamer was immobilized onto streptavidin-coated MBs via streptavidin–biotin interaction to capture the aptamer. The amount of biotin-DNA1 loading on the MBs was determined by the decrease of biotin-DNA1’s characteristic absorbance at 260 nm in the supernatant before and after immobilization (Supporting Information Figure S3a). Around 3.53 pmol biotin-DNA1 was loaded onto 1 mg of MBs based on the calculation. The purified DNA–invertase conjugates obtained through the method described in the previous section were hybridized with the aptamer through another 12 base pairs. In the presence of cocaine, the DNA–invertase conjugates were released because of the cocaine-induced structure switching of the aptamer. After magnetic separation, the released conjugates catalyzed the hydrolysis of PGM-inert sucrose into PGM-detectable glucose, establishing the relationship between PGM signal readout and the concentration of cocaine in the samples (Figure 5). A detection limit of 1.8 μM was achieved according to the definition of 3σb/slope (σb, standard deviation of the blank samples). This detection limit is better than the test under the same condition using unpurified DNA–invertase conjugates instead of purified conjugates for sensor preparation, which resulted in a higher detection limit of 4.2 μM (similar to that of 3.4 μM in our previous work,64 using DNA–invertase conjugates without the biotin displacement approach). The enhanced performance of the detection using purified DNA–invertase conjugates over unpurified ones (1.8 vs 4.2 μM), although moderate (2.3-fold), was most probably due to the presence of unreacted invertase at high concentrations in the conjugates that cause nonspecific adsorption of unreacted invertase on the MBs. These adsorbed unreacted invertase molecules could leak during cocaine detection and thus increased the blank signals, reducing the signal-to-noise ratio and lowering the performance. As shown in the inset of Figure 5, at low analyte concentrations, the detection using purified DNA–invertase conjugates did display a lower blank signal, higher signal-to-noise ratio (ratio of signal enhancement over blank) and higher sensitivity according to the slope of calibration curves. On the other hand, the detection was selective to cocaine, because adenosine at high concentrations did not give detectable PGM signal changes (Supporting Information Figure S4), suggesting the selectivity of the aptamer was well preserved in the sensor design using purified DNA–invertase conjugates.
Figure 4.

Structure-switching assay of cocaine by purified DNA–invertase conjugates using a PGM.
Figure 5.
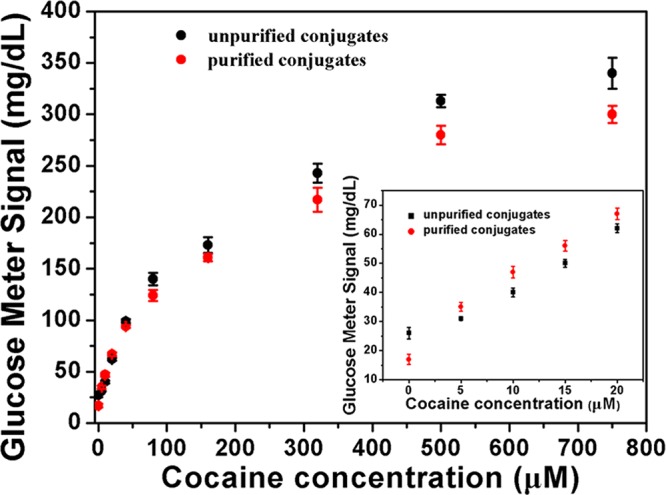
Performance of cocaine detection in buffer using the PGM. The calibration curve of cocaine detection (inset).
Application in Competitive Hybridization Assay for DNA Detection
In the above sensor design for cocaine detection, there is a washing step during sensor preparation to remove most unreacted invertase to ensure that only DNA–invertase conjugates were immobilized on MBs by DNA hybridization with the biotin-DNA1. Therefore, the use of purified DNA–invertase conjugates gave only moderate performance enhancement (detection limit of 1.8 μM) compared to using unpurified conjugates (detection limit of 4.2 μM). To further demonstrate the advantage of using purified conjugates, the DNA–invertase conjugates after purification by the biotin displacement method were applied in a competitive DNA hybridization assay, and the performance was then compared with the same assay using unpurified conjugates instead. In contrast to sandwich assays, competitive assays has the advantage of requiring no washing steps during the detection, thus making the detection more simple and efficient.66 In a typical competitive assay (Figure 6a), target DNA and purified DNA–invertase conjugates were added simultaneously to MBs immobilized with capturing biotin-DNA2. Around 3.85 pmol biotin-DNA2 was loaded onto 1 mg of MBs based on the calculation (Supporting Information Figure S3b). When the target DNA was present, it competed with the purified DNA–invertase conjugates in hybridizing with the capturing DNA on MBs. Therefore, less purified DNA–invertase conjugates were captured on the MBs in the presence of more target DNA. After magnetic separation to remove MBs, the conjugates remaining in solution catalyzed the hydrolysis of sucrose into glucose for PGM measurement (Figure 6a). For samples without target DNA, most of the purified DNA–invertase conjugates were captured by MBs, giving a very low blank signal (26 mg/dL, Supporting Information Figure S5), while the signal increased to 114 mg/dL in the presence of 1 μM target DNA. In contrast, if unpurified DNA–invertase conjugates were used for the same assays, the blank signal was found about 8 times as high as that when using purified conjugates (213 mg/dL, Supporting Information Figure S5), which was because large amounts of unreacted invertase remained in solution after magnetic separation and caused efficient glucose production even if no target DNA was present. The signal enhancement ratio reached 4.4-fold when using the purified conjugates in the assays while only 1.3-fold for the unpurified conjugates (Figure 6b). The detection limit of the competitive assays using the purified conjugates was about 2.1 nM (Supporting Information Figure S6a) compared with 65.2 nM (Supporting Information Figure S6b) for the unpurified conjugates, indicating a significant performance enhancement by lower the detection limit from 65.2 to 2.1 nM (more than 30-fold improvement). The selectivity of this competitive assay toward target DNA over other three types of DNA molecules (single mismatch, double mismatch and random DNAs) was also carried out. Addition of these control DNA sequences produced very low glucose signal enhancement over the blank (Figure 6c).
Figure 6.

(a) Competitive assay of DNA by purified DNA–invertase conjugates using a PGM. (b) PGM signal enhancement of DNA detection with purified DNA–invertase conjugates (red line) and unpurified conjugates (black line). (c) Selectivity of competitive DNA assay with the purified conjugates using a PGM. DNA concentration is 1 μM.
In summary, a simple and efficient method to purify DNA–invertase conjugates from the mixture containing the conjugates and unreacted invertase and DNA was developed in this study. The method was based on a biotin displacement strategy to release desthiobiotinylated DNA–invertase conjugates from streptavidin-coated MBs for purification. The applications of these purified DNA–invertase conjugates in a structure-switching aptamer sensor for cocaine and a competitive hybridization assay for DNA were also demonstrated, and both experiments showed enhanced analytical performance compared with the same assays using unpurified DNA–invertase conjugates. Because the amine and thiol modifications on DNA are commercially available and nearly all proteins have surface reactive amines for conjugation, the method reported in this study can be generally applied for the preparation and purification of almost any DNA–protein conjugates for various applications.
Acknowledgments
This work was supported by the U.S. National Institute of Health (ES016865) and the National Science Foundation (CTS-0120978) and the National Natural Science Foundation of China with the Grant (21175079 and 21375074).
Supporting Information Available
Additional material as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhang H.; Li F.; Dever B.; Li X. F.; Le X. C. Chem. Rev. 2012, 113, 2812–2841. [DOI] [PubMed] [Google Scholar]
- Sassolas A.; Leca-Bouvier B. D.; Blum L. J. Chem. Rev. 2008, 108, 109–139. [DOI] [PubMed] [Google Scholar]
- Breaker R. R.; Joyce G. F. Chem. Biol. 1994, 1, 223–229. [DOI] [PubMed] [Google Scholar]
- Cuenoud B.; Szostak J. W. Nature 1995, 375, 611–634. [DOI] [PubMed] [Google Scholar]
- Wang W.; Billen L. P.; Li Y. Chem. Biol. 2002, 9, 507–517. [DOI] [PubMed] [Google Scholar]
- Schlosser K.; Li Y. Chem. Biol. 2009, 16, 311–322. [DOI] [PubMed] [Google Scholar]
- Silverman S. K. Acc. Chem. Res. 2009, 42, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M. M.; Aguirre S. D.; Lazim H.; Li Y. Angew. Chem., Int. Ed. 2011, 50, 3751–3754. [DOI] [PubMed] [Google Scholar]
- Huizenga D. E.; Szostak J. W. Biochemistry 1995, 34, 656–665. [DOI] [PubMed] [Google Scholar]
- Famulok M.; Mayer G.; Blind M. Acc. Chem. Res. 2000, 33, 591–599. [DOI] [PubMed] [Google Scholar]
- Stojanovic M. N.; de Prada P.; Landry D. W. J. Am. Chem. Soc. 2001, 123, 4928–4931. [DOI] [PubMed] [Google Scholar]
- Lee J. F.; Hesselberth J. R.; Meyers L. A.; Ellington A. D. Nucleic Acids Res. 2004, 32, D95–D100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Wang F.; Liu X. J. Am. Chem. Soc. 2008, 130, 3244–3245. [DOI] [PubMed] [Google Scholar]
- Wen Y.; Pei H.; Wan Y.; Su Y.; Huang Q.; Song S.; Fan C. Anal. Chem. 2011, 83, 7418–7423. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhong X.; Cheng F.; Zhang J. R.; Jiang L. P.; Zhu J. J. Anal. Chem. 2012, 84, 4140–4146. [DOI] [PubMed] [Google Scholar]
- Pei H.; Li J.; Lv M.; Wang J.; Gao J.; Lu J.; Li Y.; Huang Q.; Hu J.; Fan C. J. Am. Chem. Soc. 2012, 134, 13843–13849. [DOI] [PubMed] [Google Scholar]
- Hesselberth J.; Robertson M. P.; Jhaveri S.; Ellington A. D. Rev. Mol. Biotechnol. 2000, 74, 15–25. [DOI] [PubMed] [Google Scholar]
- Rajendran M.; Ellington A. D. Comb. Chem. High Throughput Screening 2002, 5, 263–270. [DOI] [PubMed] [Google Scholar]
- Liu J.; Cao Z.; Lu Y. Chem. Rev. 2009, 109, 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Gold L. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Willner I.; Shlyahovsky B.; Zayats M.; Willner B. Chem. Soc. Rev. 2008, 37, 1153–1165. [DOI] [PubMed] [Google Scholar]
- Xing H.; Wong N. Y.; Xiang Y.; Lu Y. Curr. Opin. Chem. Biol. 2012, 16, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G.; Ye M.; Donovan M. J.; Song E.; Zhao Z.; Tan W. Chem. Commun. 2012, 48, 10472–10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L. M.; Zhang X. B.; Kong R. M.; Yang B.; Tan W. J. Am. Chem. Soc. 2011, 133, 11686–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave N.; Liu J. Chem. Commun. 2012, 48, 3718–3720. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhong X.; Zhang H.; Le X. C.; Zhu J. J. Anal. Chem. 2012, 84, 5170–5174. [DOI] [PubMed] [Google Scholar]
- Liu X.; Freeman R.; Willner I. Chem.—Eur. J. 2012, 18, 2207–2211. [DOI] [PubMed] [Google Scholar]
- Tan X.; Chen T.; Xiong X.; Mao Y.; Zhu G.; Yasun E.; Li C.; Zhu Z.; Tan W. Anal. Chem. 2012, 84, 8622–8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus S. A.; Li Y. J. Am. Chem. Soc. 2013, 135, 7181–7186. [DOI] [PubMed] [Google Scholar]
- Zhao X. H.; Gong L.; Zhang X. B.; Yang B.; Fu T.; Hu R.; Tan W.; Yu R. Anal. Chem. 2013, 85, 3614–3620. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. J. Am. Chem. Soc. 2005, 127, 12677–12683. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Angew. Chem., Int. Ed. 2006, 45, 90–94. [Google Scholar]
- Liu J.; Lu Y. Chem. Commun. 2007, 4872–4874. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Chiuman W.; Lam J. C. F.; McManus S. A.; Chen W.; Cui Y.; Pelton R.; Brook M. A.; Li Y. J. Am. Chem. Soc. 2008, 130, 3610–3618. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Gao X.; Liu Q.; Lin Z.; Qiu B.; Chen G. Chem. Commun. 2011, 47, 7437–7439. [DOI] [PubMed] [Google Scholar]
- Li J.; Jia Y.; Zheng J.; Zhong W.; Shen G.; Yang R.; Tan W. Chem. Commun. 2013, 49, 6137–6139. [DOI] [PubMed] [Google Scholar]
- Du Y.; Li B.; Wei H.; Wang Y.; Wang E. Anal. Chem. 2008, 80, 5110–5117. [DOI] [PubMed] [Google Scholar]
- Du Y.; Chen C.; Yin J.; Li B.; Zhou M.; Dong S.; Wang E. Anal. Chem. 2010, 82, 1556–1563. [DOI] [PubMed] [Google Scholar]
- Du Y.; Chen C.; Zhou M.; Dong S.; Wang E. Anal. Chem. 2011, 83, 1523–1529. [DOI] [PubMed] [Google Scholar]
- Sheng Q. L.; Liu R. X.; Zheng J. B.; Zhu J. J. Nanoscale 2013, 5, 7505–7511. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Chai Y.; Yuan R.; Zhuo Y.; Gan X. Chem. Commun. 2013, 49, 7328–7330. [DOI] [PubMed] [Google Scholar]
- Chen Y.; O’Donoghue M. B.; Huang Y. F.; Kang H.; Phillips J. A.; Chen X.; Estevez M. C.; Yang C. J.; Tan W. J. Am. Chem. Soc. 2010, 132, 16559–16570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L.; Wu C.; You M.; Han D.; Chen T.; Zhu G.; Jiang J.; Yu R.; Tan W. J. Am. Chem. Soc. 2013, 135, 12952–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Zhu G.; Li Y.; Li C.; You M.; Chen T.; Song E.; Yang R.; Tan W. ACS Nano 2013, 7, 6545–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit M. V.; Mazumdar D.; Lu Y. Bioconjugate Chem. 2008, 19, 412–417. [DOI] [PubMed] [Google Scholar]
- Chen T.; Shukoor M. I.; Wang R.; Zhao Z.; Yuan Q.; Bamrungsap S.; Xiong X.; Tan W. ACS Nano 2011, 5, 7866–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhou H. S. Anal. Chem. 2008, 80, 7174–7178. [DOI] [PubMed] [Google Scholar]
- Pelossof G.; Tel-Vered R.; Willner I. Anal. Chem. 2012, 84, 3703–3709. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Feng F.; Zhao L.; Wang C.; Wang H.; Tian M.; Qin J.; Duan Y.; He X. Biosens. Bioelectron. 2013, 47, 265–270. [DOI] [PubMed] [Google Scholar]
- Niemeyer C. M. Angew. Chem., Int. Ed. 2010, 49, 1200–1216. [DOI] [PubMed] [Google Scholar]
- Sacca B.; Niemeyer C. M. Chem. Soc. Rev. 2011, 40, 5910–5921. [DOI] [PubMed] [Google Scholar]
- Yan H.; Park S. H.; Finkelstein G.; Reif J. H.; LaBean T. H. Science 2003, 301, 1882–1884. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Stanzel M.; Gumbrecht W.; Humenik M.; Sprinzl M. Biosens. Bioelectron. 2007, 22, 1798–1806. [DOI] [PubMed] [Google Scholar]
- Freeman R.; Sharon E.; Tel-Vered R.; Willner I. J. Am. Chem. Soc. 2009, 131, 5028–5029. [DOI] [PubMed] [Google Scholar]
- Schröder H.; Hoffmann L.; Müller J.; Alhorn P.; Fleger M.; Neyer A.; Niemeyer C. M. Small 2009, 5, 1547–1552. [DOI] [PubMed] [Google Scholar]
- Wang Z. G.; Wilner O. I.; Willner I. Nano Lett. 2009, 9, 4098–4102. [DOI] [PubMed] [Google Scholar]
- You M.; Wang R. W.; Zhang X.; Chen Y.; Wang K.; Peng L.; Tan W. ACS Nano 2011, 5, 10090–10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Liu M.; Liu Y.; Woodbury N. W.; Yan H. J. Am. Chem. Soc. 2012, 134, 5516–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Xiang Y.; Lu Y.; Crooks R. M. Angew. Chem., Int. Ed. 2012, 51, 6925–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L.; Zhou C.; Yang Z.; Liu D. Small 2013, 9, 3088–3091. [DOI] [PubMed] [Google Scholar]
- Teller C.; Willner I. Trends Biotechnol. 2010, 28, 619–628. [DOI] [PubMed] [Google Scholar]
- Kynclova E.; Hartig A.; Schalkhammer T. J. Mol. Recognit. 1995, 8, 139–145. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Lu Y. Nat Chem 2011, 3, 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Lu Y. Anal. Chem. 2012, 84, 1975–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Lu Y. Anal. Chem. 2012, 84, 4174–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Jiang B.; Xie J.; Xiang Y.; Yuan R.; Chai Y. Chem. Commun. 2012, 48, 10733–10735. [DOI] [PubMed] [Google Scholar]
- Mohapatra H.; Phillips S. T. Chem. Commun. 2013, 49, 6134–6136. [DOI] [PubMed] [Google Scholar]
- Pang H.; Du L.; Pei J.; Wei Y.; Du Q.; Huang R. Curr. Top. Med. Chem. 2013, 13, 1234–1241. [DOI] [PubMed] [Google Scholar]
- Xiang Y.; Lu Y. Chem. Commun. 2013, 49, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Zhu Z.; Zou Y.; Huang Y.; Liu D.; Jia S.; Xu D.; Wu M.; Zhou Y.; Zhou S.; Yang C. J. J. Am. Chem. Soc. 2013, 135, 3748–3751. [DOI] [PubMed] [Google Scholar]
- Clark L. C.; Lyons C. Ann. N.Y. Acad. Sci. 1962, 102, 29–45. [DOI] [PubMed] [Google Scholar]
- Heller A.; Feldman B. Chem. Rev. 2008, 108, 2482–2505. [DOI] [PubMed] [Google Scholar]
- Montagnana M.; Caputo M.; Giavarina D.; Lippi G. Clin. Chim. Acta 2009, 402, 7–13. [DOI] [PubMed] [Google Scholar]
- Niemeyer C. M.; Sano T.; Smith C. L.; Cantor C. R. Nucleic Acids Res. 1994, 22, 5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan N.; Kim B. H. Chem. Rev. 2006, 106, 3712–3761. [DOI] [PubMed] [Google Scholar]
- Goodman R. P.; Erben C. M.; Malo J.; Ho W. M.; McKee M. L.; Kapanidis A. N.; Turberfield A. J. ChemBioChem 2009, 10, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Green N. M. Adv. Protein Chem. 1975, 29, 85–133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.