

Abstract
Oligomers of the amyloid-β (Aβ) peptide have been implicated in the neurotoxicity associated with Alzheimer’s disease. We have used single-molecule techniques to examine quantitatively the cellular effects of adding well characterized Aβ oligomers to primary hippocampal cells and hence determine the initial pathway of damage. We found that even picomolar concentrations of Aβ (1–40) and Aβ (1–42) oligomers can, within minutes of addition, increase the levels of intracellular calcium in astrocytes but not in neurons, and this effect is saturated at a concentration of about 10 nM of oligomers. Both Aβ (1–40) and Aβ (1–42) oligomers have comparable effects. The rise in intracellular calcium is followed by an increase in the rate of ROS production by NADPH oxidase in both neurons and astrocytes. The increase in ROS production then triggers caspase-3 activation resulting in the inhibition of long-term potentiation. Our quantitative approach also reveals that only a small fraction of the oligomers are damaging and that an individual rare oligomer binding to an astrocyte can initiate the aforementioned cascade of responses, making it unlikely to be due to any specific interaction. Preincubating the Aβ oligomers with an extracellular chaperone, clusterin, sequesters the oligomers in long-lived complexes and inhibits all of the physiological damage, even at a ratio of 100:1, total Aβ to clusterin. To explain how Aβ oligomers are so damaging but that it takes decades to develop Alzheimer’s disease, we suggest a model for disease progression where small amounts of neuronal damage from individual unsequestered oligomers can accumulate over time leading to widespread tissue-level dysfunction.
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the loss of brain tissue and impaired cognitive function.1 One of the pathological hallmarks of AD is the aggregation of amyloid-β (Aβ) peptides into β-sheet rich fibrils and plaques.2 It is of vital importance for the development of therapeutic strategies for AD to determine the key factors that lead to the initial development of the disease and also those factors that inhibit its progression. Although plaques containing Aβ fibrils have been viewed as the conventional hallmark of AD, recent research has implicated small oligomeric species formed during the aggregation of Aβ in the neuronal toxicity and cognitive deficits associated with diseases such as AD.2−5 A large number of studies has been performed in which mixtures of Aβ monomers and oligomers have been added to cells and brain slices and the consequent physiological changes monitored;6,7 the results of these studies have identified oligomers of Aβ, not the monomers, to be the most damaging species. In the majority of such experiments, however, only the total monomer concentration is known accurately, and in several cases, it is higher than the endogenous concentrations of Aβ monomers in CSF, typically 1–10 nM.8 Therefore, the important questions concerning the specific mechanisms by which oligomers initially damage cells and the concentration of oligomers that is required to elicit such damage remain to be answered.
Our approach to addressing these questions is to determine directly the monomer and oligomer concentrations and indeed the complete distribution of oligomer sizes of preparations of Aβ40 and Aβ42, using single molecule fluorescence techniques, immediately prior to applying these preparations to primary neuronal cultures and brain slices. All of our measurements were completed 10–30 min following the addition of Aβ to the cellular cultures. This experimental procedure is designed to determine the earliest detectable physiological response at the lowest possible concentration of oligomers and how this response varies with oligomer concentration, and hence to identify the initial events that lead to cellular damage. A defining objective of the present study is to conduct a highly quantitative analysis of the concentration and time dependence of the physiological effects of Aβ40 and Aβ42 oligomers on primary neuroglial cultures, ensuring that the Aβ oligomer concentration is defined in every experiment. In order to achieve this objective, we have monitored a variety of key indicators of cellular health and homeostasis, notably intracellular calcium levels, the release of reactive oxygen species, and the activation of caspase-3. All of these metrics have been previously suggested to be affected by exposure to Aβ species in previous studies using higher Aβ concentrations.9−15
Experimental Procedures
Aβ40 and Aβ42 Peptide Preparation and Characterization
Monomeric solutions of HiLyteFluor488- and HiLyteFluor647-labeled Aβ40 or Aβ42 (Anaspec, San Jose, CA) were prepared by dissolving the lyophilized peptides in SSPE buffer (150 mM NaCl, 10 mM Na2H2PO4 × H2O, and 10 mM Na2EDTA, adjusted to pH 12 using NaOH), followed by sonication over ice for 30 min (Bandelin Sonorex, Berlin, Germany), and subsequently flash freezing into 5 μL aliquots.16 Prior to each of the incubations, aliquots of each peptide were diluted into SSPE buffer (pH adjusted to 7.4 using HCl) to the desired concentration and placed under conditions for aggregation (e.g., 37 °C, agitation). The concentration of each labeled peptide was measured before mixing the two different fluorophore-labeled samples using cTCCD as previously described.16
For each experiment, Aβ monomers (Aβ40 at 20 μM and Aβ42 at 10 μM) were incubated in SSPE buffer (defined above) at 37 °C with agitation (200 rpm on a rotary shaker). After 1 h of aggregation, the samples were placed at 4 °C and used within 10 h of preparation. Monomeric solutions were kept frozen at −80 °C until use. For experiments with clusterin, the chaperone was added at a 1:1 molar ratio to Aβ (unless otherwise stated) and incubated for 30 min at 25 °C.
For each preparation of Aβ40 or Aβ42, the number and size distributions of oligomers were determined using the single molecule cTCCD method. The instrumentation and methodology required for this characterization have been described in detail previously.16
Preparation and Labeling of Human Clusterin
Clusterin was extracted from human serum from Wollongong Hospital (Wollongong, NSW, Australia), as described previously.17 Labeling of clusterin was carried using lysine conjugation of succinimidyl ester-functionalized AlexaFluor647 (Molecular Probes, Grand Island, NY) using previously described protocols.16
Cell Cultures
Mixed cultures of neurons and glial cells were prepared as described previously with modifications, from Sprague–Dawley rat pups 2–4 days postpartum (UCL breeding colony).18 Experimental procedures were performed in full compliance with the United Kingdom Animal (Scientific Procedures) Act of 1986. The hippocampus and cortex were removed and placed in ice-cold PBS (Ca2+ and Mg2+-free, Invitrogen, Paisley, UK). The tissue was then minced and trypsinized (0.25% for 5 min at 37 °C), triturated, and plated on poly-d-lysine-coated coverslips, and cultured in Neurobasal A medium (Invitrogen, Paisley, UK) supplemented with B-27 (Invitrogen) and 2 mM l-glutamine. Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air, and the medium was in each case replaced twice a week and maintained for 12–15 days before experimental use to ensure the expression of glutamate and other receptors. Neurons were easily distinguishable from glia using microscopy: they appeared phase bright, had smooth rounded somata and distinct processes, and lay just above the focal plane of the glial layer.
Measurements of [Ca2+]c and ROS
For measurements of [Ca2+]c, cells were loaded for 30 min at room temperature with 5 μM fura-2 AM and 0.005% pluronic acid in a HEPES-buffered salt solution (HBSS) containing 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES; the pH of each solution was adjusted to 7.35 with NaOH, and the fluorescence of 488 nM-excitable fura-2 was measured as a function of time.
For measurement of ROS production, dihydroethidium (2 μM HEt) was added into the solutions during the experiments. No preincubation (loading) was used for HEt to limit the intracellular accumulation of oxidized products. Measurements monitored the ratio of two fluorescent wavelengths, representing the oxidized and nonoxidized form, as a function of time (see Microscopy).10,19,20
Large fields containing 100–200 cells were imaged at a given time. These images were taken alternating between phase-contrast and fluorescence images for the duration of the experiment (∼10 −30 min).
Caspase-3 Activation Assay
For measurements of caspase-3 activation, cells were loaded for 15 min at room temperature with 10 μM NucView 488 caspase-3 substrate (Biotium, Hayward, CA) in HBSS. NucView 488 is a member of a novel class of enzyme substrates for real-time detection of caspase-3 activity in live cells. The substrate can rapidly cross cell membranes to enter the cytoplasm, where it is cleaved by caspase-3 to release the high-affinity DNA dye, which fluoresces when excited by a 488 nm laser. The released DNA dye migrates to the cell nucleus. Therefore, cells having undergone caspase-3 activation are distinguishable from all other cells by visualizing their bright-green nuclei. The NucView 488 substrate was used according to the manufacturer’s specifications.
Microscopy
All microscopy protocols used for monitoring intracellular Ca2+ and ROS have been described previously.10,19 Fluorescence measurements were obtained on an epi-fluorescence inverted microscope equipped with a 20× (0.5 NA) fluorite objective. [Ca2+]c was monitored in single cells using excitation light provided by a xenon arc lamp, the beam passing through a monochromator centered sequentially at 340 and 380 nm (Cairn Research, Kent, UK). The emitted fluorescence was reflected through a 515 nm long-pass filter to a cooled CCD camera (Retiga, QImaging, Canada). All imaging data for Ca2+ and ROS experiments were collected every 15–30 s and analyzed using software from Andor (Belfast, UK). Each coverslip on which cells were cultured was placed in an AttoFluor Cell Culture chamber (Molecular Probes, Grand Island, NY) in 300 μL of Hank’s Balanced Salt Solution (HBSS). Solutions containing Aβ and clusterin were added for specific experiments to the HBSS surrounding the cells. The fura-2 data were not calibrated in terms of [Ca2+]c because of the difficulty in deconvoluting the strength of the signal from the amount of dye taken up by each cell. The fura-2 ratio is defined as the ratio of fluorescence emission upon excitation at 340 nm (Ca2+-bound form) to emission upon excitation at 380 nm (unbound fluorophore). For HEt measurements, a 543 nm excitation and 560 nm long pass filter were used to quantify oxidized HEt and excitation at 355 nm, and measurement at 405–470 nm was used to detect nonoxidized HEt. The HEt ratio is defined as the ratio of fluorescence emission upon excitation at 543 nm to emission upon excitation at 355 nm. All of the data shown were obtained from at least 5 coverslips and 2–3 different cell preparations. We measured between 100 and 200 cells per coverslip.
Confocal images were obtained using a Zeiss (Oberkochen, Germany) 710 confocal laser scanning microscope and a 40× oil immersion objective. A 488 nm argon laser was used to excite NucView 488, and the resulting fluorescence was measured using a bandpass filter from 510 and 560 nm. Images were acquired at 10 frames s–1 for 30 min.
Electrophysiology
All the protocols used are as described previously.21 Acute hippocampal slices were prepared from 26 to 32 day-old male Wistar rats. Experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act of 1986. Animals were sacrificed by dislocation of the neck followed by decapitation. The brains were rapidly removed and placed in ice-cold artificial CSF (aCSF) containing (in mM) 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, and 10 d-glucose (bubbled with 95% O2/5% CO2). Transverse hippocampal slices (400 μm thick) were prepared using a McIllwain tissue chopper (Mickle Laboratory Engineering, Gomshall, UK). Hippocampal slices were stored in aCSF (between 20–25 °C) for 1–2 h before transferring to the recording chamber, in which they were submerged in aCSF (30 °C) flowing at 2 mL/min. Extracellular field potentials were recorded in the CA1 region of the hippocampus using glass electrodes containing 3 M NaCl. Stimulating electrodes were placed in the Subiculum and CA2 (Schaffer Collateral pathway) of the hippocampus. Stimuli (constant voltage) were delivered alternately to the two electrodes (each electrode at 0.016 Hz). LTP was induced by two trains of tetanic stimuli (each 100 Hz, 1 s; repeated after a 30 s interval). The slopes of the evoked field potential responses were measured and expressed relative to the normalized preconditioning baseline.
Statistical Methods
All statistical analyses were performed using both Origin 8 (OriginLab) and Prism 6.00 (GraphPad, La Jolla, CA). Nonparametric tests were performed to avoid assumptions of normality.
Results
In Vitro Characterization of Aβ40 and Aβ42 Oligomers
We used a single-molecule fluorescence method, confocal two-color coincidence detection (cTCCD),16,22 to characterize the quantities of oligomers and monomers of both Aβ40 and of Aβ42 in our samples. Aβ42 has a higher aggregation propensity and is the primary isoform found in AD-associated amyloid plaques. Although Aβ40 is less-aggregation prone than Aβ42, it is the more abundant isoform in cerebrospinal fluid and has been found to form oligomers with similarly cytotoxic properties to those of Aβ42.16,23,24 The results of these experiments indicate that 3.9 ± 0.4% of the Aβ42 molecules were present as oligomers in the preparations used for addition to cells, corresponding to a concentration of ∼20 nM oligomers in a 500 nM solution of Aβ42 monomers. Aβ40, with its reduced propensity to aggregate relative to Aβ42, was found to have a maximum of 0.3 ± 0.1% of the molecules in a 500 nM solution of monomers as oligomeric species, corresponding to a 1.5 nM concentration of oligomers in a solution of 500 nM Aβ40 monomers. Dilutions of these preparations to 5–50 nM concentrations of monomers resulted in solutions containing as little as 20 pM of oligomers. Oligomers of both Aβ isoforms in their respective preparations varied in size from dimers to 50-mers (Figure 1A,B). Since the oligomers are stable on dilution over at least 3 h,16 the distribution of oligomer sizes did not change over the course of our experiments. The concentrations of oligomers at different total peptide concentrations used in this study are given in Table 1.
Figure 1.

Characterization of Aβ40 and Aβ42 oligomers using single-molecule cTCCD. (A) The extent of fibril formation as a function of time for Aβ42 (10 μM) measured by tracking thioflavin-T fluorescence as a function of time (plotted as mean ± SD, n = 5). The right panel is zoomed in on the first 2 h of aggregation. The gray shaded area indicates the time during the aggregation reaction from which the oligomeric samples were obtained. Aβ40 oligomer-containing samples were obtained at a similar extent of reaction. (B) The distribution of sizes of the oligomers present in samples of Aβ40 and Aβ42 acquired at the time indicated in panel A, (n = 2, error bars are the range).
Table 1. Concentrations of Oligomers Present in Solutions of Aβ40 and Aβ42a.
| total peptide concentration | concn of Aβ40 oligomers | concn of Aβ42 oligomers |
|---|---|---|
| 1 μM | 15 ± 5 nM | 195 ± 20 nM |
| 500 nM | 1.5 ± 0.5 nM | 19.5 ± 2 nM |
| 50 nM | 150 ± 50 pM | 1.95 ± 0.2 nM |
| 5 nM | 15 ± 5 pM | 195 ± 20 pM |
These values were obtained by multiplying the percentage of oligomers in solution (as determined by cTCCD) by the total peptide concentration (also determined by cTCCD). Values are expressed as the mean ± SEM of at least three independent trials.
Induction of Calcium Transients in Astrocytes by Aβ Oligomers
We next examined the effects of these well-characterized, fluorescently labeled Aβ oligomers on the cytosolic calcium concentrations ([Ca2+]c) of rat primary hippocampal neuroglial cultures, using the fluorescent intracellular Ca2+-sensitive dye fura-2 and imaging a large field of cells over 30 min. We alternated between brightfield exposure and fluorescent imaging to obtain a time dependence of the Ca2+ transients. The medium in which the cells were incubated was exchanged within 5 min of the start of the experiments to eliminate effects of any proteins endogenously secreted from cells. Oligomeric preparations of Aβ42 and Aβ40 were found to induce rapidly an increase in [Ca2+]c in astrocytes) giving rise to unsynchronized oscillations (Figure 2A and Figure S1A,B (Supporting Information) though not in neurons (Figure 2B). The elevation of [Ca2+]c specific to astrocytes but not neurons was observed even at very low concentrations, down to 195 pM oligomers, of the peptide (Aβ42, Figure 2C,E and Aβ40, Figure S1C (Supporting Information)). No detectable increase in [Ca2+]c was, however, observed even at the highest concentration examined with solutions containing just monomers of Aβ40 and Aβ42 (Aβ42, Figure 2D; Aβ40, Figure S1D (Supporting Information)). In each of these cases, we ensured that the effects measured were not due to the presence of the fluorescent dye since incubating the cultures with the fluorophore alone elicited no increase in [Ca2+]c (Figure S2A, Supporting Information). Furthermore, incubation of the cultures with unlabeled versions of Aβ42 oligomers resulted in changes in intracellular calcium levels similar to those observed in experiments with the fluorescently labeled oligomers (Figure S2B, Supporting Information). These controls confirmed that the effects we observed were specific to the experiments we performed rather than effects due to the conditions of our experiments.
Figure 2.

Effects of Aβ oligomers on cytosolic calcium levels. The ratio of fluorescence emitted from excitation at 340/380 nm of the Ca2+-binding dye, fura-2, (defined to be the fura-2 ratio) correlates with the cytosolic Ca2+ concentration. For all plots displaying the change in fura-2 ratio with time, each line represents the cytosolic Ca2+ concentration within a single cell. Each experiment was performed with at least three replicates. Since the analysis was performed at the single cell level, the number of cells examined is stated. (A) Cytosolic Ca2+ concentration (as quantified by the fura-2 ratio) as a function of time in astrocytes to which 500 nM (total peptide) of Aβ42 (containing 19.5 nM oligomer) has been added. Results for experiments performed with Aβ42 are shown here. The number of astrocytes incubated with Aβ42 and examined was 549. Similar results for cells incubated with Aβ40 peptide preparations containing ∼1.5 nM oligomers can be found in Figure S1B (Supporting Information). (B) Cytosolic Ca2+ concentration (as quantified by the fura-2 ratio) as a function of time in neurons to which 500 nM (total peptide) Aβ42 containing 19.5 nM oligomers) has been added; 126 neurons were examined. (C) Cytosolic Ca2+ concentration (as quantified by the fura-2 ratio) as a function of time in astrocytes to which 50 nM (total peptide) Aβ42 (containing 1.95 nM oligomers) has been added. The number of astrocytes examined was 414. Similar results for cells incubated with Aβ40 peptide preparations containing 150 pM oligomers are shown in Figure S1C (Supporting Information). (D) Cytosolic Ca2+ concentration (as quantified by the fura-2 ratio) as a function of time in astrocytes to which 500 nM monomeric Aβ42 was added. Similar results for cells incubated with 500 nM monomeric Aβ40 peptide are shown in Figure S1D (Supporting Information). The number of astrocytes incubated with Aβ42 that were examined was 69. (E) The fraction of total astrocytes that displayed Ca2+ transients within the first 15 min following the addition of exogenously added Aβ40 or Aβ42, as a function of Aβ oligomer concentration. Each point represents the mean ± SEM of at least three biological replicates. For each concentration, from 81 to 550 cells were examined. Control experiments to verify that the observed effects on Ca2+ transients are not a consequence of fluorophores attached to the Aβ peptides are shown in Figure S2A,B (Supporting Information).
No significant difference in the average amplitude of the initial [Ca2+]c transients was observed upon incubation of the primary cultures with a 10-fold variation in concentration of either Aβ40 or Aβ42 (19.5 nM oligomer, 500 nM total Aβ42 induced a 0.67 ± 0.06 increase in the fura-2 fluorescence intensity ratio, compared to 0.71 ± 0.09 at 1.95 nM oligomer, and 0.62 ± 0.7 at 19.5 pM oligomer; similar data for the Aβ40 peptide can be found in Figure S1B,C (Supporting Information)). In contrast, the number of astrocytes that displayed a change in [Ca2+]c within 15 min increased with increasing concentration of oligomers added to the primary cultures (for both Aβ40 and Aβ42, Figure 2E). Even at low concentrations of oligomers, ∼195 pM, 16 ± 3% of astrocytes displayed [Ca2+]c transients. Increasing the concentration did not therefore increase the magnitude of the calcium response of a given astrocyte but rather increased the number of astrocytes affected.
We then performed a set of experiments designed to reveal the origin of the [Ca2+]c transients in astrocytes. When Aβ40 and Aβ42 oligomers were added to cells in a Ca2+-free medium (which included 0.5 mM of the chelating agent, EGTA), no [Ca2+]c transients were observed (Figure S1E, Supporting Information); when Ca2+ was added to the medium (at concentrations excess of EGTA), the [Ca2+]c transients could be reactivated (Figure S1F, Supporting Information). In addition, pretreatment of cells with thapsigargin (a sarcoplasmic/endoplasmic reticulum Ca2+-ATPase blocker that leads to a depletion of intracellular Ca2+) followed by subsequent extracellular addition of either Aβ40 or Aβ42 oligomers in a Ca2+-containing medium, resulted in the observation of [Ca2+]c transients (Figure S1F, Supporting Information). These results were obtained from cultures where the neuron/astrocyte ratio was consistently between 1:1 and 1:2 depending on the area and preparation used. Taken together, these experiments show that the [Ca2+]c transients in astrocytes are caused by the entry of Ca2+ from the extracellular space rather than by the release from intracellular stores.
Since we were able to determine the concentration of oligomers in each of our experiments, we can explore the dependence of the incidence of [Ca2+]c transients on the oligomer dose. As previously mentioned, we found that the number of cells displaying Ca2+ transients (rather than the magnitude of the transients themselves) correlated with the concentration of Aβ oligomers present. Therefore, we incubated cells with varying concentrations of either Aβ42 or Aβ40 oligomers and counted the number of cells that displayed Ca2+ influx within 15 min of addition of the oligomers. The regime where only a fraction of cells show calcium influx in response to the addition of oligomers can be used to roughly estimate what fraction of all oligomers can cause this influx, while at higher oligomer concentrations, all of the cells show calcium influx, and the effect saturates. For this estimation, we fitted these data in Figure 2E using the following dose–response relationship:25
In this relationship, I is the fraction of astrocytes showing cytosolic Ca2+ influx, d is the number of oligomers per cell, p is the probability that an oligomer causes Ca2+ influx, and k is a parameter determining the degree to which oligomers interact independently (k = 1), or act synergistically (k > 1) or antagonistically (k < 1). From the fit of the data (Figure S1G, Supporting Information), we extracted a k value of 0.5 ± 0.1 and a p value of 0.002 ± 003 (quoted as the mean of fit parameter ± standard error). The k value of 0.5 suggests a competitive interaction of oligomers for sites on the cell membrane. The p value of 0.002 suggests that approximately 1 in 500 oligomers is responsible for triggering the observed influx of Ca2+. While this fitting is approximate, it does clearly show that only a small fraction of the oligomers present initiate cellular damage.
Generation of Reactive Oxygen Species Induced by Aβ Oligomers
We next examined the effects of Aβ oligomers on the generation of reactive oxygen species (ROS) because oxidative stress, induced by exposure to cytotoxic oligomers, has been observed to profoundly affect neuronal health and survival in the context of AD.20,26 The application of solutions containing oligomers of Aβ42 or Aβ40 resulted in a significant increase in the rate of ROS production in astrocytes (by a factor of 3.5 ± 0.3 for Aβ42 and a factor of 2.0 ± 0.05 for Aβ40, Figure 3A,H). As the monomeric peptides had no significant effect on the rate of ROS production (the ratio of rates was 1.1 ± 0.1 for monomeric Aβ42 and 0.97 ± 0.02 for monomeric Aβ40), we can attribute the increase in ROS generation specifically to oligomers of Aβ40 and Aβ42 (Figure 3A,H). This control also verified that the media exchange just prior to the experiments had no detectable effect on cell physiology.
Figure 3.
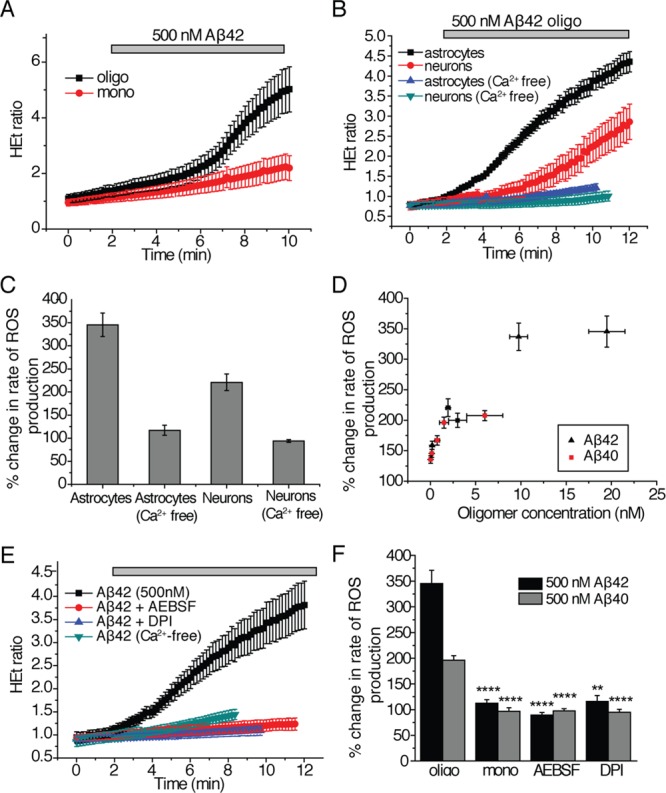
Effects of Aβ species on ROS production. The ratio of fluorescence emitted from excitation at 543/355 nm of the fluorescent dye dihydroethidium (HEt) that is sensitive to oxidation correlates with the amount of cytosolic reactive oxygen species (ROS) present in the cell. The HEt ratio is defined as the ratio of fluorescence emission upon excitation at 543 nm to emission upon excitation at 355 nm. All plots are displayed as the mean ± SEM of all cells measured in at least three independent trials. (A) The HEt ratio as a function of time upon the addition of Aβ42 (500 nM total peptide) either containing only monomers (mono) or 19.5 nM oligomers (oligo). Similar data for Aβ40 is summarized in (H). The number of cells examined for experiments involving oligomers of Aβ42 was 163 and 181 for those involving oligomers of Aβ40; 201 cells were examined in experiments involving monomeric Aβ42 and 156 for Aβ40. (B) The HEt ratio as a function of time upon the addition of Aβ42 (500 nM total peptide) containing 19.5 nM oligomers to astrocytes and neurons in mixed primary cultures, where the data are separated into ROS production in astrocytes and neurons. The experiment was conducted in media with and without Ca2+. For experiments in Ca2+-containing media, 256 astrocytes and 131 neurons were examined. For experiments in Ca2+-free media, 106 astrocytes and 72 neurons were examined. (C) The change in rate of ROS production (%) in astrocytes and neurons in media with and without Ca2+. Data from B are replotted here for clarity. (D) The change in rate of ROS production (%) in astrocytes and neurons as a function of oligomer concentration. Data for experiments with both Aβ42 and Aβ40 are shown here as the mean ± SEM of at least three trials per concentration. (E) The HEt ratio as a function of time upon the addition of Aβ42 (500 nM total peptide), containing 19.5 nM oligomers, to astrocytes and neurons in mixed primary cultures treated with NADPH-oxidase inhibitors, AEBSF (20 μM), or DPI (0.5 μM). The results of experiments without inhibitors and in Ca2+-free medium are shown for comparison; similar data for Aβ40 are summarized in H. In experiments with DPI, 134 cells were examined with Aβ42 and 62 with Aβ40. In experiments with AEBSF, 164 cells were examined with Aβ42 and 121 cells with Aβ40. (F) The percent change in the rate of ROS production in astrocytes and neurons in mixed cultures upon the addition of either 500 nM Aβ42 containing 19.5 nM oligomers (termed “oligo”) or 500 nM Aβ40 containing 1.5 nM oligomers (“oligo”) or 500 nM monomeric solutions (“mono”) in the presence of NADPH oxidase inhibitors (AEBSF or DPI). The data here are replotted from parts A and E. ** represents a p-value <0.01, and **** represents a p-value <0.0001 using a Kruskal–Wallis test followed by Dunn’s post-test. Comparisons were performed with the “oligo” sample of the same Aβ isoform. Control experiments to verify that the observed effects on ROS production are not affected by the fluorophores attached to the Aβ peptides are shown in Figure S2C,D (Supporting Information).
Importantly, the rate of ROS production also increased in neurons upon the addition oligomers of Aβ42 (by a factor of 2.3 ± 0.4). This increase, however, occurred after a longer length of time following the initial addition of the oligomers than was observed for the corresponding increase in astrocytes (Figure 3B,C). In astrocytes, where large increases in ROS production were observed upon the addition of Aβ oligomers, the quantity of oligomers (whether of Aβ42 or of Aβ40) correlated directly with the increase in the rate of ROS production (Figure 3D) suggesting that the difference in toxicity between solutions of Aβ42 and Aβ40 is directly related to their oligomer content rather than to inherent differences in their amino acid sequences. We also observed that the increased rate of ROS production in astrocytes instigated by Aβ oligomers does not occur without calcium in the extracellular medium. This finding suggests a direct relationship between the calcium transients observed in astrocytes and the increased ROS production (Figure 3B,C). Although no calcium transients were observed in neurons, ROS production was still inhibited in neurons in Ca2+-free medium (Figure 3B,C) indicating that the neuronal ROS production in these hippocampal samples occurs as a result of calcium entry into astrocytes. Previous work has suggested that these results do not depend on the ratio of astrocytes to neurons in the preparation.10,11
In order to probe the mechanism of ROS production, we examined the effects of a range of small molecule compounds and found that incubating the hippocampal cultures for 20 min with NADPH oxidase inhibitors (0.5 μM diphenyleneiodonium chloride or DPI, or 20 μM aminoethyl-benzenesulfonylfluoride or AEBSF) prior to exposing the cultures to solutions of Aβ40 or Aβ42 oligomers inhibited the increase in rate of ROS production (AEBSF, 0.89 ± 0.02-fold change for Aβ42 and 0.97 ± 0.03-fold change for Aβ40; DPI, 1.2 ± 0.02-fold change for Aβ42 and 0.94 ± 0.01-fold change for Aβ40; Figure 3E,H). The effects of these inhibitors suggest that the Aβ40 and Aβ42 induce ROS production in astrocytes through activation of NADPH oxidase.
Aβ Oligomer-Induced Activation of Caspase-3 in Both Astrocytes and Neurons
Next, we examined the effects of Aβ42 on the activation of caspase-3. Caspase 3 activation has been associated not only with apoptosis but also with nonapoptotic pathways of synaptic pruning and LTP deficits as shown in recent work on rodent models and primary culture models of Alzheimer’s disease biology.21,27 The extracellular application of 500 nM Aβ42 (19.5 nM oligomers) induced activation of caspase-3 in 78 ± 9.7% of both neurons and astrocytes within 30 min of observation (Figure 4A,C). Strikingly, even small quantities of oligomeric Aβ42 (50 nM total peptide concentration and 1.95 nM oligomers) activated caspase-3 in 36 ± 2.7% of the cells (Figure 4C). We verified that the activation of caspase-3 by Aβ42 was specific to oligomers since monomeric Aβ42 (500 nM) only activated 4.1 ± 1.2% of cells during the time of observation (Figure 4B,C).
Figure 4.

Effects of Aβ oligomers on the activation of caspase-3. The NucView488 substrate used to monitor the activation of caspase-3 in real time becomes fluorescent when cleaved by caspase-3 and subsequently migrates to the nucleus. The kinetics of caspase-3 activation were, therefore, monitored by measuring the fluorescence within each cell as a function of time. In the kinetic traces shown here, each line represents a single cell with data for neurons in red and for astrocytes in black. Additionally, end point measurements of net caspase-3 activation after 30 min of observation were taken by counting the number of cells observed with fluorescent nuclei as a fraction of total cells observed. The experiments were performed on at least 4 different samples. (A) Fluorescence of the NucView488 caspase-3 substrate as a function of time following treatment of primary cultures containing both neurons and astrocytes with 500 nM Aβ42 (containing 19.5 nM oligomers); 370 cells were monitored over 6 samples. (B) Fluorescence of the NucView488 caspase-3 substrate as a function of time following the treatment of primary cultures containing both neurons and astrocytes with 500 nM of monomeric Aβ42; 187 cells were monitored over 4 samples. (C) Proportion of cells (%) that experience activation of caspase-3 following 30 min of incubation with monomeric Aβ42 (500 nM) or oligomeric Aβ42 solutions (total peptide concentrations of 50 nM and 500 nM with oligomer concentrations of 1.95 nM and 19.5 nM). These data are the end points (after 30 min) of experiments presented in A and B. For experiments using 50 nM of Aβ42, 231 cells were monitored over 4 different samples. ** signifies a p-value <0.01 by a Mann–Whitney nonparametric rank correlation. Comparisons were performed relative to the data obtained with 500 nM of Aβ42 oligomers. (D) Fluorescence of the NucView488 caspase-3 substrate as a function of time following the treatment of primary hippocampal cultures containing both neurons and astrocytes with 500 nM Aβ42 (containing 19.5 nM oligomers). Cultures in this experiment had been pretreated with the NADPH-oxidase inhibitor, AEBSF (20 μM), for 30 min prior to the addition of Aβ42; 154 cells were monitored in a single sample. (E) Fluorescence of the NucView488 caspase-3 substrate as a function of time following the treatment of primary hippocampal cultures containing both neurons and astrocytes with 500 nM Aβ42 (containing 19.5 nM oligomers). The cultures in this experiment had been pretreated with the NADPH-oxidase inhibitor, DPI (0.5 μM), for 30 min prior to the addition of Aβ42; 138 cells were monitored on a single sample. Control experiments to verify that the observed effects on caspase-3 activation are not affected by the fluorophores attached to the Aβ peptides are reported in Figure S2E,F, Supporting Information.
Aβ42-Induced Inhibition of Long-Term Potentiation in Hippocampal Slices
We then explored the relationship between these changes in cellular pathology and the inhibition of long-term potentiation (LTP) by Aβ42 oligomers.7,21 Using systematic electrical stimulation patterns on hippocampal brain slices, we found in accord with the results of cellular experiments that a concentration of 500 nM (total peptide concentration) of Aβ42 (containing 19.5 nM oligomers) inhibits LTP in hippocampal slices (Figure 5A,B). In order to investigate whether or not the NADPH-oxidase inhibitors prevent the downstream detrimental effects caused by Aβ42 oligomers, we incubated the hippocampal slices with the NADPH oxidase inhibitor, AEBSF, which, as we have described above, inhibited an increase in ROS production in our cellular studies. When we measured the LTP levels following the addition of Aβ42 to the hippocampal slices, we found that the presence of AEBSF prevented the Aβ-induced inhibition of LTP (Figure 5C) as has been reported previously for other ROS inhibitors.28 There was no detectable cell death over the duration of this experiment.
Figure 5.

Aβ oligomers induce the inhibition of LTP at the CA1-Shaffer collateral synapse in the hippocampus. The CA1 regions of hippocampal slices from rats were stimulated in the Schaffer-collateral pathway (test input) of the hippocampus, while another region, the subiculum, served as an internal control (control input). The summation of the evoked excitatory field potentials (fEPSP) was quantified by measuring the peak amplitude and the rate of voltage change (slope = mV/ms) before and after high frequency electrical pulse stimulation (HFS). The slopes of the fEPSP measurements are greater upon the induction of long-term potentiation of synaptic efficacy (LTP). HFS (100 Hz) was applied twice for 1 s each with a 30 s gap in between. After this stimulation, LTP is visible in the test input as an increased fEPSP slope. This value is normalized to the internal control input. All values are plotted as the means ± SEM. (A) The fEPSP slope as a function of time following HFS in untreated slices showing the induction of LTP. Data were recorded for 8 slices. (B) The fEPSP slope as a function of time in hippocampal slices incubated prior to the experiment for 2 h with 500 nM Aβ42 (containing 19.5 nM oligomers). No induction of LTP is observed after HFS. Data were recorded for 7 slices. (C) The fEPSP slope as a function of time in hippocampal slices incubated prior to the experiment for 30 min with the NADPH oxidase inhibitor, AEBSF (20 μM), followed by a 2 h incubation with 500 nM Aβ42 (containing 19.5 nM oligomers). Induction of LTP is observed after HFS. Data were recorded for 7 slices.
Clusterin Prevents Damage Induced by Aβ Oligomers
Clusterin is an extracellular chaperone that has been genetically associated with AD at the level of genome-wide significance29,30 and that is present in human cerebrospinal fluid (CSF) at endogenous concentrations similar to those of the Aβ peptides.8,31 In previous work, we have shown that, in vitro, clusterin suppresses Aβ40 aggregation and binds selectively to Aβ40 oligomers,16 and we found that a similar phenomenon occurs for the Aβ42 peptide (Figure S3A, Supporting Information). Clusterin prevented calcium influx, ROS production, and caspase 3 activation (Figure 6A–C). At substoichiometric levels relative to the total Aβ42 monomer, clusterin protected both neurons and astrocytes from Aβ-oligomer-induced ROS supporting the conclusion that clusterin forms complexes, specifically with the oligomers, to inhibit their toxicity (Figure 6B) and revealing how much clusterin is needed for this inhibition. Clusterin could also prevent the Aβ42-induced inhibition of LTP; when we exposed the hippocampal slices to Aβ42 oligomers that had been preincubated with clusterin, we found that the latter, like AEBSF, prevented the Aβ42-induced inhibition of LTP (Figure 6D). This finding directly relates our in vitro level experiments with the tissue-level processes of LTP. It also provides strong evidence that oligomers are the key inducers of synaptic dysfunction and that the sequestration of these cytotoxic species can effectively prevent their damaging effects.
Figure 6.

Clusterin inhibits physiological damage caused by Aβ oligomers. (A) (i) The cytosolic Ca2+ concentration is shown as a function of time in astrocytes to which 500 nM (total peptide) Aβ42 (containing 19.5 nM oligomers), incubated with 500 nM clusterin, has been added; 327 astrocytes and 187 neurons were examined. (ii) The same experiment with 500 nM Aβ40 (1.5 nM oligomers). Each line represents the intracellular calcium within a single cell; 69 cells were examined, and 158 astrocytes and 84 neurons were examined. (B) (i) The percent change in rate of ROS production in astrocytes and neurons in mixed cultures upon the addition of either 500 nM Aβ42 containing 19.5 nM oligomers (termed “oligo”) or 500 nM Aβ40 containing 1.5 nM oligomers (“oligo”) or 500 nM monomeric solutions (“mono”) in the presence or absence of a 1:1 molar ratio of clusterin to Aβ (“clusterin”). ** represents a p-value <0.01, and **** represents a p-value <0.0001 using a Kruskal–Wallis test followed by Dunn’s post-test. Comparisons were performed with the “oligo” sample of the same Aβ isoform. (ii) The HEt ratio as a function of time upon the addition of Aβ42 (50 nM total peptide), Aβ42 (50 nM total peptide) mixed with 50 nM clusterin, or clusterin only (50 nM total peptide) to astrocytes and neurons in mixed primary cultures. Experiments examined 61 cells with Aβ42 and 59 with Aβ40. (iii) The HEt ratio as a function of time upon the addition of Aβ42 (500 nM total peptide containing 19.5 nM oligomers) mixed with 0, 0.5, 50, or 500 nM of clusterin and then added to astrocytes and neurons in mixed primary cultures. For the various ratios of (Aβ/clusterin), the following number of cells was examined: 10:1, 111; 100:1, 99; and 1000:1, 114. (C) (i) Fluorescence of the NucView488 caspase-3 substrate as a function of time following the treatment of primary cultures containing both neurons and astrocytes with a mixture of 500 nM Aβ42, containing 19.5 nM oligomers, and 500 nM clusterin; 215 cells were monitored over 4 different samples. (ii) The proportion of cells (%) that experience activation of caspase-3 following 30 min of incubation with either oligomeric Aβ42 solutions (total peptide concentrations of 50 nM and 500 nM with oligomer concentrations of 1.95 nM and 19.5 nM) or Aβ42 solutions (500 nM total peptide, 19.5 nM oligomers) incubated with 500 nM clusterin. These data are the end points (after 30 min) of the experiments presented in Figure 4A and panel C, i. ** signifies a p-value <0.01 by a Mann–Whitney nonparametric rank correlation. Comparisons were performed relative to data acquired with 500 nM oligomers of Aβ42. (D) (i) The fEPSP slope as a function of time in hippocampal slices incubated prior to each experiment for 2 h with a mixture of 500 nM Aβ42 (containing 19.5 nM oligomers) and 500 nM clusterin. The induction of LTP is observed after HFS. Data were recorded for 9 slices. (ii) The fEPSP slope as a function of time following tetanus in hippocampal slices incubated prior to the experiment for 2 h with a mixture of 500 nM Aβ42 (containing 19.5 nM oligomers). This experiment was conducted as a biological control for the results presented in panel D, i. No induction of LTP is observed, and 4 slices were studied.
Discussion
Physiological Responses Can Be Initiated by Individual Aβ Oligomers at Low Concentrations
We have examined the initial physiological responses in both neurons and astrocytes to solutions containing known quantities of both Aβ oligomers and monomers and then observed how the extracellular chaperone clusterin inhibits these responses. While these pathways induced by the addition of Aβ oligomers have been previously identified in other work, how the response depends on the oligomer concentration has not been determined to date. We were able to observe rises in intracellular Ca2+, increased generation of ROS, and activation of caspase-3 in response to physiologically relevant concentrations of both total Aβ40 and Aβ42 (in the 1–10 nM range).8 First, the Aβ40 and Aβ42 oligomer-mediated increase in [Ca2+]c, resulting from the entry of extracellular Ca2+ through the plasma membrane of astrocytes, activates NADPH oxidase. This increases ROS production in astrocytes and then neurons, which then initiates caspase-3 cleavage in both cell types. Finally, in brain slices, Aβ oligomers, acting via caspase-3 cleavage,21 impair LTP, a cellular analogue of memory. These effects are all observed upon incubation of the cells and tissue with oligomeric but not monomeric Aβ species. Since similar concentrations of Aβ40 and Aβ42 oligomers induce similar degrees of [Ca 2+]c transients and ROS production (Figures 2E and 3D), it appears that the main difference in the context of cytotoxicity between preparations of Aβ40 and Aβ42 containing the same total peptide concentration is in the oligomerization propensity of the two peptides to form oligomeric species. In vitro work has corroborated such observations suggesting a secondary nucleation mechanism used by Aβ42 to catalyze oligomer formation.32
The lack of observable [Ca2+]c transients when Aβ oligomers are added to cultures in Ca2+-free medium and the observation of [Ca 2+]c transients even after depletion of Ca2+ from the endoplasmic reticulum stores using thapsigargin suggest that the origin of the observed Ca2+ transients is from the extracellular medium. In combination with previously published results,9,12,13,15,33,34 our data suggest that individual oligomeric Aβ40 and Aβ42 can disrupt the plasma membranes of astrocytes, but not of neurons, thereby leading to the entry of extracellular Ca2+ into the former but not the latter. A possible mechanism for this phenomenon could be through the formation of pore-like structures that have previously been observed in studies of Aβ oligomer toxicity.13,ref35 Specific binding to any ion transporter is unlikely due to the low oligomer concentrations at which these effects still occur. In the light of previous observations, the differences in the nature and composition of astrocytic and neuronal membranes provide one possible explanation for the observation that the effects of the Aβ oligomers differ between cell types.9,35 Subtle differences in membrane lipid composition, perhaps, could lead to differential binding of Aβ oligomers to these cell types and could be an explanation of the cell-type-specific effects observed.
ROS production in neurons appears to be induced by Aβ oligomers in a different manner from that in astrocytes, as the entry of extracellular Ca2+ triggers increased ROS production in astrocytes, but no Ca2+ transients are observed in neurons. One possible mechanism by which ROS production could be stimulated in neurons is by exposure to pro-inflammatory factors such as cytokines, which are known to be released by astrocytes that have been exposed to Aβ oligomers.36,37 Exposure of neurons to such factors have been shown to induce ROS production and caspase-3 activation.37 While damage to both glia and neurons occurs in Alzheimer’s disease, it is unclear as to which cell type is damaged first. This work suggests that the initial damage is to astrocytes in agreement with previous work that made similar observations with cell cultures using higher concentrations of oligomerized Aβ fragments.10,20 Moreover, there has been an increasing amount of evidence of damage to astrocytes in the onset and progression of tissue damage in cellular and organismal models of AD.37−39
The calcium influx and ROS production increase smoothly with oligomer concentration and then saturate at about 10 nM oligomer concentration. There is no evidence for a sigmoidal curve that might suggest specific binding to a specific target, for example, a cell surface receptor. At higher oligomer concentrations, the effects we have observed are already saturated but other specific interactions may start to occur, turning on additional pathways and giving rise to other physiological changes. This conclusion makes interpretation of such experiments more complex. Furthermore, in many experiments the oligomer concentration is not measured and will depend on the preparation method used. Therefore, even at the same total monomer concentration, the fraction of oligomers could be significantly higher than the preparations used in this work and have a different distribution of oligomer sizes. Our work reveals, for the first time, however, that the initial calcium influx in an astrocyte can be caused by an individual oligomer and that several oligomers do not need to bind the cell at the same time. This is an important observation since it limits the possible mechanism by which the calcium influx may be induced. Furthermore, at low concentrations the fraction of astrocytes showing calcium influx will depend linearly with oligomer concentration, so a small fraction of astrocytes will still be subjected to calcium influx at subpicomolar oligomer concentrations.
Model for the Onset of Neuroglial Damage in Response to Aβ Oligomers
Quantitative analysis of the results described in this article suggests that even picomolar concentrations of Aβ40 or Aβ42 oligomers can cause Ca2+ influx in astrocytes, which leads to ROS production and then caspase-3 activation not just in astrocytes but also neurons. By incubating the Aβ oligomers with clusterin prior to their addition to the primary hippocampal cultures, we were able to inhibit all of the aforementioned physiological responses. In a previous study,16 we found that clusterin forms long-lived complexes with both Aβ40 and Aβ42. In this work, we have determined how much clusterin is required to prevent cellular damage. The primary mechanism of the protective effect of clusterin appears to be binding to Aβ oligomers of either isoform, thereby preventing detrimental interactions between oligomers and hippocampal cells. In our experiments, clusterin was found to be an effective inhibitor of the cellular damage caused by Aβ oligomers at concentrations as low as 5 nM, indicating that the dissociation constant of an Aβ oligomer–clusterin complex is at least in the low nanomolar range, a finding consistent with recent in vitro studies.16 Similar instances of sequestration by binding proteins in vivo have been previously observed to inhibit toxicity and increase degradation propensity of the sequestered species.40
AD takes decades to develop, but our data shows that an individual oligomer, unsequestered by chaperones, can cause neuronal damage within 30 min. These apparently conflicting observations lead us to suggest the following model for the onset of AD. Extracellular chaperones (such as clusterin) are present in the CSF of healthy individuals at sufficient levels to protect against the cytotoxic effects by a dual mode of action, namely, inhibiting the formation of Aβ40 and Aβ42 oligomers and sequestering them when formed. Even with high binding affinities, however, there is a finite probability that individual oligomers will, from time to time, escape such protective mechanisms and interact with cells to initiate the cascade of calcium influx, ROS production, and caspase-3 activation in astrocytes and neurons on a local cellular level, resulting in altered synaptic function. The accumulation of damage to neurons over long periods of time could potentially result in tissue-level symptoms including the loss of cognitive function. Similar stochastic models for other neurodegenerative diseases, where random events lead to cell death, have been proposed for the pathologies associated with Parkinson’s disease41 and huntingtin-aggregate-induced toxicity.42
The extent and rate of neuronal damage, as seen in our data, increases with the oligomer concentration and decreases with the clusterin concentration. Therefore, any genetic or epigenetic perturbations that result in lowered protection against aggregate-induced damage such as lower clusterin concentrations, reduced efficacy of clusterin binding, or increased production of Aβ oligomers could manifest themselves in a greater rate of Aβ-oligomer-induced cellular damage and earlier onset of disease. Such situations could occur in early onset, inheritable forms of AD where higher populations of toxic oligomers are likely to be present at any given time and as a result of aging where relative or absolute decrease in the efficacy of clearance is likely to occur.43,44
Supporting Information Available
Further control experiments including those testing effects of fluorescent dyes used for the single-molecule characterization of Aβ oligomers. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
◆ (P.N.) Whitehead Institute for Biomedical Research, 9 Cambridge Center, Cambridge, MA, USA.
Author Present Address
¶ (K.M.H.) National Institutes of Health (NHLBI-CMM), 10 Center Drive, Bldg. Ten CRC, Bethesda MD, USA.
Author Contributions
▽ P.N. and K.M.H. contributed equally to this work.
This work was supported by the Wellcome Trust/MRC Parkinson’s Disease Consortium Grant to UCL/IoN, the Wellcome Trust/MRC Alzheimer’s Disease Consortioum Grant to the University of Cambridge/Bristol, Korea-UK Alzheimer’s Disease Consortium Grant by the Korean Ministry of Health and Welfare, and the Augustus Newman Foundation. P.N. is supported by the Marshall Aid Commemoration Commission (Marshall Scholarship) and the National Science Foundation (Graduate Research Fellowship). K.M.H and A.Y.A were supported by the Wellcome Trust, MRC Parkinson’s Consortium Grant. M.R.W. acknowledges the support of the Australian Research Council (DP0773555 and DP0984341). A.Y.A. is a Parkinson’s UK Senior Research Fellow. D.H.K. is supported by the Korea-UK Alzheimer’s Disease Consortium Grant by Korean Ministry of Health and Welfare. K.C. is supported by the Wolfson Research Merit Award and Royal Society, London.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Selkoe D. J. (2001) Alzheimer’s disease: genes, proteins, and Therapy. Physiol. Rev. 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S. M.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Bucciantini M.; Giannoni E.; Chiti F.; Baroni F.; Formigli L.; Zurdo J. S.; Taddei N.; Ramponi G.; Dobson C. M.; Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. [DOI] [PubMed] [Google Scholar]
- Cleary J. P.; Walsh D. M.; Hofmeister J. J.; Shankar G. M.; Kuskowski M. A.; Selkoe D. J.; Ashe K. H. (2005) Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nature Neurosci. 8, 79–84. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2008) Soluble oligomers of the amyloid-β protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P. D.; Pirttila T.; Mehta S. P.; Sersen E. A.; Aisen P. S.; Wisniewski H. M. (2000) Plasma and cerebrospinal fluid levels of amyloid-β proteins 1–40 and 1–42 in Alzheimer disease. Arch. Neurol. 57, 100–105. [DOI] [PubMed] [Google Scholar]
- Abramov A. Y.; Ionov M.; Pavlov E.; Duchen M. R. (2011) Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: implications for Alzheimer’s disease. Aging Cell 10, 595–603. [DOI] [PubMed] [Google Scholar]
- Abramov A. Y.; Canevari L.; Duchen M. R. (2004) β-Amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov A. Y.; Canevari L.; Duchen M. R. (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 23, 5088–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark R. J.; Hensley K.; Butterfield D. A.; Mattson M. P. (1995) Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 15, 6239–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A.; Smith M.; Parker I. (2011) Single-channel Ca2+ imaging implicates Aβ1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell. Biol. 195, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz J. C.; Simakova O.; Jacobson K. A.; Arispe N.; Pollard H. B. (2009) Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 3348–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M.; Kuroda Y. (2000) Molecular mechanism of neurodegeneration induced by Alzheimer’s β-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res. Bull. 53, 389–397. [DOI] [PubMed] [Google Scholar]
- Narayan P.; Orte A.; Clarke R. W.; Bolognesi B.; Hook S.; Ganzinger K. A.; Meehan S.; Wilson M. R.; Dobson C. M.; Klenerman D. (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β1–40 peptide. Nature Struct. Mol. Biol. 19, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. R.; Easterbrook-Smith S. B. (1992) Clusterin binds by a multivalent mechanism to the Fc and Fab regions of IgG. Biochim. Biophys. Acta 1159, 319–326. [DOI] [PubMed] [Google Scholar]
- Vaarmann A.; Gandhi S.; Gourine A. V.; Abramov A. Y. (2010) Novel pathway for an old neurotransmitter: Dopamine-induced neuronal calcium signalling via receptor-independent mechanisms. Cell Calcium 48, 176–182. [DOI] [PubMed] [Google Scholar]
- Cremades N.; Cohen S. I.; Deas E.; Abramov A. Y.; Chen A. Y.; Orte A.; Sandal M.; Clarke R. W.; Dunne P.; Aprile F. A.; Bertoncini C. W.; Wood N. W.; Knowles T. P.; Dobson C. M.; Klenerman D. (2012) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov A. Y.; Duchen M. R. (2005) The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R. Soc., B 360, 2309–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J.; Whitcomb D. J.; Olsen K. M.; Kerrigan T. L.; Lo S.-C.; Bru-Mercier G.; Dickinson B.; Scullion S.; Sheng M.; Collingridge G.; Cho K. (2011) Aβ1–42 inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nature Neurosci. 14, 545–547. [DOI] [PubMed] [Google Scholar]
- Orte A.; Birkett N. R.; Clarke R. W.; Devlin G. L.; Dobson C. M.; Klenerman D. (2008) Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 105, 14424–14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde T. E.; Eckman C. B.; Younkin S. G. (2000) Biochemical detection of Aβ isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim. Biophys. Acta 1502, 172–187. [DOI] [PubMed] [Google Scholar]
- Gravina S. A.; Ho L.; Eckman C. B.; Long K. E.; Otvos L.; Younkin L. H.; Suzuki N.; Younkin S. G. (1995) Amyloid β protein (Aβ) in Alzheimer’s disease brain. J. Biol. Chem. 270, 7013–7016. [DOI] [PubMed] [Google Scholar]
- Zwart M. P.; Daròs J.-A.; Elena S. F. (2011) One is enough in vivo effective population size is dose-dependent for a plant RNA virus. PLoS Pathogens 7, e1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A.; Rottkamp C. A.; Nunomura A.; Raina A. K.; Perry G. (2000) Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta 1502, 139–144. [DOI] [PubMed] [Google Scholar]
- Li Z.; Jo J.; Jia J.-M.; Lo S.-C.; Whitcomb D. J.; Jiao S.; Cho K.; Sheng M. (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Rowan M. J.; Anwyl R. (2004) β-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J. Neurosci. 24, 6049–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D.; Abraham R.; Hollingworth P.; Sims R.; Gerrish A.; Hamshere M. L.; Pahwa J. S.; Moskvina V.; Dowzell K.; Williams A.; Jones N.; Thomas C.; Stretton A.; Morgan A. R.; Lovestone S.; Powell J.; Proitsi P.; Lupton M. K.; Brayne C.; Rubinsztein D. C.; Gill M.; Lawlor B.; Lynch A.; Morgan K.; Brown K. S.; Passmore P. A.; Craig D.; McGuinness B.; Todd S.; Holmes C.; Mann D.; Smith A. D.; Love S.; Kehoe P. G.; Hardy J.; Mead S.; Fox N.; Rossor M.; Collinge J.; Maier W.; Jessen F.; Schurmann B.; van den Bussche H.; Heuser I.; Kornhuber J.; Wiltfang J.; Dichgans M.; Frolich L.; Hampel H.; Hull M.; Rujescu D.; Goate A. M.; Kauwe J. S. K.; Cruchaga C.; Nowotny P.; Morris J. C.; Mayo K.; Sleegers K.; Bettens K.; Engelborghs S.; De Deyn P. P.; Van Broeckhoven C.; Livingston G.; Bass N. J.; Gurling H.; McQuillin A.; Gwilliam R.; Deloukas P.; Al-Chalabi A.; Shaw C. E.; Tsolaki M.; Singleton A. B.; Guerreiro R.; Muhleisen T. W.; Nothen M. M.; Moebus S.; Jockel K.-H.; Klopp N.; Wichmann H. E.; Carrasquillo M. M.; Pankratz V. S.; Younkin S. G.; Holmans P. A.; O’Donovan M.; Owen M. J.; Williams J. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J.-C.; Heath S.; Even G.; Campion D.; Sleegers K.; Hiltunen M.; Combarros O.; Zelenika D.; Bullido M. J.; Tavernier B.; Letenneur L.; Bettens K.; Berr C.; Pasquier F.; Fievet N.; Barberger-Gateau P.; Engelborghs S.; De Deyn P.; Mateo I.; Franck A.; Helisalmi S.; Porcellini E.; Hanon O.; de Pancorbo M. M.; Lendon C.; Dufouil C.; Jaillard C.; Leveillard T.; Alvarez V.; Bosco P.; Mancuso M.; Panza F.; Nacmias B.; Bossu P.; Piccardi P.; Annoni G.; Seripa D.; Galimberti D.; Hannequin D.; Licastro F.; Soininen H.; Ritchie K.; Blanche H.; Dartigues J.-F.; Tzourio C.; Gut I.; Van Broeckhoven C.; Alperovitch A.; Lathrop M.; Amouyel P. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. [DOI] [PubMed] [Google Scholar]
- Wilson M. R., Yerbury J. J., and Poon S. (2008) Extracellular Chaperones and Amyloids, in Heat Shock Proteins and the Brain: Implications for Neurodegenerative Diseases and Neuroprotection (Asea A. A. A., and Brown I. R., Eds.), pp 283–315, Springer: The Netherlands. [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N. (2004) Architecture of the Alzheimer’s AßP ion channel pore. J. Membr. Biol. 197, 33–48. [DOI] [PubMed] [Google Scholar]
- Chow S.-K.; Yu D.; MacDonald C. L.; Buibas M.; Silva G. A. (2010) Amyloid β-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro 2, e00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Sokolov Y.; Edmonds B.; McIntire T. M.; Milton S. C.; Hall J. E.; Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366. [DOI] [PubMed] [Google Scholar]
- Saridaki T.; Zampagni M.; Mannini B.; Evangelisti E.; Taddei N.; Cecchi C.; Chiti F. (2012) Glycosaminoglycans (GAGs) suppress the toxicity of HypF-N prefibrillar aggregates. J. Mol. Biol. 421, 616–630. [DOI] [PubMed] [Google Scholar]
- Bhat R.; Crowe E. P.; Bitto A.; Moh M.; Katsetos C. D.; Garcia F. U.; Johnson F. B.; Trojanowski J. Q.; Sell C.; Torres C. (2012) Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS One 7, e45069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwood C.; Pooler A.; Atherton J.; Hanger D.; Noble W. (2011) Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2, e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A.; Jimenez S.; Mecha M.; Dávila D.; Guaza C.; Vitorica J.; Torres-Aleman I. (2012) Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 17, 705–718. [DOI] [PubMed] [Google Scholar]
- Furman J. L.; Sama D. M.; Gant J. C.; Beckett T. L.; Murphy M. P.; Bachstetter A. D.; Van Eldik L. J.; Norris C. M. (2012) Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luheshi L. M.; Hoyer W.; de Barros T. P.; van Dijk Härd I.; Brorsson A.-C.; Macao B.; Persson C.; Crowther D. C.; Lomas D. A.; Ståhl S.; Dobson C. M.; Härd T. (2010) Sequestration of the Aβ peptide prevents toxicity and promotes degradation in vivo. PLoS Biol. 8, e1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G.; Lumsden C. J.; McInnes R. R. (2001) Inherited neurodegenerative diseases: the one-hit model of neurodegeneration. Hum. Mol. Genet. 10, 2269–2275. [DOI] [PubMed] [Google Scholar]
- Colby D. W.; Cassady J. P.; Lin G. C.; Ingram V. M.; Wittrup K. D. (2006) Stochastic kinetics of intracellular huntingtin aggregate formation. Nature Chem. Biol. 2, 319–323. [DOI] [PubMed] [Google Scholar]
- Luheshi L. M.; Tartaglia G. G.; Brorsson A.-C.; Pawar A. P.; Watson I. E.; Chiti F.; Vendruscolo M.; Lomas D. A.; Dobson C. M.; Crowther D. C. (2007) Systematic in vivo analysis of the intrinsic determinants of amyloid-β pathogenicity. PLoS Biol. 5, e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega K. G.; Sigurdson W.; Ovod V.; Munsell L.; Kasten T.; Morris J. C.; Yarasheski K. E.; Bateman R. J. (2010) Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.