

Abstract
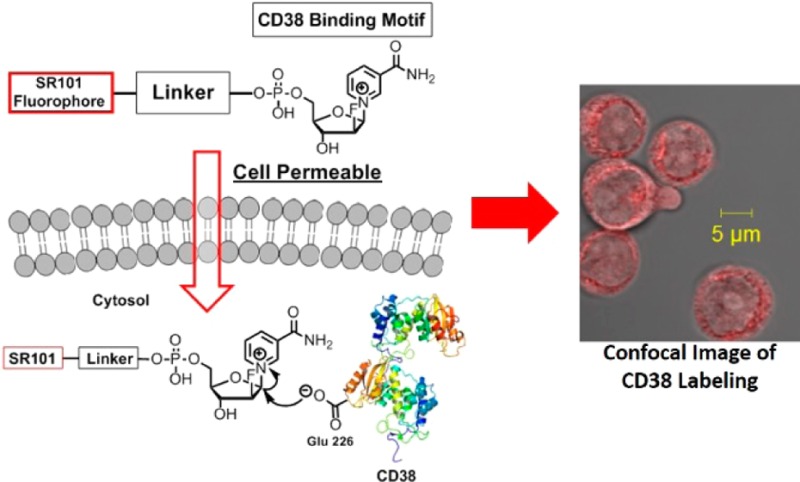
Nicotinamide adenine dinucleotide (NAD) is increasingly recognized as an important signaling molecule that affects numerous biological pathways. Thus, enzymes that metabolize NAD can have important biological functions. One NAD-metabolizing enzyme in mammals is CD38, a type II transmembrane protein that converts NAD primarily to adenosine diphosphate ribose (ADPR) and a small amount of cyclic adenosine diphosphate ribose (cADPR). Localization of CD38 was originally thought to be only on the plasma membrane, but later reports showed either significant or solely, intracellular CD38. With the efficient NAD-hydrolysis activity, the intracellular CD38 may lead to depletion of cellular NAD, thus producing harmful effects. Therefore, the intracellular localization of CD38 needs to be carefully validated. Here, we report the synthesis and application of a cell permeable, fluorescent small molecule (SR101–F-araNMN) that can covalently label enzymatically active CD38 with minimal perturbation of live cells. Using this fluorescent probe, we revealed that CD38 is predominately on the plasma membrane of Raji and retinoic acid (RA)-treated HL-60 cells. Additionally, the probe revealed no CD38 expression in K562 cells, which was previously reported to have solely intracellular CD38. The finding that very little intracellular CD38 exists in these cell lines suggests that the major enzymatic function of CD38 is to hydrolyze extracellular rather than intracellular NAD. The fluorescent activity-based probes that we developed allow the localization of CD38 in different cells to be determined, thus enabling a better understanding of the physiological function.
Introduction
Nicotinamide adenine dinucleotide (NAD) is an important cofactor used in many metabolic reactions. Recently, it has been increasingly recognized that, in addition to serving as a cofactor, it also serves as an important signaling molecule by effecting protein posttranslational modifications, such as NAD-dependent deacylation and ADP-ribosylation.1,2 Thus, enzymes that metabolize NAD can have important biological functions. One mammalian enzyme that metabolizes NAD is cluster of differentiation 38 (CD38). CD38, a type II membrane protein, has important physiological functions, demonstrated by the compromised immune response and social memory defect in CD38 knockout mice.3,4 In addition, its expression is associated with a poor prognosis in chronic lymphocytic leukemia.5 However, the molecular mechanism underlying its physiological functions is still not well understood. It is reported to function as both an enzyme and a receptor. CD38 has 69% overall homology to an Aplysia cyclase, which converts NAD to cyclic adenosine diphosphate-ribose (cADPR), a molecule that is reported to be a calcium mobilizing messenger.6 CD38 was thus considered to have a similar enzymatic function.6 However, CD38 catalyzes the formation of mainly adenosine diphosphate ribose (ADPR) and only a minute amount of cADPR.7−10 Under certain conditions, CD38 can also catalyze a base-exchange reaction converting nicotinamide adenine dinucleotide phosphate (NADP) to nicotinic acid adenine dinucleotide phosphate (NAADP), which is also a calcium mobilizing messenger.11 As a receptor, it was reported that CD38 can initiate transmembrane signaling in response to antibody binding.12
The cellular localization of CD38 has also been perplexing. CD38 was originally identified as a cell surface protein, but was later reported to be present in intracellular compartments, such as the mitochondria, Golgi, and ER, with the highest levels of expression in the nuclear membranes.13−20 In addition, a recent report suggested the existence of type III CD38 on the plasma membrane with the catalytic domain facing the cytosol.21 However, conflicting results on CD38 cellular localization have been reported, with some showing only plasma membrane CD38 and others showing only nuclear localized CD38.15,22 Because of the efficient NAD-hydrolysis activity of CD38, the intracellular CD38 may lead to depletion of cellular NAD, thus producing detrimental effects. Therefore, the intracellular localization of CD38 merits careful investigation. Methods used to study cellular localization of CD38 include the use of antibodies for confocal immunofluorescence after cell fixation and permeabilization, subcellular fractionation, and less frequently, CD38–GFP fusion proteins.13−20,22 To mitigate the possibility of artifacts from these methods, there is a need for new methods with minimal perturbation of live cells and minimal interference with CD38 signaling. In addition, previously used labeling methods are typically followed by a separate step of organelle isolation to check CD38 enzymatic activity. A method to both localize and demonstrate activity will be helpful.
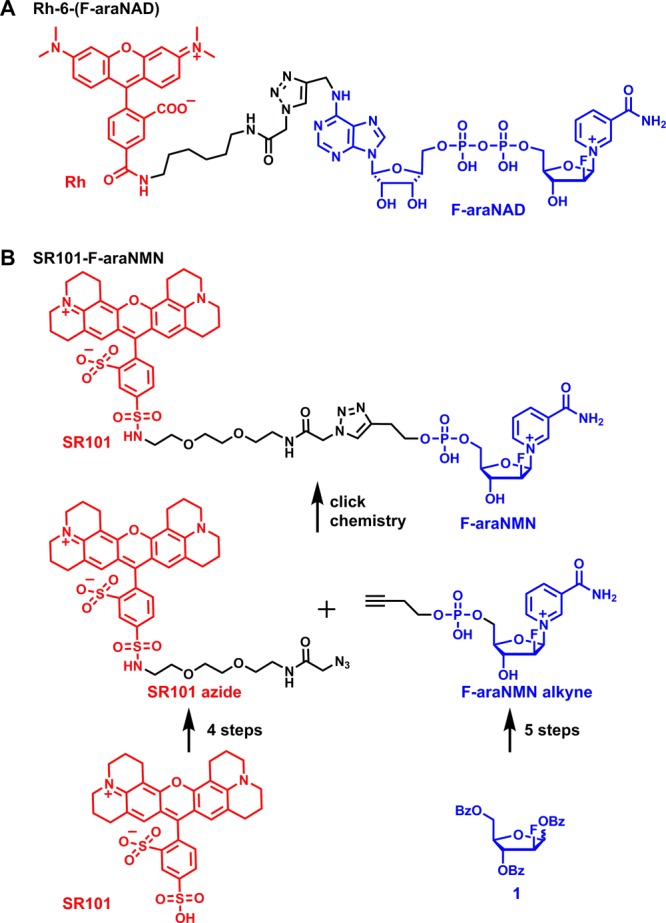
Previously, we developed a CD38-labeling method that uses a suicide substrate, 2′-deoxy-2′-fluoro arabinosyl NAD conjugated with tetramethylrhodamine, Rh-6-(F-araNAD) (Scheme 1A). Rh-6-(F-araNAD) can covalently label CD38 on the key catalytic residue, Glu226, forming a stable covalent intermediate.23 This method was successful in labeling CD38 while not interfering with antibody-induced CD38 signaling events. It was mechanism-based labeling and thus only labeled catalytically active CD38. However, this molecule was not cell permeable; therefore, it cannot be used to label intracellular CD38. In the present study, we developed a cell permeable fluorescent activity-based small-molecule probe for labeling CD38 in live cells. This probe allowed detection of catalytically active CD38 both on the plasma membrane and inside the cells. Using this probe, we investigated the cellular distribution of CD38 in two leukemia (HL-60 and K562) and one lymphoma (Raji) cell lines that were previously reported to have intracellular CD38.15,16 Our results showed CD38 is localized mainly at the plasma membrane with very little intracellular CD38. In particular, our results showed no pronounced nuclear localization as previously reported, which may have been an artifact of the immunofluorescence method. The clarification of CD38 intracellular localization will facilitate the understanding of its physiological function.
Scheme 1. (A) Molecular Structure of Rh-6-(F-araNAD); (B) Synthesis of the Cell-Permeable Fluorescent CD38 Probe, SR101–F-araNMN.
Experimental Section
General Methods
Reagents, unless specified otherwise, were purchased from commercial suppliers in the highest purity available and used as supplied. Antibodies for detection of human CD38 (mouse) were bought from BD Biosciences (San Jose, California, USA, cat. #: 611114), while antibodies for detection of human GAPDH (rabbit), as well as horseradish peroxidase-linked antimouse and antirabbit antibodies were bought from Cell Signaling (Danvers, MA, USA). Mammalian protein extraction reagent (M-PER) was bought from Pierce (Rockford, IL, USA). All-trans retinoic acid (RA), protease and phosphatase inhibitors were bought from Sigma (St. Louis, MO, USA). ECL was bought from GE Healthcare (Pittsburgh, PA, USA). Both sulforhodamine and tetramethylrhodamine fluorescence signals from protein gels were recorded by Typhoon 9400 Variable Mode Imager (GE Healthcare Life Sciences). For visualization of tetramethylrhodamine, a green laser at 532 nm was used for excitation and a collection filter of 580 nm with band-pass of 30 nm was used for emission collection. For visualization of sulforhodamine, the 532 nm laser was used for excitation and 610BP30 filter for emission collection. Detection of fluorescence was done using a PMT at 650 V and normal sensitivity. Images collected were analyzed by ImageQuant TL v2005.
Labeling of Purified CD38 (wt and E226D, E226Q mutants) with SR101–F-araNMN In Vitro
CD38 (wt or E226D and E226Q mutants, 8 μM, expressed and purified as reported earlier24,25) and sulforhodamine 101-F-araNMN (SR101–F-araNMN) (20 μM) in 10 μL reaction buffer (25 mM HEPES, 50 mM NaCl, pH 7.4) were incubated at 37 °C for 30 min, then mixed with 2 μL 6× protein loading buffer. The samples were heated at 100 °C for 7 min and then resolved by SDS-PAGE. Before staining with Coomassie blue, the gel was irradiated under UV light (Transillum, Fisher Scientific, model DLT-A), and the fluorescence image was recorded with a digital camera (Nikon Coolpix L22).
Labeling of CD38 in Live Cells
HL-60 cells were treated with 1 μM RA in cell culture media (GIBCO RPMI Medium 1640 with 10% GIBCO Heat-inactivated Fetal Bovine Serum) for 24 h in a 5% CO2 incubator at 37 °C. Untreated HL-60, Raji, and K562 cells were cultured using the same media without RA. Then the cells were harvested from 4 mL cell culture (1 × 106 cells/mL) by centrifugation at 25 °C, 1200 rpm for 5 min. The cells were initially washed once using 500 μL PBS. Cells were resuspended in 100 μL PBS (reaction volume). In one experimental procedure, these cells were directly labeled with SR101–F-araNMN as detailed below to label all the CD38 molecules. In another experimental procedure, the cells were first treated with a nonfluorescent, cell-impermeable probe, 6-alkyne-F-araNAD, to block the cell surface CD38 before labeling intracellular CD38 with SR101–F-araNMN. 6-Alkyne-F-araNAD was added to a final concentration of 10 μM. After incubation at RT for 8 min, the cells were washed once with 500 μL PBS. Cells were resuspended in 100 μL PBS (reaction volume), and SR101–F-araNMN was added to a final concentration of 10 μM and allowed to incubate at RT for 8 min. The cells were then washed once with 500 μL cold PBS (PBS at 4 °C), followed by resuspending the cells in 1 mL cold methanol (methanol stored in −20 °C for at least 1 h prior to use), and samples were held at −20 °C for 10 min. Methanol was removed after centrifugation at 4 °C, 2000 rpm to pellet the cells. Cells were then resuspended in a fresh 1 mL of cold methanol (methanol at −20 °C) and incubated on ice for 40 min. Again, methanol was removed and cells were washed once with 500 μL fresh, cold PBS (PBS at 4 °C) to ensure removal of methanol. Finally, cells were resuspended in 100 μL of PBS. Then, 10 μL PBS containing the cell suspension was applied onto a microscope slide and covered with a micro cover glass. Confocal images (8 line average) of cells were acquired with a Zeiss LSM 710 confocal microscope with a 63×/1.4 oil immersion objective. Green 561 nm (15mW DPSS laser, laser power percentage given in the figure captions) was used for sulforhodamine (SR) fluorescence. Emission signal in the range of 566–717 nm (SR emission) was detected.
In-Gel Fluorescence Analysis of CD38 Intracellular Localization
Lysis buffer recipe: Tris-HCl pH 7.9 (25 mM), NaCl (150 mM), glycerol (10%), Igepal (1%), 25 μL protease inhibitor cocktail (PIC, Sigma-Aldrich, #P8340)/500 uL, PMSF (0.5 mM), EDTA pH 8.0 (5 mM). HL-60 cells were treated with 1 μM RA in cell culture media for 24 h in a 5% CO2 incubator at 37 °C. Raji and K562 cells were not treated with RA. Untreated HL-60, Raji, and K562 cells were cultured using the same media except for being used without any added treatment. Similar amounts of cells were used for both live cell and whole cell lysate in-gel fluorescent labeling. To do this, cells were counted to make sure the same number of cells were used in the live cell and the whole cell lysate labeling. Cells were harvested by centrifugation at 25 °C, 1200 rpm, for 5 min. Cells were washed twice with 1 mL PBS. From this point, one batch of cells was lysed to obtain whole cell lysate, and then the fluorescent labeling was carried out, and another batch of cells was fluorescently labeled first, and then whole cell lysate was collected. To label live cells, the cells were first suspended in 100 μL PBS (reaction volume) and fluorescent molecule—either SR101–F-araNMN or Rh-6-(F-araNAD)—was added to a final concentration of 10 μM. Then the sample was kept at RT for about 8 min, followed by centrifugation at 4 °C, 1500 rpm, for 3 min. Cells were washed twice using 500 μL cold PBS (PBS at 4 °C). PBS was removed prior to addition of about 30 μL lysis buffer followed by freeze/thaw lysis (the samples were frozen at −80 °C and then removed from −80 °C and thawed on ice for ∼30 min while vortexing briefly every 5 min). Once samples were fully thawed on ice, they were centrifuged at 4 °C, 14,000 rpm, for 6 min to collect the supernatant (proteins solubilized by detergent in lysis buffer). Protein concentration in the cell lysate was determined using the Bradford assay, and 25 μg of lysate was resolved by SDS-PAGE. In the case of collecting whole cell lysate followed by fluorescent labeling, the cells were washed with PBS and resuspended in 30 μL lysis buffer, and then the whole cell lysates were collected as described above. Protein concentration in the cell lysate was determined using the Bradford assay. Once protein concentration was determined, stock lysate was diluted to 2.5 μg/μL, and 10 μL of the diluted lysates was mixed with the fluorescent molecule to give a final 10 μM labeling concentration. The fluorescent labeling reaction was quenched using 2 μL of 6× SDS containing protein loading buffer. The samples were heated at 100 °C for 7 min and then resolved by SDS-PAGE. Before staining with Coomassie blue, the fluorescence image of the gel was recorded by a Typhoon 9400 Variable Mode Imager with settings of green laser at 532 nm and emission collection filter of 580BP30 (rhodamine) and 610BP30 (sulforhodamine). Detection was done using PMT650 V (normal sensitivity), and data were analyzed by ImageQuant TL v2005.
Western Blot Analysis
Cell cultures were seeded at 0.2 × 106 cells/mL, and those that received RA were treated at a concentration of 1 μM. After 48 h, cells were washed twice with PBS before being lysed by adding 400 μL of a lysis buffer consisting of M-PER with 1:100 (v/v) dilutions of protease inhibitors and phosphatase inhibitors, and the cells were placed on ice for 30 min. The lysate was then spun at 4 °C, 13,000 rpm, for 30 min, and supernatant was saved and used for analysis. Then, 25 μg of protein lysate was loaded per lane and resolved by SDS-PAGE analysis and transferred to PVDF membrane. Membranes were then blocked for 1 h in a solution of 5% dry nonfat milk in PBS-Tween before probing with 1:1000 (v/v) dilutions of antibody in 5% BSA in PBS-Tween overnight at 4 °C. Membranes were probed with 1:1000 (v/v) dilutions of secondary antibody in 5% BSA in PBS-Tween for 1 h at RT before visualizing with ECL. Blots shown are representative of at least three independent repeats.
Results
Design and Synthesis of a Cell-Permeable CD38 Probe
Our strategy for making a cell-permeable CD38 probe was to first choose a cell-permeable fluorophore and then conjugate it to F-araNMN instead of F-araNAD (Scheme 1). F-araNMN is smaller and has one fewer negative charges than F-araNAD, and thus may be more cell permeable. Among several fluorescent dyes that we checked, sulfurhodamine 101 (SR101) showed the desired cell permeability. This was interesting as SR101 has size, charge, and overall structure similar to those of tetramethylrhodamine, yet SR101 is more cell permeable. To conjugate SR101 to F-araNMN, we decided to use the copper-catalyzed Huisgen 1,3-dipolar cycloaddition between alkyne and azide, commonly known as click chemistry (Scheme 1B).26 We designed an alkyne-containing F-araNMN compound (F-araNMN alkyne) and SR101 azide compound. To obtain F-araNMN alkyne, we began with a 2′-fluoroarabinoside (1) where all hydroxyl groups were protected with benzoyl groups. The anomeric O-benzoyl group was first replaced with bromine and then by nicotinamide. The benzoyl groups were removed using potassium carbonate in methanol. Then in a one-pot reaction, the 5′-hydroxyl group was phosphorylated and then connected to 3-butyn-1-ol via a phosphodiester bond to give the desired F-araNMN alkyne compound. To make the SR101 azide compound, we first made the sulfonyl chloride derivative of SR101 and then attached a linker with an amino group and an azido group at opposite ends. Finally, the SR101 azide was conjugated to F-araNMN alkyne via click chemistry to obtain the desired SR101–F-araNMN (Scheme 1B). The detailed synthesis is shown in Scheme S1 in the Supporting Information (SI).
In Vitro Labeling of Purified CD38 by SR101–F-araNMN
To test whether SR101–F-araNMN can covalently label CD38 at the catalytic E226 residue, we used purified, wild-type, and catalytic mutants (E226Q and E226D) of CD38 extracellular catalytic domain. Both wild-type and mutants were incubated with SR101–F-araNMN for 10 min. The reaction mixtures were resolved by SDS-PAGE and visualized by fluorescence and then stained with Coomassie blue. Wild-type CD38 was fluorescently labeled by SR101–F-araNMN, but the CD38 catalytic mutants were not (Figure 1). These results demonstrated that SR101–F-araNMN is an activity-/mechanism-based probe for CD38. We compared the labeling efficiency of Rh-6-(F-araNAD) and SR101–F-araNMN. With CD38 at 1 μM and the probes at 10 μM, the labeling reactions were complete within 0.5 min for Rh-6-(F-araNAD) and within 5 min for SR101–F-araNMN (Figure S1 in SI). Furthermore, the addition of NAD (1 mM) or nicotinamide (100 μM) to the labeling reaction had very little effect on labeling efficiency (Figure S1). Thus, SR101–F-araNMN is slightly less efficient than Rh–F-araNAD, but still labels CD38 very efficiently.
Figure 1.
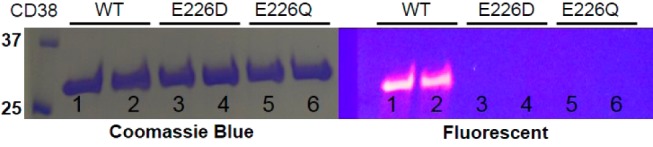
In-vitro labeling of purified CD38 with SR101–F-araNMN. Lanes 1 and 2, CD38 wt; lanes 3 and 4, CD38 E226D; lanes 5 and 6, CD38 E226Q. Ladder is on the left, listed first.
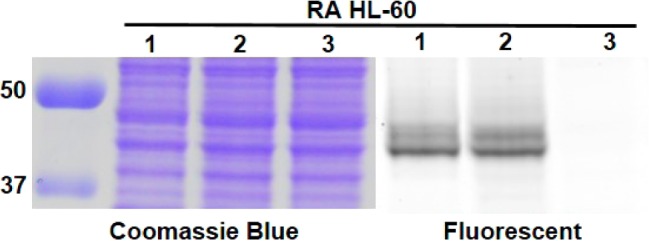
To confirm that this probe is specific for CD38 in cells, we used the human leukemia cell line, HL-60. HL-60 was chosen because these cells have a very low levels of CD38 but could be induced to express higher levels of CD38 with RA.27 Consequently, untreated HL-60 cells were used as the negative control. In one experiment, we labeled live cells, followed by collecting whole cell lysates and then resolved the whole cell lysates with SDS-PAGE (lanes 1, Figure 2). In another experiment, we collected the whole cell lysate, incubated with the probe, and then resolved the lysates by SDS-PAGE (lanes 2, Figure 2). In both cases, only one major fluorescent band, which corresponds to the size of CD38, was detected in RA-treated cells; while no major fluorescent bands were detected in untreated cells (Figure 2). The weak fluorescent band around 20 kD was shown to be from a cleaved form of CD38 (Figure S2 in SI). A weak fluorescent band at about 70 kD was also detected in both RA treated and untreated HL-60 cells in the live-cell labeling but not in the whole-cell lysate labeling (Figure 2). By optimizing the experimental conditions, we were able to partially or completely eliminate this band (Figure S2 and S6 in SI). Altogether, this result demonstrated that SR101–F-araNMN is specific for CD38 in cells. The higher CD38 expression in RA-treated cells was also confirmed by Western blot using a monoclonal anti-CD38 antibody (see Figure 7c).
Figure 2.
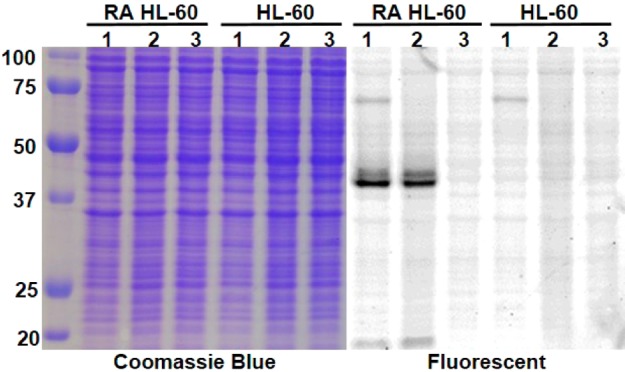
In-gel fluorescence analysis of CD38 in HL-60 cells (RA and untreated). Lanes 1, live-cell labeling with SR101–F-araNMN; lanes 2, whole cell lysate labeled with SR101–F-araNMN; lanes 3, whole cell lysate with no CD38 probe. Ladder is on the left, listed first.
Figure 7.
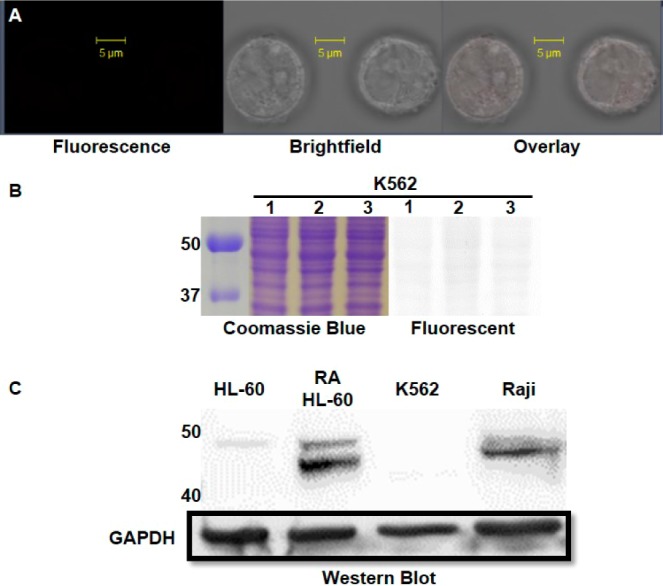
(A) Confocal image of K562 cells with SR101–F-araNMN labeling. Confocal microscope settings: laser power: 4.5%, pinhole: 1.1 airy units, master gain for PMT: 803. (B) K562 in-gel fluorescence analysis: lanes 1, live cell labeling with SR101–F-araNMN followed by in-gel fluorescence; lanes 2, whole cell lysate was obtained first followed by labeling with SR101–F-araNMN (labeling all catalytically active CD38); lanes 3, whole cell lysate without CD38 probe. Ladder is on the left, listed first. The full gel images are shown in Figure S7 in SI. (C) Western blot analysis for detection of CD38 in all cell lines.
Confocal Microscopy Imaging for Labeling of CD38 in Live Cells with SR101–F-araNMN
The ability of SR101–F-araNMN to enter live cells and label CD38 was next tested using HL-60 cells and visualized using confocal microscopy. We first incubated cells with either 10 μM SR101–F-araNMN or 10 μM Rh-6-(F-araNAD) for 8 min at RT followed by washing with PBS to remove excess unbound dye. The incubation time was determined to be sufficient to allow for the binding of the probe to CD38 as determined by an in vitro labeling experiment (Figure S1 in SI). We indeed found that SR101–F-araNMN was cell permeable as strong fluorescence was observed inside HL60 cells (Figure 3A); conversely, HL-60 cells labeled with Rh-6-(F-araNAD) showed no fluorescence inside (Figure 3B). However, washing with PBS alone could not remove excess SR101–F-araNMN molecules in the cells (even HL-60 cells without RA treatment had strong fluorescence inside), making it difficult to differentiate free vs CD38-bound SR101–F-araNMN. To circumvent this problem, we used methanol to wash away the unbound SR101–F-araNMN as it was more soluble in methanol. This was proven successful as HL-60 cells treated with fluorescent molecule alone, SR101, then washed with methanol showed essentially no fluorescence (Figure 3C). In contrast, RA-treated HL-60 cells were strongly fluorescently labeled with SR101–F-araNMN (Figure 3D) on the plasma membrane and only weakly inside the cells.
Figure 3.
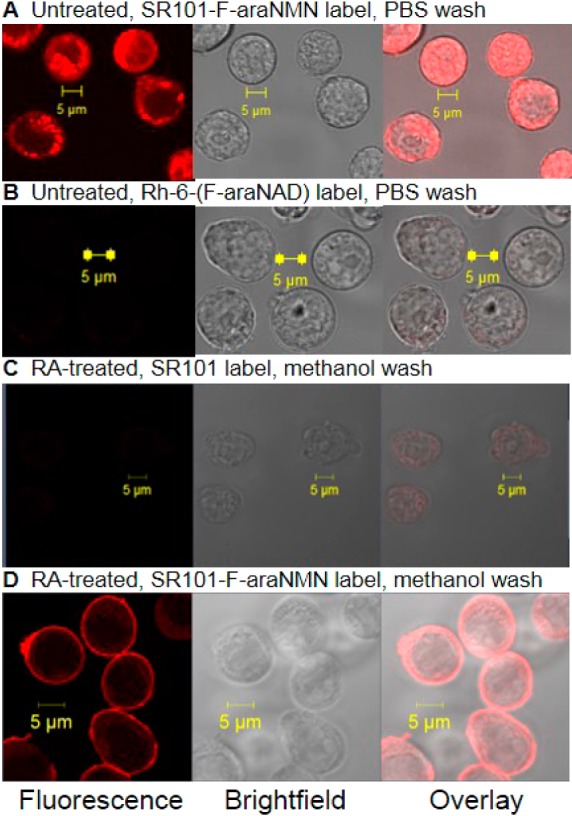
Confocal images of HL-60 cells (with or without RA-treatment): (A) Cells without RA-treatment labeled with SR101–F-araNMN and washed with PBS. (B) Cells without RA-treatment labeled with Rh-6-(F-araNAD) and washed with PBS. (C) RA-treated cells labeled with SR101 (negative control) and washed with methanol. (D) RA-treated cells labeled with SR101–F-araNMN and washed with methanol. Confocal microscope settings: laser power: 4.5%, pinhole: 1.1 airy unit, master gain for PMT: 875.
Then, we turned our focus to locating the intracellular CD38 with SR101–F-araNMN. Although there was some weak fluorescence inside the RA-treated HL-60 cells, it was difficult to visualize the intracellular signal because the fluorescence on the plasma membrane was too strong. To overcome this, we used 6-alkyne-F-araNAD to first block plasma membrane CD38. The 6-alkyne-F-araNAD was not cell permeable and had no fluorescent molecule attached. HL-60 cells (RA treated and untreated) were first incubated with 6-alkyne-F-araNAD to block plasma membrane CD38, followed by incubation either with SR101 as negative control or SR101–F-araNMN to label intracellular CD38. This allowed us to clearly see the intracellular fluorescently labeled CD38, after optimizing the confocal microscope settings to maximize detection. HL-60 cells without RA treatment also showed very weak fluorescent signal. This represented the low levels of CD38 present, which was further confirmed by Western blot data shown later (see Figure 7C). Importantly, an increase in fluorescence (∼2.5-fold) was observed in RA-treated cells compared to HL-60 cells without RA treatment, confirming that the probe was labeling CD38 (Figure 4).
Figure 4.
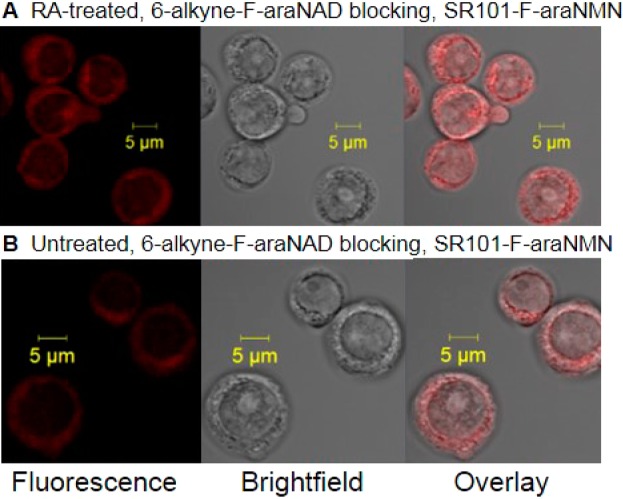
Confocal images of HL-60 cells (RA or untreated) blocked with 6-alkyne-(F-araNAD) followed by intracellular CD38 labeling with SR101–F-araNMN. (A) RA treated HL-60 cells. (B) Untreated HL-60 cells. Confocal microscope settings: laser power: 11.0%, pinhole: 1.3 airy units, Master gain for PMT: 940. With laser settings kept constant between the two images, higher fluorescence indicates higher amount of active CD38.
It has been reported that cell fixation with methanol alone can cause loss of cytosolic and nuclear proteins.28 To rule out that the low detection of intracellular CD38 was not due to the loss of CD38 during the methanol wash, we also used a combination of paraformaldehyde (PFA) fixation with methanol permeabilization after SR101–F-araNMN labeling of CD38 in RA-treated HL-60 cells.28 This method preserves both cell-surface and intracellular proteins.28 The data from this combined PFA fixation and methanol permeabilization led to the same conclusion that CD38 was mainly concentrated on the plasma membrane (Figure S4 in SI). In addition, subcellular fractionation of RA-treated HL-60 cells was done to obtain nucleus, membrane, and cytosolic fractions.29,30 Using a monoclonal anti-CD38 antibody that would presumably detect both catalytically active and inactive CD38, the Western blot data again showed that the nucleus fraction contained very low levels of CD38, while the membrane fraction contain very high levels of CD38 (Figure S5 in SI).
Thus, SR101–F-araNMN was cell permeable and capable of labeling CD38 within live cells without perturbation of the cell before labeling. In addition, it signifies that the intracellular CD38 is in fact catalytically active. Despite the intracellular presence, our results revealed that CD38 was mainly concentrated on the plasma membrane as the plasma membrane had the strongest fluorescence.
In-Gel Fluorescence of Whole Cell Lysate Using CD38 Probes to Label and Quantify Intracellular CD38
The weak intracellular labeling diverged from previous reports regarding pronounced intracellular CD38. We thus decided to quantify the relative amount of intracellular vs plasma membrane CD38. We split the cells from the same culture into two equal portions. One portion was treated with the impermeable Rh-6-(F-araNAD) and then the cells were lysed after washing away excess probes (live cell labeling). The other portion was lysed first and then the total cell lysate was incubated with Rh-6-(F-araNAD) (whole cell lysate labeling). The two batches of cell lysates were then resolved by SDS-PAGE and analyzed by fluorescence and Coomassie blue staining. If a significant amount of CD38 is intracellular, then we expected that the live cell labeling intensity would be weaker than the whole cell lysate labeling intensity with the impermeable Rh-6-(F-araNAD). Contrary to this, we saw essentially the same labeling intensities with the live cell labeling and the whole cell lysate labeling (after correcting for protein loading using the Coomassie blue staining) with Rh-6-(F-araNAD) (Figure 5). In addition, to more accurately quantify the percentage of intracellular CD38, we used another approach with the small-molecule probes. We first blocked cell surface CD38 with 6-alkyne-F-araNAD and then labeled intracellular CD38 with SR101–F-araNMN. In the control experiment, we labeled the total CD38 using SR101–F-araNMN without blocking of the cell surface CD38. We then collected the whole cell lysates and used in-gel fluorescence to quantify the intracellular CD38 vs total CD38 (Figure S3 in SI). Quantification using this method showed that about 6.5% of CD38 molecules were intracellular in RA-treated HL-60 cells. This result further supported the previous confocal imaging results with SR101–F-araNMN and suggested that the amount of intracellular and catalytically active CD38 was very little.
Figure 5.
In-gel fluorescence analysis showed that most CD38 was present on plasma membrane. Lanes 1, live cell labeling with Rh-6-(F-araNAD) followed by in-gel fluorescence (labeling plasma membrane CD38 only); lanes 2, whole cell lysate was obtained first followed by labeling with Rh-6-(F-araNAD) (labeling all catalytically active CD38); lanes 3, whole cell lysate without CD38 probes. Protein ladder is on the left, listed first. The full gel images are shown in Figure S7 in SI.
CD38 Labeling in Raji and K562 Cells
Once cell permeability and CD38 specific labeling was confirmed for SR101–F-araNMN in HL-60 cells, we investigated the intracellular distribution of CD38 in Raji and K562, which are lymphoma and leukemia cell lines, respectively. Both types of cells were reported to have intracellular CD38.15 In particular, in Raji cells, it was reported that CD38 was present in a special subnuclear location called the Cajal body. In K562 cells, it was reported that there was only intracellular CD38 and no plasma membrane CD38.15
Raji cells showed strong plasma membrane fluorescent labeling by SR101–F-araNMN similar to RA-treated HL-60 cells (Figure 6A). To better visualize intracellular labeling, we first blocked plasma membrane CD38 with the nonfluorescent, cell impermeable probe, 6-alkyne-F-araNAD. Then, the cells were incubated with SR101–F-araNMN. We also used DAPI to stain the nucleus. Very weak fluorescence was observed in the nucleus and most of the label localized in the cytoplasm when methanol was used to wash off the probe (Figure 6B). Similar results were obtained when we used PFA and methanol to treat cells (Figure S4 in SI). We again used the in-gel fluorescence analysis, which showed essentially the same labeling intensities in the live cell labeling and the whole cell lysate labeling with Rh-6-(F-araNAD), after correcting for protein loading using the Coomassie blue staining (Figure 6C). This result further supported the previous confocal imaging results with SR101–F-araNMN and suggested that the amount of intracellular CD38 was very little. Also, quantification using 6-alkyne-F-araNAD and SR101–F-araNMN as described above for HL-60 cells, revealed that about 4.2% of catalytically active CD38 in Raji cells is intracellular (Figure S3 in SI).
Figure 6.
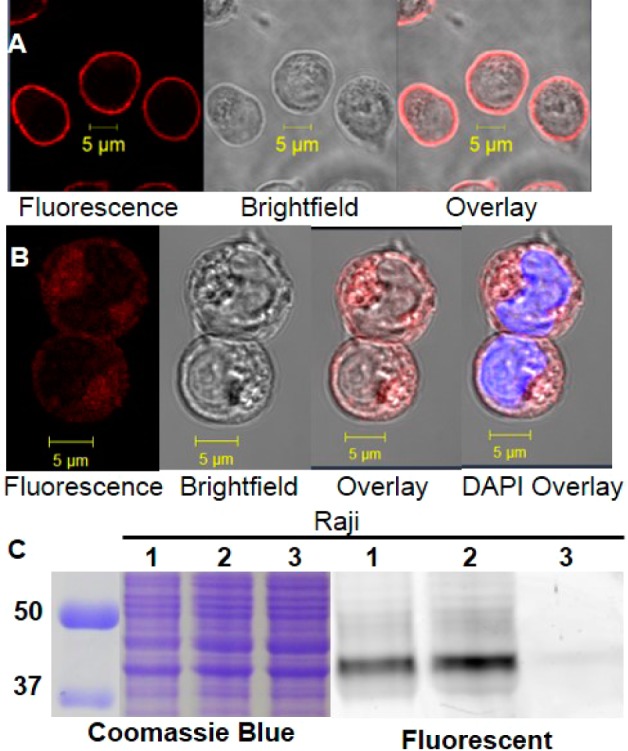
Labeling of CD38 in Raji cells. (A) Confocal image of Raji cells labeled with SR101–F-araNMN. Confocal microscope settings for (A): laser power: 5.5%, pinhole: 1.1 airy units, master gain for PMT: 866. (B) Confocal image of Raji cells blocked with 6-alkyne-(F-araNAD) then labeled with SR101–F-araNMN (visualization of intracellular CD38 only). DAPI (blue) staining dsDNA showing nucleus. Confocal microscope settings for (B): laser power: 10%, pinhole: 1.1 airy units, master gain for PMT: 866. An increase in laser power and master gain was necessary in order to have enough fluorescence emission to see the signal. This indicates a low amount of intracellular active CD38. (C) In-gel fluorescence analysis: lanes 1, live cell labeling with Rh-6-(F-araNAD) followed by in-gel fluorescence (labeling plasma membrane CD38 only); lanes 2, whole cell lysate was obtained first followed by labeling with Rh-6-(F-araNAD) (labeling all catalytically active CD38); lanes 3, whole cell lysate without CD38 probes. Ladder is on the left, listed first. The full gel images are shown in Figure S7 in SI.
As for the K562 cells, no SR101–F-araNMN fluorescent labeling was detected from confocal imaging (Figure 7A). Consistent with this, labeling of cell lysate with SR101–F-araNMN also failed to detect the presence of CD38 (Figure 7B). This result was contrary to the reported result showing that K562 had only intracellular CD38. One possibility for the discrepancy was that intracellular CD38 was present but that it was not catalytically active for unknown reasons. To rule out this possibility, we further performed Western blot analysis on whole cell lysate to detect CD38. The Western blot again showed that K562 cells did not express CD38, while CD38 was detected in RA-treated HL-60 and Raji whole cell lysates, along with low expression in untreated HL-60 cells (Figure 7c). Therefore, our data collectively demonstrated that K562 cells did not express CD38.
Discussion
We developed a cell-permeable CD38 probe, SR101–F-araNMN, by choosing a more cell-permeable fluorescent dye (SR101) and by using F-araNMN instead of F-araNAD. Using purified CD38, SR101–F-araNMN could label wild type CD38 but not the catalytic mutant of CD38, confirming that it was an activity-based probe. Using three human blood cancer cell lines, we further confirmed that SR101–F-araNMN was cell permeable and was able to specifically label CD38. Combined with the previously developed impermeable CD38 probes, Rh-6-(F-araNAD) and 6-alkyne-F-araNAD, we were able to better visualize intracellular CD38 in these cell lines and obtained CD38 localization information that could correct several misconceptions about the intracellular distribution, which are discussed below.
CD38 Is Highly Concentrated in the Plasma Membranes of Raji and RA-Treated HL-60 Cells
It was reported that CD38 was present mainly on the plasma membrane with the catalytic domain facing outside.22 Consistent with this, when we labeled CD38 in RA-treated HL-60 cells or untreated Raji cells with SR101–F-araNMN, the strongest fluorescence resided on the plasma membrane based on confocal imaging. When limiting the fluorescence detection to intracellular CD38, HL-60 cells showed nearly a 2.5-fold increase of intracellular CD38 level upon RA treatment. However, the CD38 expression on the plasma membrane had a much greater increase based upon fluorescence detected. Additionally, our probe was activity-based; therefore, the intracellular CD38 we detected in RA-treated HL-60 and untreated Raji cells should be catalytically active. As a consequence, if the intracellular CD38 had access to cellular NAD, it might lead to decreased intracellular NAD levels. On the basis of confocal imaging, intracellular CD38 was found to be present in punctate bodies in the cytosol, which were likely membrane organelles, such as ER, Golgi, or mitochondria. At this point, we do not know the exact identities of the intracellular organelles that contain CD38, nor do we know the topology of CD38 (facing the cytosol or the matrix) on these intracellular organelles.
Raji and RA-Treated HL-60 Cell Nuclei Have Little CD38
A previous report on CD38 localization used confocal immunofluorescence to show the presence of CD38 primarily within the nucleus of Raji cells.15 In fact, it was shown that CD38 colocalized in nuclear Cajal bodies, which are small subnuclear membraneless organelles present either free in the nucleoplasm and/or physically associated to specific regions of chromatin. However, using the activity-based probes for CD38, we found very little CD38 labeled within the nucleus, and certainly no concentrated areas of fluorescence that would represent active CD38 in Cajal bodies. In fact, the nucleus had the weakest CD38 labeling in both Raji cells and RA-treated HL-60 cells. Although we could not rule out the presence of catalytic inactive CD38, the results obtained with the K562 cells suggested that this was unlikely (discussed below). Our results thus suggested that very little CD38 is present in the nuclei of Raji and RA-treated HL-60 cells.
K562 Cells Do Not Have Detectable CD38 Expression
It was reported on the basis of immunofluorescence that K562 cells express intracellular CD38 but no plasma membrane CD38.15 When labeling K562 cells with SR101–F-araNMN, we saw no fluorescence from confocal microscopy imaging on either the plasma membrane or within the cell. To rule out that SR101–F-araNMN was not permeable to K562 cells, we also labeled K562 whole cell lysates with SR101–F-araNMN. No fluorescent CD38 band was observed on SDS-PAGE gel, consistent with the confocal imaging results. This result was contrary to the reported intracellular only localization of CD38. To resolve the conflicting observations, we first considered the possibility that K562 cells had CD38 but that CD38 was catalytically inactive; and thus, CD38 could not be labeled with our probe. However, Western blot showed that K562 cells had no detectable CD38 expression. Therefore, the labeling result with SR101–F-araNMN in K562 cells was reliable and revealed the absence of CD38 in K562 cells.
Antibodies vs Activity-Based Probes for Detecting CD38 Intracellular Localization
The most commonly used methods to detect CD38 cellular localization are confocal microscopy coupled with immunofluorescence or GFP fusion. Using GFP fusion proteins requires the overexpression of the fusion proteins, which may cause artifacts due to the overexpression or the fusion. Immunofluorescence has the advantages of detecting endogenous proteins but it requires the use of antibodies. CD38 antibodies are convenient tools to use in most applications. However, for detecting the intracellular localization of CD38, antibodies have some limitations. First, antibodies are not cell permeable; thus, the cells have to be fixed and permeablized before labeling with antibodies. Second, washing conditions have to be controlled well to wash away unbound antibodies but not antibodies that bind to the protein target. The potential problems can be fixed but require extra efforts to optimize experimental conditions. The reported prominent nuclear CD38 in Raji and K562 cells is possibly a false positive caused by insufficient washing, which could have been avoided if proper negative and positive controls were used to help find the optimal washing conditions.15 When inspecting the data on the immunofluorescence detection of CD38 in Raji and K562 cells, we noticed that there were no negative or positive controls provided. Nuclear membrane localization was also reported in other cell types.20,31 Immunofluorescence was the major method used in these studies reporting the nuclear membrane localization of CD38. These studies may also suffer from similar problems. Compared with immunofluorescence, our permeable CD38 probe has certain advantages that can complement the use of antibodies. First, the probe identifies catalytically active CD38 because labeling proceeds in an activity-dependent way while immunofluorescence can be used to reveal total protein levels. Second, the probe is cell permeable and thus does not require detergent permeabilization of cells. Thus, the labeling can be done under less perturbing conditions. Unfortunately, we had to fix cells to wash away excess probe molecule. Optimization of the probes to allow aqueous buffer wash will be required for true live-cell imaging. However, because it forms a rather stable covalent linkage with CD38, the washing step can be done very extensively to make sure all excess CD38 probes are washed away. This way, fewer false positives will be observed. This is probably an important feature that allowed us to conclude that there is very little intracellular CD38. The availability of both a permeable and impermeable CD38 probe greatly facilitated the quantification of intracellular versus cell surface CD38, which is otherwise not easy to accurately quantify.
Conclusion
NAD is increasingly recognized as an important signaling molecule that regulates many physiologically important processes. Understanding the function of NAD-metabolizing enzymes, such as CD38, is thus important and can impact a variety of different areas. Ensuring proper cellular function requires the spatial distribution of different proteins to be delicately regulated. In the case of CD38, localization is similarly important to its biological function. Investigating its correct cellular localization will help to understand several unaddressed questions about the function of CD38. Early studies on CD38 focused on its ability to make cADPR and NAADP, which are considered important second messengers capable of releasing intracellular stored calcium. However, in vitro, the most efficient activity of CD38 is the hydrolysis of NAD, with kcat of 96 s–1 and Km of 16 μM.32 If CD38 is present in the nucleus as previously reported, it will likely deplete nuclear NAD. Using the permeable and impermeable CD38 probes, we found that the nucleus actually contained the least amount of fluorescence, which we interpreted as the lack of CD38 in the nucleus. Considering the efficient NAD hydrolysis activity, we think the lack of nuclear CD38 actually makes sense; otherwise, intracellular NAD may be depleted. CD38-catalyzed formation of cADPR accounted for less than 2% of the total product. If this activity is physiologically important, it is possible that a certain intracellular pool of CD38 may have higher cyclase activity. CD38-catalyzed formation of NAADP in vitro requires low pH and high concentration of nicotinic acid. The low pH condition is only possible in certain acidic organelles, such as the lysosome. Thus, investigating the exact intracellular localization of CD38 will likely provide insights into the physiological relevance of different enzymatic activities of CD38. The activity-based probes we developed will allow the determination of intracellular localization of CD38 in different cells and thus enable a better understanding of the physiological function of CD38.
Acknowledgments
This work is supported by NIH R01GM086703 (H.L.) and R01CA1525870 (A.Y.). The confocal imaging was enabled by the Cornell University Biotechnology Resource Center that is supported by NIH 1S10RR025502-01 for all data collected on the Zeiss LSM 710 Confocal.
Supporting Information Available
Details of experimental procedures, acquired and measured NMR, and LC–MS spectra for the synthesis of the CD38 probe, SR101–F-araNMN. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Houtkooper R. H.; Cantó C.; Wanders R. J.; Auwerx J. Endocr. Rev. 2010, 31, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. Org. Biomol. Chem. 2007, 5, 2541. [DOI] [PubMed] [Google Scholar]
- Partida-Sanchez S.; Cockayne D. A.; Monard S.; Jacobson E. L.; Oppenheimer N.; Garvy B.; Kusser K.; Goodrich S.; Howard M.; Harmsen A.; Randall T. D.; Lund F. E. Nat. Med. 2001, 7, 1209. [DOI] [PubMed] [Google Scholar]
- Jin D.; Liu H. X.; Hirai H.; Torashima T.; Nagai T.; Lopatina O.; Shnayder N. A.; Yamada K.; Noda M.; Seike T.; Fujita K.; Takasawa S.; Yokoyama S.; Koizumi K.; Shiraishi Y.; Tanaka S.; Hashii M.; Yoshihara T.; Higashida K.; Islam M. S.; Yamada N.; Hayashi K.; Noguchi N.; Kato I.; Okamoto H.; Matsushima A.; Salmina A.; Munesue T.; Shimizu N.; Mochida S.; Asano M.; Higashida H. Nature 2007, 446, 41. [DOI] [PubMed] [Google Scholar]
- D’Arena G.; Musto P.; Cascavilla N.; Dell’Olio M.; Di Renzo N.; Perla G.; Savino L.; Carotenuto M. Leuk. Lymphoma 2001, 42, 109. [DOI] [PubMed] [Google Scholar]
- States D. J.; Walseth T. F.; Lee H. C. Trends Biochem. Sci. 1992, 17, 495. [DOI] [PubMed] [Google Scholar]
- Howard M.; Grimaldi J. C.; Bazan J. F.; Lund F. E.; Santos-Argumedo L.; Parkhouse R. M.; Walseth T. F.; Lee H. C. Science 1993, 262, 1056. [DOI] [PubMed] [Google Scholar]
- Kim H.; Jacobson E. L.; Jacobson M. K. Science 1993, 261, 1330. [DOI] [PubMed] [Google Scholar]
- Lee H. C.; Zocchi E.; Guida L.; Franco L.; Benatti U.; De Flora A. Biochem. Biophys. Res. Commun. 1993, 191, 639. [DOI] [PubMed] [Google Scholar]
- Takasawa S.; Tohgo A.; Noguchi N.; Koguma T.; Nata K.; Sugimoto T.; Yonekura H.; Okamoto H. J. Biol. Chem. 1993, 268, 26052. [PubMed] [Google Scholar]
- Chini E. N.; Chini C. C.; Kato I.; Takasawa S.; Okamoto H. Biochem. J. 2002, 362, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubiaur M.; Fernandez O.; Ferrero E.; Salmeron J.; Malissen B.; Malavasi F.; Sancho J. J. Biol. Chem. 2002, 277, 13. [DOI] [PubMed] [Google Scholar]
- Khoo K. M.; Chang C. F. Int. J. Biochem. Cell Biol. 2002, 34, 43. [DOI] [PubMed] [Google Scholar]
- Yalcintepe L.; Albeniz I.; Adin-Cinar S.; Tiryaki D.; Bermek E.; Graeff R. M.; Lee H. C. Exp. Cell Res. 2005, 303, 14. [DOI] [PubMed] [Google Scholar]
- Trubiani O.; Guarnieri S.; Eleuterio E.; Di Giuseppe F.; Orciani M.; Angelucci S.; Di Primio R. J. Cell Biochem. 2008, 103, 1294. [DOI] [PubMed] [Google Scholar]
- Orciani M.; Trubiani O.; Guarnieri S.; Ferrero E.; Di Primio R. J. Cell Biochem. 2008, 105, 905. [DOI] [PubMed] [Google Scholar]
- Ceni C.; Pochon N.; Brun V.; Muller-Steffner H.; Andrieux A.; Grunwald D.; Schuber F.; De Waard M.; Lund F.; Villaz M.; Moutin M. J. Biochem. J. 2003, 370, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinherz E. L.; Kung P. C.; Goldstein G.; Levey R. H.; Schlossman S. F. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M.; Mizuguchi M.; Otsuka N.; Ikeda K.; Takahashi H. Brain Res. 1997, 756, 52. [DOI] [PubMed] [Google Scholar]
- Adebanjo O. A.; Anandatheerthavarada H. K.; Koval A. P.; Moonga B. S.; Biswas G.; Sun L.; Sodam B. R.; Bevis P. J.; Huang C. L.; Epstein S.; Lai F. A.; Avadhani N. G.; Zaidi M. Nat. Cell Biol. 1999, 1, 409. [DOI] [PubMed] [Google Scholar]
- Zhao Y. J.; Lam C. M.; Lee H. C.. Sci. Signal. 2012, 5, ra67. [DOI] [PubMed]
- Moreno-Garcia M. E.; Sumoza-Toledo A.; Lund F. E.; Santos-Argumedo L. Mol. Immunol. 2005, 42, 703. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Congleton J.; Liu Q.; Merchant P.; Malavasi F.; Lee H. C.; Hao Q.; Yen A.; Lin H. J. Am. Chem. Soc. 2009, 131, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Kriksunov I. A.; Graeff R.; Munshi C.; Lee H. C.; Hao Q. Structure 2005, 13, 1331. [DOI] [PubMed] [Google Scholar]
- Munshi C.; Aarhus R.; Graeff R.; Walseth T. F.; Levitt D.; Lee H. C. J. Biol. Chem. 2000, 275, 21566. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- Lamkin T. J.; Chin V.; Varvayanis S.; Smith J. L.; Sramkoski R. M.; Jacobberger J. W.; Yen A. J. Cell Biochem. 2006, 97, 1328. [DOI] [PubMed] [Google Scholar]
- Schnell U.; Dijk F.; Sjollema K. A.; Giepmans B. N. Nat. Methods 2012, 9, 152. [DOI] [PubMed] [Google Scholar]
- Rockstroh M.; Muller S.; Jende C.; Kerzhner A.; Bergen M.; Tomm J. J. Integr. Omics 2011, 1, 135. [Google Scholar]
- Cox B.; Emili A. Nat. Protoc. 2006, 1, 1872. [DOI] [PubMed] [Google Scholar]
- Khoo K. M.; Han M. K.; Park J. B.; Chae S. W.; Kim U. H.; Lee H. C.; Bay B. H.; Chang C. F. J. Biol. Chem. 2000, 275, 24807. [DOI] [PubMed] [Google Scholar]
- Sauve A. A.; Munshi C.; Lee H. C.; Schramm V. L. Biochemistry 1998, 37, 13239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.