

Abstract
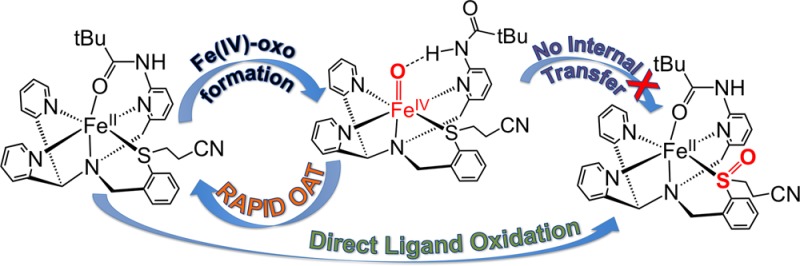
The new ligand N3PyamideSR and its FeII complex [FeII(N3PyamideSR)](BF4)2 (1) are described. Reaction of 1 with PhIO at −40 °C gives metastable [FeIV(O)(N3PyamideSR)]2+ (2), containing a sulfide ligand and a single amide H-bond donor in proximity to the terminal oxo group. Direct evidence for H-bonding is seen in a structural analogue, [FeII(Cl)(N3PyamideSR)](BF4)2 (3). Complex 2 exhibits rapid O-atom transfer (OAT) toward external sulfide substrates, but no intramolecular OAT. However, direct S-oxygenation does occur in the reaction of 1 with mCPBA, yielding sulfoxide-ligated [FeII(N3PyamideS(O)R)](BF4)2 (4). Catalytic OAT with 1 was also observed.
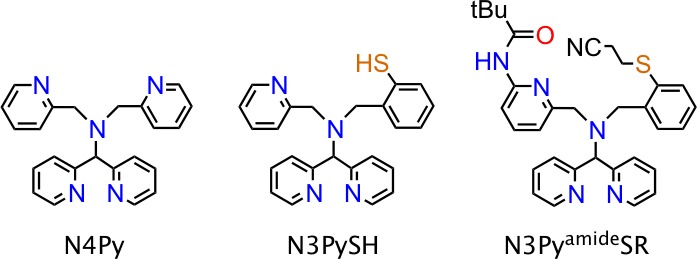
Determining the influence of both first- and second-coordination-sphere elements on non-heme iron-Ox species has been the focus of much recent effort.1 Well characterized, high-valent FeIV(O) complexes are known, but most of these systems contain structurally similar polyamino/pyridyl ligands.1a Very few FeIV(O) complexes have been reported with heteroatoms other than nitrogen in the first coordination sphere. The inclusion of sulfur donors is important for modeling key metal–sulfur interactions in non-heme enzymes, but is challenging because of the inherent problems associated with the facile oxidation of sulfur.2 In earlier work we described an FeII complex with a thiolato donor built into the first coordination sphere, N3PyS– (Figure 1), which undergoes facile O2-mediated S-oxygenation.2b This complex provided a functional model of the non-heme iron enzyme cysteine dioxygenase (CDO), but no FeIV(O) intermediate was detected. In contrast, the FeIV(O) complex of the all-nitrogen analogue N4Py is well-characterized.3
Figure 1.
Ligands discussed in the present study.
Encouraged by our success in ligand design to incorporate an S-bound substrate mimic, we have made further efforts to control the reactivity of non-heme Fe centers by tuning both the first and second coordination spheres through ligand modification.4 In the present work, a new thioether, amide-appended ligand N3PyamideSR (Figure 1) has been prepared from the N3PyS scaffold. The thiolato donor was converted to a thioether group to discourage S oxidation in the first coordination sphere and potentially allow for the stabilization of an FeIV(O) complex, similar to the proposed protective function of N–H---S hydrogen bonds in thiolate-ligated metalloenzymes.5 In addition, a pendant amide group was installed in the second coordination sphere to examine the influence of a single H-bond donor on the reactivity of Fe/Ox intermediates, including FeIV(O) species. The influence of multiple H-bond donors on sterically encumbered metal–oxygen species has been examined, but there remains relatively little known about the influence of a single H-bond donor on sterically accessible MOx sites.6
The new ligand N3PyamideSR was used to prepare the FeII complex [FeII(N3PyamideSR)](BF4)2 (1). Complex 1 reacts with PhIO to give a metastable ferryl complex at low temperature, [FeIV(O)(N3PyamideSR)]2+ (2). This species is a rare example of a well characterized FeIV(O) complex with sulfur ligation.7 Remarkably, complex 2 shows no propensity for intramolecular sulfoxidation, but instead exhibits rapid intermolecular O-atom transfer (OAT) to external thioether substrates. In addition, the rates of OAT are dramatically accelerated when compared to the same reaction exhibited by other FeIV(O) complexes, and this increase in reactivity may be due, at least in part, to the presence of the designed NH---O=Fe hydrogen bond.
Reaction of N3PyamideSR with FeII(BF4)2 in MeCN led to the isolation of crystalline 1 (88%). The X-ray structure of 1 at 110 K (Figure 2) reveals an octahedral FeII complex with the amide group coordinated in the open site. Bond distances (Table S1) for 1 are consistent with low-spin (ls) FeII (S = 0), and Mössbauer spectroscopy at 5 K reveals a sharp, well-resolved doublet (δ = 0.47 mm/s; ΔEQ = 0.77 mm/s, Figure 3) typical for ls-FeII. However, the room-temperature 1H NMR spectrum for 1 reveals paramagnetically shifted peaks, but this is likely due to a small paramagnetic impurity and further work is ongoing to definitively assign the spin state at higher temperatures for this complex.
Figure 2.
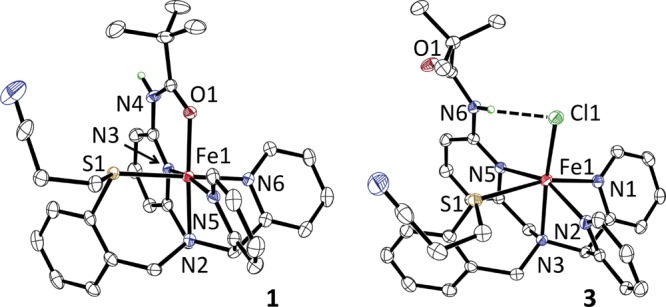
Displacement ellipsoid plots (50% probability level) of the dication of 1 (left) and the cation of 3 (right). H-atoms omitted for clarity, except for the amide N–H.
Figure 3.
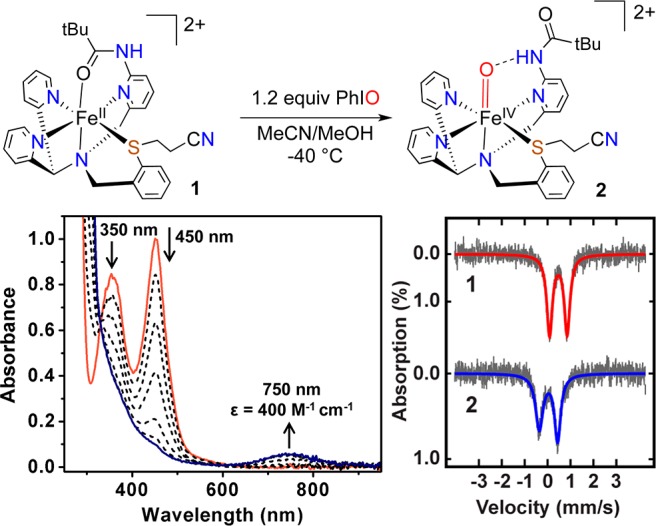
Synthesis of 2 (top); UV–vis spectral changes for the conversion of 1 to 2 (bottom, left); Mössbauer spectra (47 mT) for 1 and 2 (bottom, right) at 5 K in CH3CN.
Reaction of 1 with PhIO (1.2 equiv, dissolved in minimal MeOH) in MeCN at −40 °C leads to the rapid decay of the peaks for 1 at 350 and 450 nm (ε = 5 × 103 and 6 × 103 M–1 cm–1, respectively), and the isosbestic production of a new peak at 750 nm (ε = 400 M–1 cm–1, Figure 3). This broad, weak band in the near-IR region is characteristic of non-heme FeIV(O) complexes.1 The new species with λmax = 750 nm is stable at −40 °C for hours but immediately decays upon warming above −30 °C. This behavior contrasts that of [FeIV(O)(N4Py)]2+, which is stable at room temperature (t1/2 = 60 h).3a Analysis of the reaction mixture by low-temperature, high-resolution ESI-MS (+ mode) (−40 °C) shows a parent ion at m/z 311.0914 and isotope pattern corresponding to the dicationic [Fe(O)(N3PyamideSR)]2+ (Figure S3). The 750 nm species is EPR silent (X-band, 15 K), consistent with an integer-spin FeIV(O) complex. Confirmation of the nature of the new species from 1 + PhIO was obtained through Mössbauer spectroscopy, which shows a well-resolved doublet (δ = 0.04 mm/s, ΔEQ = 0.80 mm/s, Figure 3). These parameters, especially the low isomer shift, are definitive for an FeIV(O) complex.1 The lack of peaks for 1 indicates that the FeII complex has been quantitatively converted to the FeIV(O) complex (2).
A structural analogue of metastable 2 was synthesized from FeCl2, N3PyamideSR, and anion metathesis with NaBF4, affording yellow, crystalline [FeII(Cl)(N3PyamideSR)](BF4) (3). The X-ray structure of 3 (Figure 2) shows that the carbonyl group has been displaced by Cl–. Importantly, the free amide N–H group is clearly oriented for H-bonding to the terminal Cl– (Cl---H–N = 2.497 Å; Cl–H–N = 167.53°).8 The structure of 3 fully supports the proposed structure for 2, in which the pendant N–H amide forms a stable, 6-membered H-bond ring with the terminal oxo ligand.
The structures and Mössbauer parameters for 1 and 2 were examined by density functional theory (DFT) methods. Assuming a ground state singlet for 1, the optimized geometry (B3LYP) (Figure S14, Table S1) and calculated Mössbauer parameters (δ = 0.39 mm/s, ΔEQ = 0.82 mm/s) are in good agreement with the experimental data, providing a calibration of our theoretical methods. Geometry optimization for 2 confirms the proposed structure (Figure S15), including the amide NH---O H-bond, and a triplet ground state is found for 2 (32) with a close-lying quintet (52) excited state 2.3 kcal/mol higher in energy (Table S2). The experimental isomer shift for 2 falls within the range for 3[FeIV(O)] complexes as well as the low end of the range for 5[FeIV(O)] complexes (Table S4), and thus the Mössbauer data alone do not conclusively show a triplet ground state. However, the DFT-derived Mössbauer parameters for 32 are a good match for the experimental values (calcd: δ = 0.03 mm/s, ΔEQ = 0.55 mm/s, Table S3), whereas the calculated parameters for 52 deviate significantly from experiment. Moreover almost all well-defined 5[FeIV(O)] species are 5-coordinate, whereas the optimized geometry for 2 is 6-coordinate. The experimental data, together with DFT calculations and reactivity studies (vide infra), provide strong evidence that the ground state for 2 is most likely a triplet state.
We have previously shown that the thiolato donor in [FeII(N3PyS)]+ is S-oxygenated by O2, as is the coordinated S-Cys donor in CDO.2b,9 Thus it is notable that protection of the thiolato donor to a thioether allows for a metastable FeIV(O) complex 2 to be trapped at low temperature, exhibiting no intramolecularS-oxygenation. However, when 2 is exposed to external thioether substrates, rapid OAT occurs. Reaction of 2 with PhSMe at −40 °C leads to the fast decay of the 750 nm band and isosbestic return of the starting FeII complex 1 (82% recovery in 30 min) (Figure S7). Analysis by 1H NMR showed PhS(O)Me was produced in 80% yield (eq 1).
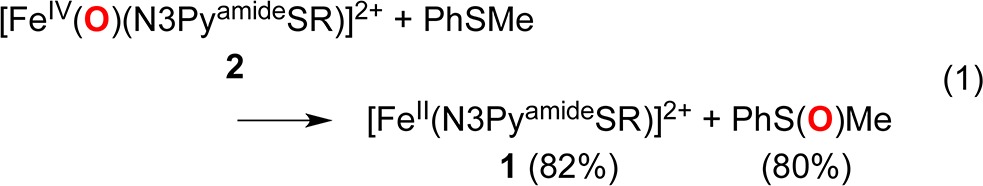 |
1 |
The kinetics of intermolecular sulfoxidation were monitored by UV–vis under pseudo-first-order conditions, and could be well fit to a single exponential model (Figure S8). A linear dependence was found for kobs versus [PhSMe], yielding a second-order rate constant of k = 4.3 M–1 s–1 (Figure S9). The rate constants for other non-heme FeIV(O) complexes are given in Table 1. Complex 2 shows dramatically enhanced OAT reactivity, exhibiting a nearly 70-fold rate enhancement at −40 °C vs the parent [FeIV(O)(N4Py)]2+ at 0 °C.10b The reaction between [FeIV(O)(N4Py)]2+ and PhSMe at −40 °C was run as a direct comparison, but we observed no spectroscopic change under these conditions over 40 min. The only example in Table 1 that also exhibits significantly increased reactivity is found in row 3, in which the strong proton donor HClO4 is postulated to activate the FeIV(O) complex through monoprotonation of the oxo ligand ([(N4Py)FeIV(OH)]3+).10a We suggest that the unique amide H-bond donor in 2 may activate the terminal oxo ligand in an analogous fashion, increasing the electrophilicity of the oxo ligand.
Table 1. Second-Order Rate Constants for OAT of Various FeIV(O) Complexes to Thioanisole.
The choice of oxidant for 1 is critical to the reaction pathway. If PhIO is replaced with the more powerful oxidant mCPBA, the UV–vis signature for the ferryl complex is not obtained, but instead an intense band at 700 nm (1000 M–1 cm–1) is produced. This new species is thermally stable, unlike 2, and an X-ray structure of this product (Figure 4) reveals that the ligated sulfide has been S-oxygenated to sulfoxide, affording [FeII(N3PyamideS(O)R)]2+(BF4)2 (4). The major isomer of 4 is S-bound, while a minor isomer is O-bound to iron (Figure S6). Complex 4 also provides an authentic iron(II)-sulfoxide complex with a characteristic UV–vis band at λmax = 700 nm. The complete absence of this peak during the formation of 2 or its subsequent reactions with external substrates confirms that intramolecular S-oxygenation does not occur for 2. The susceptibility of the coordinated thioether to sulfoxidation with mCPBA further emphasizes the remarkable stability of 2 toward intramolecular OAT.
Figure 4.
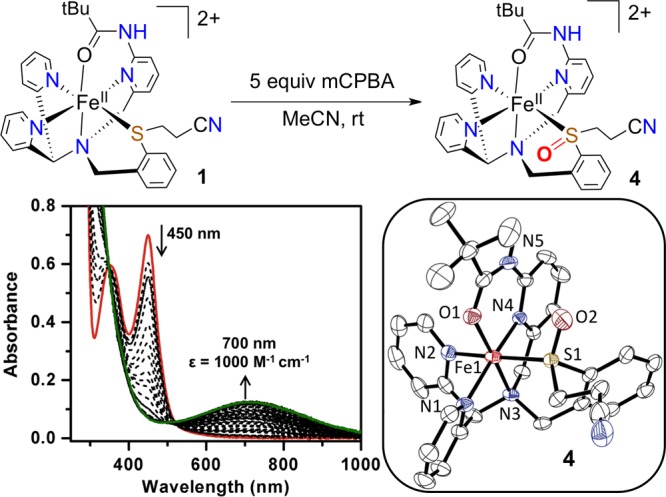
Synthesis of sulfoxide complex 4 (top); UV–vis spectral changes for the formation of 4 (bottom left); and displacement ellipsoids (50% probability level) for the dication of 4 showing the major S-bound isomer, with H-atoms omitted for clarity, except for the amide N–H (bottom right).
A preliminary examination of 2 for its ability to abstract hydrogen atoms reveals strongly muted reactivity. Addition of the H-atom donor 2,4-di-tert-butylphenol to 2 at −40 °C leads to the slow decay of the 750 nm band and return of the peaks for 1 (Figure S11). The reaction rate with a large excess of phenol (500 equiv) leads to a kobs of only 4.0 × 10–3 s–1 (Figure S12), which is 2.5 times slower than H-atom transfer (HAT) to [FeIV(O)(TMC)(NCCH3)]2+ at 0 °C for the same phenol.11 This observation is in line with calculations that predict that H-bonds in non-heme FeIV(O) and MnIV(O) complexes should lower H-atom abstraction reactivity.12 Given that only one tBu group on the substrate is in proximity to the O–H bond, it is also unlikely that sterics play a significant role in lowering the reaction rate. These results also argue against the involvement of a high-spin 5[FeIV(O)] ground state (or accessible quintet excited state) in the reactivity of 2, because a 5[FeIV(O)] species should exhibit accelerated HAT.4,13
Finally, the efficient intermolecular OAT facilitated by 2 to PhSMe can be exploited for catalytic sulfoxidation. Complex 1 was combined with excess PhSMe (100 equiv) at room temperature and then treated with PhIO (50 equiv). A transient color change signaling the formation of 2 was observed, followed by an immediate return of the orange color for 1. Product analysis by 1H NMR gave 40 turnovers of PhS(O)Me in 2 h (Figure S13), before catalyst decomposition. These results demonstrate that complex 1 is able to function as a potent catalyst for the oxidation of thioethers.
The new findings reported here are summarized schematically in Figure 5. We have shown that a rare, metastable non-heme FeIV(O) complex can be generated with a pendant thioether donor in the first coordination sphere. Despite the fact that 2 does not undergo intramolecular S-oxygenation, it does facilitate the rapid sulfoxidation of external thioether substrates. Although more work is needed to determine the origins of this high selectivity, coordination of the thioether ligand to iron may constrain the orientation of the S donor, and/or raise its redox potential, such that oxidation is discouraged. The dramatic rate enhancement in OAT facilitated by 2 is similar to that seen for [FeIV(O)(N4Py)]2+ upon addition of strong H+ donors, suggesting that the inclusion of the amide H-bond donor in the second coordination sphere may enhance OAT reactivity through a similar increase in electrophilicity of the terminal oxo ligand. However other factors may contribute to the strong increase in reactivity seen for 2, including the accessibility of a high-spin quintet state, which cannot be completely ruled out by the available data. The metal-oxo group may also be more exposed in 2 as compared to [FeIV(O)(N4Py)]2+ because of replacement of an equatorial py donor with a less sterically encumbered sulfide donor, allowing for easier approach of substrate. The novel reactivity seen for 2 provides motivation for further examination of first- and second-coordination-sphere effects in this, and other non-heme iron model complexes.
Figure 5.
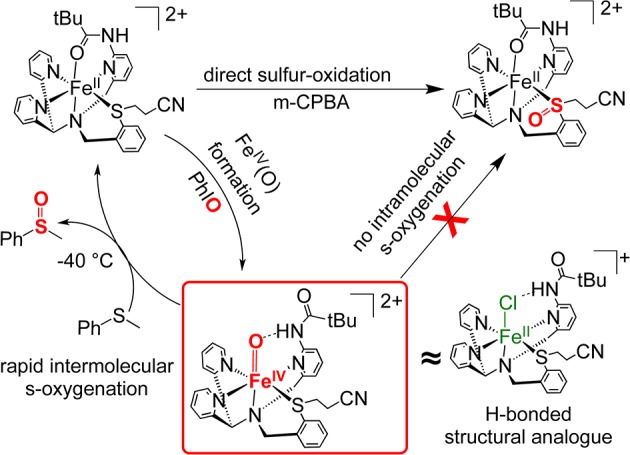
Summary of reactivity for 1 and 2.
Acknowledgments
The NIH (D.P.G., GM62309 and GM101153) is gratefully acknowledged for financial support. G.N.L.J. thanks the Marsden Fund and The International Mobility Fund administered by Royal Society of New Zealand. I. I.-B. and O. T. gratefully acknowledge support through the “Solar Technologies Go Hybrid” initiative of the German Federate State of Bavaria.
Supporting Information Available
Detailed experimental procedures, ESI-MS data, crystallographic information for 1, 3, and 4, Mössbauer spectroscopy, details for kinetic studies, and computational investigations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a McDonald A. R.; Que L. Jr. Coord. Chem. Rev. 2013, 257, 414. [Google Scholar]; b Shook R. L.; Borovik A. S. Inorg. Chem. 2010, 49, 3646. [DOI] [PMC free article] [PubMed] [Google Scholar]; c de Visser S. P.; Rohde J. U.; Lee Y. M.; Cho J.; Nam W. Coord. Chem. Rev. 2013, 257, 381. [Google Scholar]
- a McQuilken A. C.; Goldberg D. P. Dalton Trans. 2012, 41, 10883. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McQuilken A. C.; Jiang Y.; Siegler M. A.; Goldberg D. P. J. Am. Chem. Soc. 2012, 134, 8758. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jiang Y.; Widger L. R.; Kasper G. D.; Siegler M. A.; Goldberg D. P. J. Am. Chem. Soc. 2010, 132, 12214. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sallmann M.; Siewert I.; Fohlmeister L.; Limberg C.; Knispel C. Angew. Chem., Int. Ed. 2012, 51, 2234. [DOI] [PubMed] [Google Scholar]; e Badiei Y. M.; Siegler M. A.; Goldberg D. P. J. Am. Chem. Soc. 2011, 133, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Liu T. B.; Li B.; Singleton M. L.; Hall M. B.; Darensbourg M. Y. J. Am. Chem. Soc. 2009, 131, 8296. [DOI] [PubMed] [Google Scholar]; g O’Toole M. G.; Kreso M.; Kozlowski P. M.; Mashuta M. S.; Grapperhaus C. A. J. Biol. Inorg. Chem. 2008, 13, 1219. [DOI] [PubMed] [Google Scholar]; h Lugo-Mas P.; Taylor W.; Schweitzer D.; Theisen R. M.; Xu L.; Shearer J.; Swartz R. D.; Gleaves M. C.; DiPasquale A.; Kaminsky W.; Kovacs J. A. Inorg. Chem. 2008, 47, 11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kaizer J.; Klinker E. J.; Oh N. Y.; Rohde J. U.; Song W. J.; Stubna A.; Kim J.; Münck E.; Nam W.; Que L. Jr. J. Am. Chem. Soc. 2004, 126, 472. [DOI] [PubMed] [Google Scholar]; b Klinker E. J.; Kaizer J.; Brennessel W. W.; Woodrum N. L.; Cramer C. J.; Que L. Jr. Angew. Chem., Int. Ed. 2005, 44, 3690. [DOI] [PubMed] [Google Scholar]
- Sahu S.; Widger L. R.; Quesne M. G.; de Visser S. P.; Matsumura H.; Moënne-Loccoz P.; Siegler M. A.; Goldberg D. P. J. Am. Chem. Soc. 2013, 135, 10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zheng P.; Takayama S. I. J.; Mauk A. G.; Li H. B. J. Am. Chem. Soc. 2012, 134, 4124. [DOI] [PubMed] [Google Scholar]; b Dey A.; Okamura T.; Ueyama N.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2005, 127, 12046. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chiou S. J.; Innocent J.; Riordan C. G.; Lam K. C.; Liable-Sands L.; Rheingold A. L. Inorg. Chem. 2000, 39, 4347. [DOI] [PubMed] [Google Scholar]
- a Lacy D. C.; Mukherjee J.; Lucas R. L.; Day V. W.; Borovik A. S. Polyhedron 2013, 52, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gupta R.; Lacy D. C.; Bominaar E. L.; Borovik A. S.; Hendrich M. P. J. Am. Chem. Soc. 2012, 134, 9775. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lacy D. C.; Gupta R.; Stone K. L.; Greaves J.; Ziller J. W.; Hendrich M. P.; Borovik A. S. J. Am. Chem. Soc. 2010, 132, 12188. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sen Soo H.; Komor A. C.; Iavarone A. T.; Chang C. J. Inorg. Chem. 2009, 48, 10024. [DOI] [PubMed] [Google Scholar]; e Wada A.; Harata M.; Hasegawa K.; Jitsukawa K.; Masuda H.; Mukai M.; Kitagawa T.; Einaga H. Angew. Chem., Int. Ed. 1998, 37, 798. [DOI] [PubMed] [Google Scholar]; f Kim S.; Saracini C.; Siegler M. A.; Drichko N.; Karlin K. D. Inorg. Chem. 2012, 51, 12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McDonald A. R.; Bukowski M. R.; Farquhar E. R.; Jackson T. A.; Koehntop K. D.; Seo M. S.; De Hont R. F.; Stubna A.; Halfen J. A.; Münck E.; Nam W.; Que L. Jr. J. Am. Chem. Soc. 2010, 132, 17118. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Annaraj J.; Kim S.; Seo M. S.; Lee Y. M.; Kim Y.; Kim S. J.; Choi Y. S.; Jang H. G.; Nam W. Inorg. Chim. Acta 2009, 362, 1031. [Google Scholar]; c Bukowski M. R.; Koehntop K. D.; Stubna A.; Bominaar E. L.; Halfen J. A.; Münck E.; Nam W.; Que L. Jr. Science 2005, 310, 1000. [DOI] [PubMed] [Google Scholar]
- a Sickerman N. S.; Park Y. J.; Ng G. K. Y.; Bates J. E.; Hilkert M.; Ziller J. W.; Furche F.; Borovik A. S. Dalton Trans. 2012, 41, 4358. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Aullon G.; Bellamy D.; Brammer L.; Bruton E. A.; Orpen A. G. Chem. Commun. 1998, 653. [Google Scholar]
- a Kumar D.; Sastry G. N.; Goldberg D. P.; de Visser S. P. J. Phys. Chem. A. 2012, 116, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kumar D.; Thiel W.; de Visser S. P. J. Am. Chem. Soc. 2011, 133, 3869. [DOI] [PubMed] [Google Scholar]
- a Park J.; Morimoto Y.; Lee Y. M.; Nam W.; Fukuzumi S. J. Am. Chem. Soc. 2012, 134, 3903. [DOI] [PubMed] [Google Scholar]; b Park M. J.; Lee J.; Suh Y.; Kim J.; Nam W. J. Am. Chem. Soc. 2006, 128, 2630. [DOI] [PubMed] [Google Scholar]; c Seo M. S.; Jang H. G.; Kim J.; Nam W. Bull. Korean Chem. Soc. 2005, 26, 971. [Google Scholar]
- Sastri C. V.; Lee J.; Oh K.; Lee Y. J.; Lee J.; Jackson T. A.; Ray K.; Hirao H.; Shin W.; Halfen J. A.; Kim J.; Que L. Jr.; Shaik S.; Nam W. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 19181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifi R.; Sainna M. A.; Rybak-Akimova E. V.; de Visser S. P. Chem.—Eur. J. 2013, 19, 4058. [DOI] [PubMed] [Google Scholar]
- a Wilson S. A.; Chen J.; Hong S.; Lee Y. M.; Clémancey M.; Garcia-Serres R.; Nomura T.; Ogura T.; Latour J. M.; Hedman B.; Hodgson K. O.; Nam W.; Solomon E. I. J. Am. Chem. Soc. 2012, 134, 11791. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McDonald A. R.; Guo Y. S.; Vu V.; Bominaar E. L.; Münck E.; Que L. Jr. Chem. Sci. 2012, 3, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.