

Abstract
Herein, we report a method for in vitro selection of multivalent glycopeptides, combining mRNA display with incorporation of unnatural amino acids and “click” chemistry. We have demonstrated the use of this method to design potential glycopeptide vaccines against HIV. From libraries of ∼1013 glycopeptides containing multiple Man9 glycan(s), we selected variants that bind to HIV broadly neutralizing antibody 2G12 with picomolar to low nanomolar affinity. This is comparable to the strength of the natural 2G12–gp120 interaction, and is the strongest affinity achieved to date with constructs containing 3–5 glycans. These glycopeptides are therefore of great interest in HIV vaccine design.
Antibody 2G12, isolated from an HIV positive individual, binds and neutralizes a broad range of HIV strains,1 and provides sterilizing immunity against SHIV challenge in macaque models of infection.2 2G12 recognizes an epitope comprised of 2–4 high mannose (Man9GlcNAc2) glycans on the surface of HIV envelope protein gp120,3 and glycopeptides that precisely mimic this glycan clustering and presentation may be useful as vaccines to “re-elicit” 2G12-like antibodies in vivo.4 Glycans clustered on carbohydrate,5 peptide,6 and protein scaffolds,7 as well as phage particles8 and yeast9 have been tested for this purpose, but with little success. In part, this may be due to the difficulty of designing structures in which the clustering of glycans faithfully mimics that of the 2G12 epitope on gp120. Indeed, most of these structures were recognized by 2G12 with orders of magnitude weaker affinity than was gp120, suggesting that they were not optimal mimics of the 2G12 epitope.
We have recently approached the problem of designing 2G12 epitope mimics by developing a directed evolution-based strategy, SELMA, in which a DNA backbone evolves to optimally cluster the epitope glycans.10 However, we have also been interested in the directed evolution of glycopeptides, given their relevance in both HIV and cancer vaccine design. Although many powerful methods are available for in vitro selection of peptides, comparatively little has yet been published on in vitro selection of glycopeptides. Recently, phage display with chemically modified phages enabled selection of peptide 5-mer sequences containing a single central mannose monosaccharide from ∼106 sequences.11 In an alternative approach, a single mannose was chemically attached to the N-terminal position of a 7-mer phage-displayed library of ∼108 sequences, although selections with this library have not yet been reported.12 Because carbohydrate HIV epitopes contain multiple glycans,3 it was essential that our selection method allow access to multivalent glycopeptides containing several glycans at variable positions, supported by a significant peptide framework. Herein, we report the development of such a method, based on “click”13 glycosylation of mRNA-displayed peptide libraries of ∼1013 sequences.14 We demonstrate the usefulness of this method in HIV antigen design, using it to obtain 33-mer glycopeptides containing 3–5 high-mannose nonasaccharides, which are tightly recognized by broadly neutralizing HIV antibody 2G12, with KD’s as low as 500 pM.
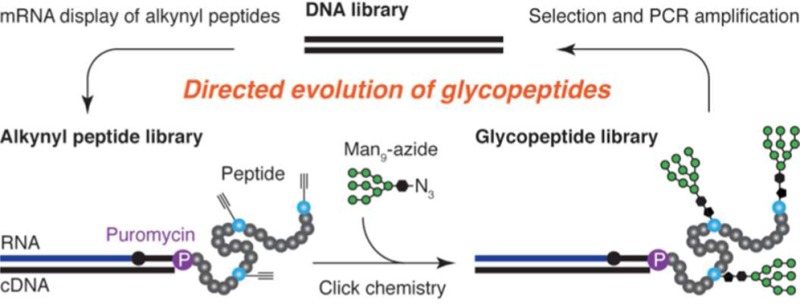
Figure 1 illustrates how glycopeptide selection can be achieved by the combination of chemical synthesis, “click” chemistry (CuAAAC, or copper-assisted azide alkyne cycloaddition),13 mRNA display selection,14 and codon reassignment15 using PURE system cell-free translation.16 In mRNA display (Figure 1A), mRNA encoding the desired peptide library is cross-linked to a 3′ puromycin oligonucleotide. The peptide library is then generated by ribosomal translation of the mRNAs, but when the ribosome stalls near the puromycin oligonucleotide, puromycin enters the ribosome active site and forms a covalent bond to the C-terminus of the nascent peptide. In this way, the peptide is covalently attached to its encoding mRNA, and peptides that survive selection can be “amplified” by (1) PCR amplification of their cDNA, followed by (2) transcription/translation of the PCR products. When translation is done with PURE system (Figure 1B), then noncanonical amino acids can be incorporated into the peptides by substituting them for canonical amino acids in the translation mixture.16b For our purposes, methionine is omitted and replaced by homopropargylglycine (HPG, Figure 1B), which is accepted by translational machinery and incorporated in every position encoded by the Met (AUG) codon of the mRNA. Alkynes in the peptides can then be “click”-glycosylated with Man9 azide10b,10c through CuAAAC. This glycosylation through a triazole linkage is non-natural; however, it is still useful for creating oligosaccharide–peptide conjugates that mimic biological functions in vitro and in vivo,17 and this linkage will be resistant to potential digestion by glycoamidases in vivo.17c Additionally, the low natural incidence of Met codons and the possibility of substitution of Met with HPG in Met-depleted cells may enable broad applications.17b,17d
Figure 1.

In vitro selection of glycopeptides. (A) Covalent linkage of nascent peptide to its mRNA, mediated by attachment to mRNA-linked puromycin inside the ribosome. (B) Use of PURE system to incorporate alkynes via the AUG codon and CuAAAC “click” chemistry glycosylation with with the synthetic Man9-azide. (C) Peptide libraries used in this study. The “fixed” library contains three constant glycosylation sites, whereas the “variable” library contains only one constant glycosylation site, at position 1. The random regions of both libraries are followed by a flexible linker and a His6 tag. Puromycin attached to mRNA is covalently linked to C-terminal arginine residues in translation.16b (D) Scheme for selection of 2G12-binding glycopeptides. The library DNA is comprised of T7 promotor (PT7), ε-enhancer followed by Shine–Dalgarno sequence (SD), the open reading frame (ORF) of the peptide, and the constant region including the sequence for annealing and photocross-linking the mRNA to a puromycin-containing oligonucleotide.
The overall selection process is illustrated in Figure 1C,D. DNA encoding the random library is transcribed and translated to generate mRNA-displayed alkynyl peptides (mRNA-peptide fusions), in which alkynes are located in very diverse arrangements. After reverse transcription to protect the mRNA as its cDNA duplex (Supporting Information Figure S1), “click” chemistry is then used to attach Man9 azides to the library alkynes,10b generating the glycopeptide library. The fraction of glycopeptides that contains the most favorable arrangements of glycans to bind to 2G12 is then selected from the library, and cDNAs of round 1 selection winners are amplified by PCR to afford the second-generation library in DNA form. The process is then repeated until multivalent glycopeptides are obtained, which have high-affinity for the target lectin.
Results and Discussion
Figure 1C shows the design of the libraries used in this study. We employed two libraries of ∼33-mer peptides with glycosylation sites located either in “fixed” or in “variable” locations. In the starting “fixed” library, every sequence contained glycosylation sites at positions 1, 12, and 23, with a 1.6% frequency of additional glycosylation at all random positions (corresponding to the unbiased statistical frequency of the AUG codon). In the “variable” library, only the first position was fixed as a glycosylation site (due to the necessity of the AUG codon for the translation start), and all other positions were random, but with codons doped (Supporting Information, note 1) to yield an increased (5%) frequency of glycosylations.
These libraries of ∼1013 sequences were then subjected in parallel to 10 rounds of selection for binding to 2G12. mRNA-displayed-glycopeptides were incubated with successively lower concentrations of 2G12, and bound complexes were retrieved from solution alternately with Protein A or Protein G magnetic beads. Bound fusions were eluted by heating, in which the gp120-binding activity of 2G12 was selectively inactivated without harming the nucleic acid tags (Supporting Information Figure S2).
Figure 2A shows the percent recovery of the library after each round of selection, as monitored by scintillation counting of radioactive 35S-cysteine or 3H-histidine. Because the recovery after round 2 was quite high (∼10%), we increased stringency by adding 100 mM mannose as a competitor in all subsequent rounds. Glycan-independent binders were removed from the library in rounds 4 and 6 by counterselection of the library prior to the CuAAAC reaction. Counterselections without 2G12 (Protein A or Protein G beads only) were used starting at round 7 to remove possible bead binders. Selection rounds 1–7 were performed at room temperature, and rounds 8–10 were performed at 37 °C, to remarkable effect (vide infra).
Figure 2.
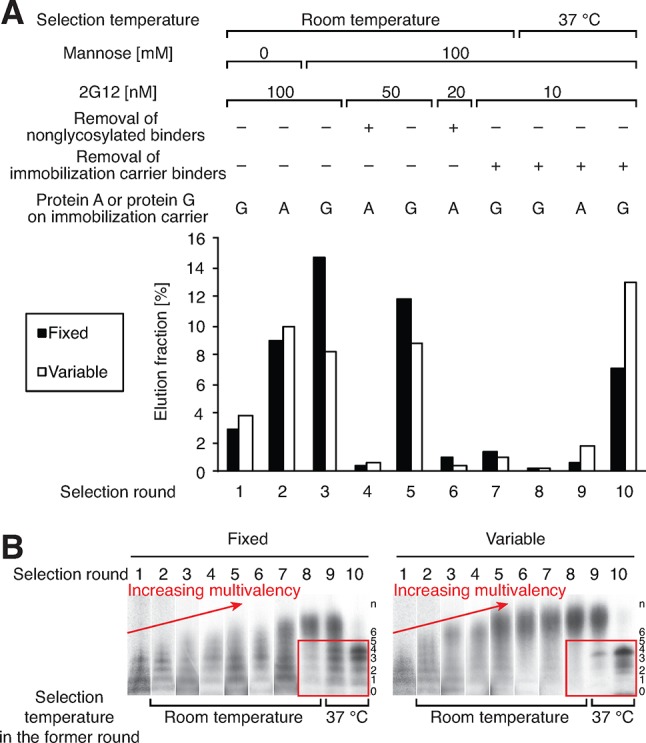
(A) Selection conditions and percentage of radioactivity (counts per min) in eluted fractions. Concentrations of the 2G12 listed for the selection are prior to addition of protein G or protein A magnetic beads. (B) Profiling of the distribution of the putative number of glycans in library peptides before selection (“n” on the right on the gel). Red boxes indicate enrichment of low-valent glycopeptides in 37 °C selection rounds.
By the end of round 7, library binding to 100 nM 2G12 had grown to a high level (Supporting Information Figure S4a). However, an undesired trend toward very high multivalency was also observed throughout these room-temperature selection rounds. This can be seen by SDS-PAGE of the nuclease P1-digested library just prior to each selection round (Figure 2B, red arrows), where separate bands are visible for library species containing different numbers of glycans. This interpretation was confirmed by sequencing of round 7 clones (Supporting Information Table S1), which showed that nearly all peptides contained 6–12 glycosylation sites. One of these, peptide 7V8 (Table 1, entry 3), exhibited a KD of 17 nM for binding to 2G12. Although this 2G12 recognition is tighter than that of most reported oligomannose clusters,5−9,18 6–12 glycans is far more than the number of gp120 glycans thought to be involved in 2G12 binding (3–4).3 Moreover, none of the sequences obtained were replicates, indicating that the library had not yet converged to the best possible sequences; therefore, we continued the selection.
Table 1. Binding Constants of Selected and Nonselected Glycopeptides.

Only the sequence of the random region (positions 1–33) is shown. All peptide sequences used in the 2G12-binding assay were followed by a linker, a His6-tag, and a FLAG-tag (GSGSLGHHHHHHRDYKDDDDK) for purification and radiolabeling purposes. Blue “m” denotes potential Man9-glycosylation sites encoded by the AUG codon. The observed consensus motif is highlighted in yellow.
In the assay, the peptides were radiolabeled with 35S-cysteine (for peptides containing cysteine) or 3H-histidine (for peptides not containing cysteine), and incubated with various concentrations of 2G12, and 2G12–peptide complexes were isolated with magnetic protein G beads. Percentages of the fractions bound were calculated from radioactivity measured by liquid scintillation counting (see Experimental Section for details). KD and Fmax (maximum fraction bound) were calculated by fitting Fbound = (Fmax [2G12])/(KD + [2G12]) to average data points. Errors reported are the standard error of the curve fit.
Not determined.
To address the high multivalency concerns, we decided to carry out subsequent selection rounds at 37 °C because of a striking temperature effect we had recently observed in related studies with our SELMA selection of glycosylated DNA libraries.10c In that work, we had found that increasing the temperature of the 2G12 selection step to 37 °C dramatically favored sequences with lower multivalency and much stronger binding. After applying this modification to the next three rounds of glycopeptide selection, we were delighted to see a parallel trend in the results: low-valent binders, barely visible in the whole-library gel at the beginning of round 8, completely took over both libraries (Figure 2B, red boxes).
Sequencing of 24 clones from each library (Table 1 and Supporting Information Table S2) confirmed the low number of glycosylations (3–4 in most sequences) and revealed a high degree of sequence convergence. Many repeat sequences were observed, as was a peptide consensus motif, mxSIP(−/x)YTY(L/xW)(−/x)P, where m denotes Man9-HPG and x denotes a variable amino acid (yellow highlights). This motif was present in some clones from both libraries, and apparently arose from convergent evolution in multiple sequence families, as it is located sometimes early, sometimes late in the sequences. Ten glycopeptides were prepared without mRNA tags by in vitro translation without the puromycin linker (Supporting Information Figures S3 and S5), and KD’s were determined by incubation of the glycopeptide with various concentrations of 2G12, followed by capture on Protein G beads and quantification of radioactivity. All 10 of the tested glycopeptides were recognized tightly by 2G12, with KD’s in the range of 0.5–5 nM, similar to the strength of 2G12–gp120 interaction (Table 1 and Supporting Information Figure S6).19 Some of these peptides lacked the peptide consensus motif, and all were dependent on glycosylation for 2G12 binding (Figure 3A), indicating that the glycans are the major element recognized by the antibody. This was further confirmed by studies showing a significant reduction in binding in the presence of 0.5 M mannose. Moreover, a reduction in binding observed with 200–800 nM recombinant gp120 (Figure 3A/B) added to the assay shows that our glycopeptides compete with gp120 for binding its site on 2G12. In contrast to round 10 selected peptides, clones picked from the libraries prior to selection showed very little binding to 2G12 at concentrations up to 128 nM (Table 1 and Supporting Information Figure S7), indicating that not all peptide backbones are suitable for highly antigenic presentation of the carbohydrates.
Figure 3.

Importance of glycans in binding of selected glycopeptides to 2G12 and competition with gp120. (A) Competition of glycopeptide binding to 2G12 with gp120 and mannose and glycosylation-dependent binding. (B) Competition of glycopeptide binding to 2G12 with varied concentrations of gp120.
To confirm that the results presented in Table 1 were not artifacts of ribosomal translation, we chemically synthesized and characterized glycopeptide 10F2 and confirmed its 2G12 binding affinity in an alternate assay, BioLayer Interferometry, (BLI).20 For preparation of this long peptide, we employed Pentelute’s new rapid flow solid-phase peptide synthesis method,21 in which activated amino acids are flowed through a thermally heated reactor containing peptide synthesis resin. In this manner, we readily obtained alkyne-containing peptide 1, in which the C-terminal His6 tag of the ribosomal peptide was replaced by an -StBu-protected cysteine (Figure 4A). CuAAAC glycosylation proceeded to near completion, and HPLC purification afforded the desired glycopeptide 2, whose identity was confirmed by mass spectrometry (Supporting Information Figures S8,S9). Reductive deprotection of the cysteine and immediate trapping with a maleimide-biotin reagent appended the biotin necessary for immobilization to the streptavidin biosensor surface used in the BLI assay. After immobilization of the biotinylated glycopeptide 3 on the sensor, 2G12 was associated to the surface at several concentrations, followed by dissociation in blank buffer (Figure 4B and Supporting Information Figure S4). The resulting response curves were fit globally to a 1:1 binding model and afforded rate constants of kon = (11.1 ± 0.4) × 104 M–1 s–1 and koff = (1.51 ± 0.02) × 10–4 s–1, corresponding to a KD of 1.37 ± 0.02 nM. This affinity measurement is in reasonable agreement with the measurement of ribosomally translated 10F2 in our bead-based assay. Moreover, this interaction is both kinetically and thermodynamically comparable to that measured for the 2G12–gp120 interaction (kon = 6.6 × 104 M–1s–1, koff = 3.8 × 10–4 s–1, KD = 5.8 nM).19
Figure 4.
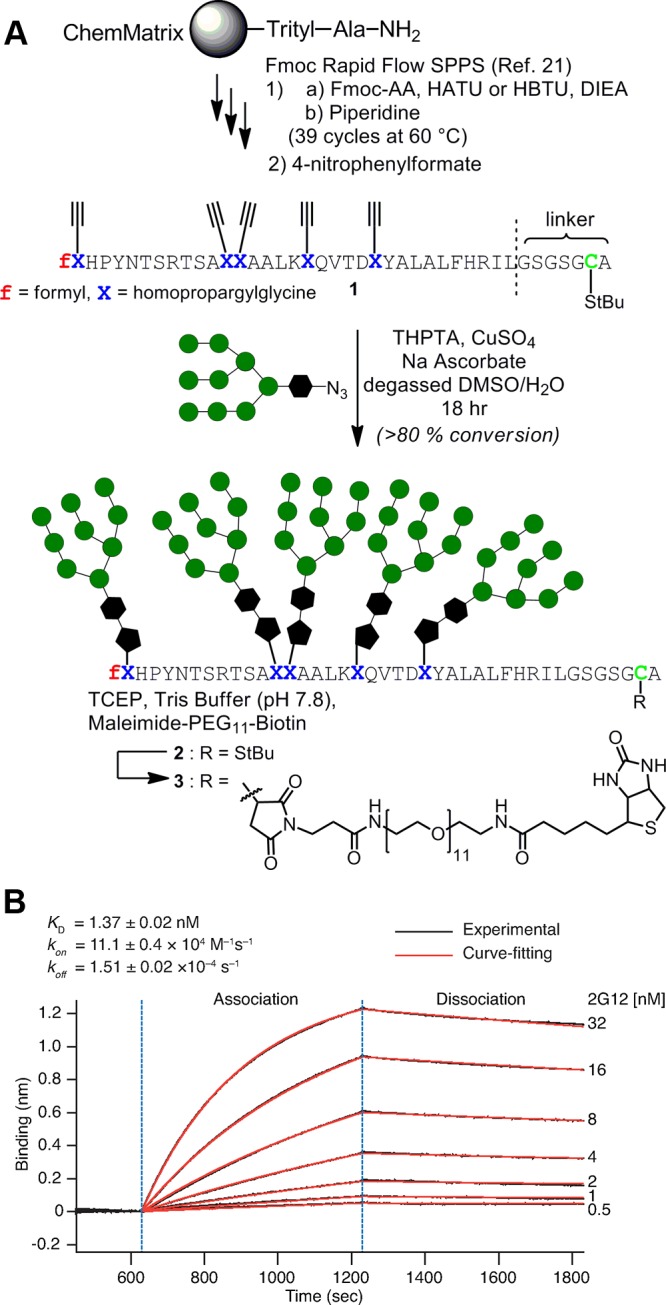
(A) Preparation of synthetic 10F2 glycopeptide and attachment of biotin for immobilization to streptavidin surface. (B) BioLayer Interferometry (BLI) measurement of 2G12 interacting with surface-immobilized synthetic 10F2 glycopeptide. kon and koff errors are standard errors of the curve fit, and the KD error is propagated from those values.
The 2G12 recognition observed for our Man9 glycopeptides represents an enhancement of up to ∼360 000-fold as compared to monovalent Man9 glycan (KD = 180 μM).18f Although Wong has prepared Man9 dendrimers that bind 2G12 with KD’s down to 3.1 nM, that level of binding was achieved only with 9- and 27-mers, whereas 610 nM binding was observed with trivalent Man9 dendrimers. Taken together, these data suggest that the clustering and/or support of Man9 by neighboring elements in our glycopeptides results in better mimicry of the 2G12 epitope than previous Man9 presentations. However, it is important to note that this high antigenicity (the ability to bind 2G12) does not necessarily ensure the desired immunogenicity (the ability to stimulate production of antibodies with 2G12-like specificity). Nevertheless, these evolved glycoclusters are extremely interesting candidates for in vivo immunogenicity studies.
Conclusion
We have successfully demonstrated the in vitro selection of multivalent glycopeptides from diverse libraries (∼1013 sequences). We have shown that the use of higher temperature in the target binding step of selection favors glycopeptides with lower multivalency, an effect that parallels what we observed in our SELMA selection of glycosylated DNAs.10 We expect that this approach can be used to design multivalent carbohydrate vaccines targeting additional HIV or cancer epitopes, as well as multivalent carbohydrate ligands for other lectins. The glycopeptides and other conjugates thus obtained will be useful tools in biological studies and potential therapeutic applications.
Experimental Section
Proteins and Ribosomes for PURE System Translation
Hexa-histidine tagged IF1, IF2, IF3, EF-Tu, EF-G, EF-Ts, RF1, RF3, RRF, MTF, MetRS, GluRS, PheRS, AspRS, SerRS, ThrRS, ArgRS, GlnRS, IleRS, LeuRS, TrpRS, AsnRS, HisRS, TyrRS, ValRS, ProRS, AlaRS, CysRS, LysRS, and GlyRS were expressed in Escherichia coli BL21 Star (DE3) (Invitrogen) and purified as previously described.16a,16b,22 Ribosomes were prepared combining the previously described protocols22a,23,24 with some modifications. E. coli A19 was grown and harvested as previously described.23 The pelleted cells were washed with ∼300 mL of suspension buffer (10 mM HEPES-KOH, pH 7.6, 10 mM magnesium acetate, 50 mM KCl, 7 mM β-mercaptoethanol) and spun at 5000g for 15 min. The pelleted cells were lysed in suspension buffer using a bead-beater, and the cleared lysate was obtained by centrifugation as previously described.23 The supernatant (∼20 mL) was mixed with the same volume of suspension buffer containing 3 M (NH4)2SO4 and centrifuged at 36 000g for 30 min. The resulted supernatant was filtered through a 0.45 μm membrane and subjected to FPLC purification to yield ribosomes as previously described.22a,24
PURE System Translation
The PURE translation system with homopropargylglycine instead of methionine was prepared as previously described16a,16b,16e,22 with slight modifications. The reaction contained 50 mM HEPES-KOH (pH 7.6), 12 mM magnesium acetate, 2 mM spermidine, 100 mM potassium glutamate, 1 mM dithiothreitol (DTT), 1X cOmplete ULTRA, EDTA-free (Roche), 1 mM ATP, 1 mM GTP, 20 mM creatine phosphate (Calbiochem), 0.01 mg/L 10-formyl-5,6,7,8-tetrahydrofolic acid, 0.04 ABS280 creatine kinase (Roche), 0.85 units/mL nucleoside 5′-diphosphate kinase from bovine liver (Sigma), 6.8 units/mL myokinase from rabbit muscle (Sigma), 100 units/mL inorganic pyrophosphatase, 48 ABS260 tRNA from E. coli MRE 600 (Roche), 20 μg/mL MTF, 10 μg/mL IF1, 40 μg/mL IF2, 10 μg/mL IF3, 10 μg/mL EF-Tu, 50 μg/mL EF-Ts, 50 μg/mL EF-G, 10 μg/mL RF1, 10 μg/mL RF3, 10 μg/mL RRF, 0.66 μM MetRS, 0.23 μM GluRS, 0.027 μM PheRS, 0.21 μM AspRS, 0.45 μM SerRS, 0.011 μM ThrRS, 0.021 μM ArgRS, 0.27 μM GlnRS, 0.11 μM IleRS, 0.093 μM LeuRS, 0.23 μM TrpRS, 0.094 μM AsnRS, 0.21 μM HisRS, 0.18 μM TyrRS, 0.089 μM ValRS, 0.031 μM ProRS, 0.070 μM AlaRS, 0.41 μM CysRS, 0.18 μM LysRS, 0.024 μM GlyRS, 1.2 μM ribosomes, a mixture of 17 natural amino acids (3 mM each), with methionine, cysteine, and histidine omitted and preadjusted pH to 7.6 with KOH, and 3 mM l-homopropargylglycine (Chiralix). To label the peptide radioisotopically, the reactions also contained l-[35S]-cysteine (Perkin-Elmer) or [2,5-3H]-l-histidine (Moravek Biochemicals) in concentrations totaling 0.002–3 mM together with nonradioactive cysteine/histidine. These reactions were assembled on ice and initiated by the addition of mRNA (0.5–1.0 μM), followed by incubation at 37 °C for 1 h for mRNA display or 2 h for individual free peptide translation.
Click Reaction (Optimized Procedure Used in Rounds 2–10 of Selection, and in Preparation of Individual Peptides for Binding Studies)
Man9-azide was synthesized as previously described.10b The click reaction was performed combining the previously described protocols10b,25 with some modifications. The dry pellets of peptides or fusions in 0.5 mL microcentrifuge tubes were redissolved in 2.5 μL of 200 mM HEPES-KOH (pH 7.6) and 10 mM aminoguanidine hemisulfate (mixture A). In the case of fusions, ∼0.05% (v/v) Triton X-100 was also added to the solution. 2.5 μL of a freshly prepared solution of 2 mM CuSO4, 2 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) ligand, and 6 mM Man9-azide was transferred to a capless 0.5 mL microcentrifuge tube (mixture B). Three microliters of a freshly prepared solution of 2.5 mM Man9-azide and 0.83 mM THPTA was added to a capless 0.5 mL microcentrifuge tube (mixture C). Sodium-l-ascorbate (less than 10 mg) was transferred to a capless 0.5 mL microcentrifuge tube. Next, the microcentrifuge tubes containing mixtures A, B, and C, and sodium-l-ascorbate were purged under argon flow in the following manner. The microcentrifuge tubes were carefully positioned at the bottom of a 25 mL two-neck pear (pointy-bottom) flask. Positive argon pressure was applied through one neck, while a rubber septum with a purge needle was used to vent the system from the other neck. After 1 h of efflux, the septum was removed, and, under Ar efflux, a pipet was inserted into the flask to add mixture B to mixture A. The sodium ascorbate was dissolved in degassed H2O to a final concentration of 100 mM, and 0.5 μL was added to the tube containing mixtures A and B. After recapping followed by 15 min of Ar purge, the vent needle was removed to keep the system under positive pressure. After an additional 1 h and 15 min, mixture C and an additional 0.25 μL of 100 mM sodium ascorbate were added to the reaction. After recapping and another 15 min of Ar purge, the vent needle was removed. After an additional 1 h and 15 min, the click reaction mixture was taken out from the flask and quenched with 1.25 μL of 10 mM EDTA (pH 8.0). At this point, the total reaction volume was reduced to ∼2–3 μL due to evaporation.
mRNA Display Selection
General Information
The libraries of glycopeptide-mRNA-DNA fusions were prepared by modifying the previously described protocol to prepare the unnatural peptide-mRNA-DNA fusions with PURE system.16e The fusions were radiolabeled with 35S-cysteine (rounds 1 and 2) or 3H-histidine (rounds 3–10), and the yields in various purification steps were monitored by liquid scintillation counting. During the procedure, the integrity of the fusion formation was checked by SDS-PAGE with visualization by autoradiography (in rounds 1 and 2) or fluorography (in rounds 3–10). Here, we describe the procedure in selection round 1 at first, and then the modifications in rounds 2–10, divided into subsections.
Preparation of Puromycin-Linked mRNA in Round 1
The puromycin-linked mRNA for selection round 1 was prepared as follows. The antisense strands of synthetic library DNA (the fixed library, 5′-CTAGCTACCTATAGCCGGTGGTGATGGTGGTGATGACCCAGAGAACCGGAGCCN30CATN30CATN30CATTTAGCTGTCCTCCTTACTAAAGTTAACCCTATAGTGAGTCGTATTA-3′, and the variable library, 5′- CTAGCTACCTATAGCCGGTGGTGATGGTGATGGTGGCCTAAGCTACCGGAGCC(SNn)32CATTTAGCTGTCCTCCTTACTAAAGTTAACCCTATAGTGAGTCGTATTA, where uppercase N is an equimolecular mixture of G, A, T, and C; S is an equimolecular mixture of G or C; lowercase n is a mixture of 40% T, 20% A, 20% G, and 20% C) were purchased from W.M. Keck Biotechnology Resource Laboratory, Yale University. The regions involved in the open reading frame in the constant regions of the two libraries were designed to have identical amino acid sequence but were not identical in nucleotide usage, so that the libraries would be PCR-amplified with different primer sets. The fixed and variable library DNAs purified by denaturing polyacrylamide gel electrophoresis (PAGE), 780 and 300 pmol, respectively, were transcribed in the presence of 1.2 equiv of the DNA containing T7 promotor sequence (5′-TAATACGACTCACTATAGGGTTAACTTTAG-3′) using MEGAshortscript kit (Ambion). The transcripts were purified by denaturing 5% PAGE and photocross-linked with puromycin-containing oligonucleotide XL-PSO, XuagccggugA15ZZACCP, where X is C6 psoralen, lowercase nucleotides have 2′OMe, uppercase A and C are DNA, Z is Spacer 9, and P is puromycin (W.M. Keck Biotechnology Resource Laboratory, Yale University), by 365 nm UV irradiation as previously described.26
Translation To Form Alkynyl Peptide–mRNA Fusions in Round 1
The radiolabeled alkynyl peptide–mRNA fusions for round 1 selection were produced as follows. The peptide–mRNA fusions were translated from 1 μM RNA, of which ∼50% was cross-linked with the puromycin-containing oligonucleotide XL-PSO, in 5.2 mL of PURE system translation reactions containing [35S]-cysteine for 1 h at 37 °C. Following translation, KCl and magnesium acetate were added to facilitate fusion formation,27 incubated for a 15 min at room temperature, and frozen as previously described.16b,16e
Purification and cDNA Synthesis of Fusions in Round 1
The mRNA–peptide fusions were captured on oligo(dT) cellulose (Ambion), washed as previously described,26b and eluted with 0.1% (v/v) Tween-20 followed by 0.22 μm-filtration and ethanol precipitation. The recovered library fusions were purified with Ni-NTA agarose (Qiagen) under a denaturing condition to remove mRNA not fused with peptide using a procedure similar to one previously described,16e and desalted by gel filtration using NAP-5 columns (GE Healthcare) with the gel filtration buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, pH 8.0, 5 mM β-mercaptoethanol, 0.2% (v/v) Triton X-100) according to the manufacturers’s protocol. The fusions were pelleted by ethanol precipitation, and cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen) with RT primers (5′-T15GTGATGGTGGTGATGACCCAGAG-3′ for the fixed library, 5′-T15GTGATGGTGATGGTGGCCTAAGC-3′ for the variable library) in the presence of Superase-In (Ambion) and 0.1% (v/v) Triton X-100 according to the manufacturer’s protocol. The reverse transcribed fusions were pelleted by ethanol precipitation.
Click Glycosylation of Fusions in Round 1
In round 1, the click reaction of fusions was not yet optimized and had to be done twice to give the desired glycosylation efficiency. The first click reaction was done under Ar with a setting as described in the section of “click reaction” but with slightly different conditions: The starting volume was ∼6 times larger, THPTA concentration was twice, and the addition of THPTA, Man9-azide, and sodium ascorbate in the middle of the reaction was not carried out. Because some insoluble pellets were observed at this point, the pellets were collected after click reaction by centrifugation and purified with Ni-NTA agarose under denaturing condition as described above. The eluted fusions were combined with the saved soluble fractions and desalted by gel filtration and ethanol precipitation. The recovered fusions were resubjected to glycosylation using a condition similar to the optimized protocol as described in the section entitled “click reaction” and then ethanol-precipitated.
Selection in Round 1
The pellets of glycosylated peptide-mRNA-cDNA fusions were redissolved in 500 μL of selection buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% v/v Triton X-100). The fixed and variable library fusions (yields of 23.0 and 14.4 pmol, equivalent to 1.4 × 1013 and 0.86 × 1013 sequences, respectively) were individually incubated with 100 nM 2G12 (Polymun Scientific) in 500 μL of selection buffer at room temperature. 100 μL of 30 mg/mL Dynabeads Protein G magnetic beads (Invitrogen) in selection buffer was added to the mixture and kept suspended by tumbling for 20 min at room temperature to capture complexes. The beads were magnetically isolated and washed with 3 × 500 μL of selection buffer. To elute the 2G12-binding fusions, the beads were resuspended in 100 μL of selection buffer, heated at 70 °C for 30 min, chilled on ice for 5 min, and incubated at room temperature for 10 min with tumbling. The supernatant was recovered, and the beads were rinsed with 2 × 100 μL of selection buffer. These solutions were combined as an eluted fraction.
PCR Amplification of cDNA of Selected Fusions in Round 1
The cDNAs of eluted fractions were amplified by PCR using Taq DNA polymerase (Roche) with the forward primer (library FP1 5′-TAATACGACTCACTATAGGGTTAACTTTAGTAAGGAGG-3′) and the reverse primer (5′-CTAGCTACCTATAGCCGGTGGTGATGGTGGTGATGACCCAGAG-3′ for the fixed library; 5′-CTAGCTACCTATAGCCGGTGGTGATGGTGATGGTGGCCTAAGC-3′ for the variable library). The amplified DNAs were purified by phenol extraction and ethanol precipitation, and used for the transcription of the next selection round.
Modification of the Procedures in Rounds 2–10 and Sequencing
The fusion preparation and purification procedures were repeated for 10 rounds except for the following changes. In rounds 3–10, the transcripts were purified using MEGAclear kit (Ambion) and cross-linked with XL-PSO. The puromycin-modified RNA was then purified with denaturing PAGE with the visualization of Gel Indicator RNA Staining Solution (Biodynamics Laboratory), and 0.5 μM was used in translation reaction in the presence of 3H-histidine. In rounds 2–10, the translation volume was reduced to 0.22–0.52 mL, and purifications and following procedures were scaled accordingly. Ni-NTA agarose affinity purification was done only once after oligo(dT) cellulose purification, except in round 2, in which fusions after reverse transcription were repurified with Ni-NTA agarose and desalted as described above. In round 2 and all subsequent rounds, the click reactions were done only once in the same or similar conditions as described in the “click reaction” section. In the selection parts, the essential differences of the conditions between selection rounds were as summarized in Figure 2A. In the rounds with Dynabeads Protein A (Invitrogen), bead amounts were twice as much as protein G beads, because the 2G12-capturing capacity of Dynabeads protein A was lower than that of Protein G. In the rounds with 100 mM mannose, binding reactions and the first two wash steps were with mannose, but not in the third wash and elution steps. In rounds 4 and 6, the unglycosylated library was negatively selected for binding to 100 nM 2G12 with protein G magnetic beads in the absence of mannose, to remove glycan-independent binders. In rounds 7–10, the negative selections were done in the presence of 100 mM mannose prior to the positive selections, to remove glycopeptides that bind to Protein A or G magnetic bead binders. For sequencing after the selection in rounds 7 and 10, the PCR-amplified DNA was cloned into pCR2.1-TOPO vector (Life Technologies) without colony color selection to avoid unintentional biases.
Nuclease-Digestion of Library Fusions
To monitor the number of glycans on the peptides in the fusions in every selection round, a part of the cDNA-RNA-glycopeptide fusions (0.05–1 pmol) was removed after the click reaction and desalted by ethanol precipitation in the presence of linear acrylamide carrier (Ambion). The recovered fusions were diluted in 6–7 μL of 200 mM ammonium acetate (pH 5.3) with 1 unit of nuclease P1 (Sigma), and incubated at 37 °C for 1 h to digest nucleic acids. The solutions then were neutralized with Tris buffer and analyzed by SDS-PAGE.
Preparation of Individual Peptides and Glycopeptides
To generate peptides from individual clones, the plasmids were used as templates for PCR with primer sets (library FP1 and 5′-CTAGCTACCTATTTGTCATCGTCGTCTTTATAATCCCGGTGGTGATGGTGGTGATGACCCAG-3′ for the fixed library members or CTAGCTACCTATTTGTCATCGTCGTCTTTATAATCCCGGTGGTGATGGTGATGGTGGCCTAA-3′ for the variable library members), and the PCR products were used for T7 transcription. The resulting mRNAs were purified by denaturing PAGE or MEGAclear kit (Ambion), and 1 μM RNA was used in PURE system translation (reaction volume of translation varied). Typically, 25 μL of translated reaction was diluted with 100 μL of binding buffer (50 mM Tris-HCl, pH 7.8, 300 mM NaCl, 5 mM β-mercaptoethanol) and 25 μL of Ni-NTA agarose suspension (Qiagen), and tumbled at room temperature for 1 h. The resins were transferred to 0.22 μm spin-filter rinsing with 100 μL of bind buffer, and washed with 3 × 200 μL of bind buffer and 2 × 200 μL of wash buffer (50 mM Tris-HCl, pH 7.8, 5 mM β-mercaptoethanol). The bound peptides were eluted with 2 × 25 μL of 0.1% TFA. The eluted peptides were analyzed by MALDI-TOF MS, using α-cyano-4-hydroxycinnamic acid matrix (Sigma), with or without desalting with ZipTip C18 resin (Millipore). For calibration of MALDI-TOF-MS, at least two of the following standards, bovine insulin, E. coli thioredoxin, and/or horse apomyoglobin, were used. To quantitate peptide yields, the radioactivities were measured by liquid scintillation counting. For the click reaction, the translated and purified peptides were mixed with 0.1% (v/v) Triton X-100, and then dialyzed against H2O containing 0.1% (v/v) Triton X-100 using Slide-A-Lyzer MINI Dialysis Devices, 3.5K MWCO (Thermo Scientific) overnight to desalt. After dialysis, the peptides were divided into two portions: one was glycosylated via the click reaction, while the other was saved as a nonglycosylated peptide control. The peptides to be glycosylated (typically less than 5 pmol) were evaporated by speedvac in a 0.5 mL microcentrifuge tube for use in click glycosylation. Because the efficiency of the click reaction was not always high with round 10 winners, crude glycosylated peptides were subjected to 2G12 affinity purification to obtain the highest-clicked fraction, as follows. The glycosylated peptide (<4 pmol) was incubated with 100 nM 2G12 in selection buffer (40 μL) at room temperature. The solution was then tumbled 30 min with 0.12 mg of equilibrated Dynabeads Protein G to capture 2G12–glycopeptide complex. Beads were then washed with 3 × 40 μL of selection buffer and resuspended in 10 μL of selection buffer. The resuspended beads were heated at 70 °C for 30 min to denature 2G12 and elute glycopeptides, chilled on ice for 5 min, and tumbled at room temperature for 10 min. The magnetically isolated supernatant then was recovered, and the beads were rinsed with 10 μL of selection buffer. The supernatant and the rinsed solution were combined as the purified glycopeptide fraction, and the yields were measured by liquid scintillation counting (the recovery of radioactivity was typically in a range of 25–55% of input radioactivity).
SDS-PAGE of Nuclease-Digested Fusions and Glycopeptides
Unless otherwise noted, SDS-PAGE of nuclease-digested fusions and glycopeptides was done as follows. A 4–20% gradient precast gel (Bio-Rad) was run using a rapid protocol (300 V for 16–20 min). Precision Plus Protein Dual Xtra Standards (Bio-Rad) were used as a molecular weight marker. To visualize the 35S-labeled peptides by autoradiography, gels were soaked in fixing solution (22.5% acetic acid and 5% ethanol) with shaking for 15 min, dried on filter paper, and exposed to a phosphorimager screen to analyze using Storm Phosphorimager (Amersham). To visualize the 35H-labeled peptides by fluorography, gels were treated with NAMP100 Amplify Fluorographic Reagent (GE Healthcare) according to the manufacture’s protocol, then dried and exposed to X-ray films at −80 °C.
Binding Curve and KD Determination of 2G12–Glycopeptide Interaction
For round 10 winners, 0.12–0.2 nM radioactive glycopeptides were incubated with 0, 0.25, 0.5, 1, 2, 4, 8, 16, 32, or 64 nM 2G12 in 40 μL of selection buffer at room temperature for 1 h. The solution then was added to 0.12 mg of pre-equilibrated Dynabeads Protein G and tumbled at room temperature for 30 min. The supernatant was removed, and the beads were washed with 3 × 40 μL of selection buffer. The radioactivities of the supernatant and wash solutions were measured by liquid scintillation counting as unbound fractions. Because the direct usage of the captured glycopeptides on the beads in liquid scintillation counting partially suppressed the radioactivity detection in the case of 3H-label, the bound glycopeptides were eluted and separated from the beads in a following manner. The beads were resuspended in 40 μL of selection buffer, heated at 70 °C for 30 min to elute the bound glycopeptides, chilled on ice for 5 min, and tumbled at room temperature for 10 min. The supernatant was removed, and the beads were washed with 40 μL of selection buffer and resuspended in 40 μL of selection buffer. The radioactivities of these solutions and suspensions were measured by liquid scintillation counting separately, and the values were combined as apparent bound fractions. The measured radioactivity of the fraction that bound to the beads without 2G12 (ranging from 0 to 6% of the total radioactivity in the assay) was subtracted as background from the radioactivity bound to the beads with 2G12, and the difference was divided by the total radioactivity to determine the percentages bound to 2G12. For glycosylated and nonglycosylated 7V8, the same procedure was done except for the following changes: the volume of each solution was reduced to 30 μL, 2 nM radioactive glycopeptide was incubated with 0, 3.125, 6.25, 12.5, 25, 50, or 100 nM 2G12, and 0.18 mg of Dynabeads Protein G was used to capture 2G12. All experiments were done at least in triplicate. KD’s were calculated as described in the footnote of Table 1.
Analysis of Competition of Glycopeptides and gp120 or Mannose for 2G12-Binding and Nonglycosylated Peptide Binding to 2G12 of Round 10 Winners
The procedure was essentially the same as described in the previous section with slight modification as follows. The volume of binding reaction was 20–30 μL, and other volumes were also adjusted accordingly. 200 nM 2G12 in selection buffer was premixed with or without 400 nM 6xHis-tagged gp120(JRFL)(HIV-1) (Immune Technology) or 1 M mannose, and further mixed with the same volume of 0.4 nM radioactive glycopeptides or nonglycosylated peptides for binding reaction. The solutions were incubated at 37 °C for 30 min to equilibrate binding competition and then incubated at room temperature for 30 min to stabilize the complexes. Pre-equilibrated protein G magnetic beads were added to give a final concentration of 6 mg/mL. The separation of unbound fractions and bound fractions was done as described above, except that 0.5 M mannose was added to the washing solution in the case of mannose competition. All experiments were done at least in triplicate.
Preparation of Synthetic Peptide 10F2 (1)
The unglycosylated peptide 10F2, fXHPYNTSRTSAXXAALKXQVTDXYALALFHRIL-GSGSGC(StBu)A (f = formyl, X = homopropargylglycine) was prepared by Fmoc solid-phase peptide synthesis using Pentelute’s recent rapid flow-based method.21 76 mg (25 μmol scale) of trityl ChemMatrix resin, loaded with 0.33 mequiv/g alanine by standard procedures,28 was subjected to 39 cycles of peptide coupling and Fmoc deprotection, with thermal heating to 60 °C (see Supporting Information Table S3 for detailed conditions). Cysteine and histidine couplings were performed with a lower base concentration to avoid racemization, and homopropargylglycine couplings were performed as batch reactions to conserve amino acid. After N-terminal formylation of p-nitrophenyl formate, the peptide was cleaved and deprotected using cleavage cocktail B (87.5/5/5/2.5 TFA/water/Phenol/iPr3SiH), and the peptide was triturated four times with cold ether to afford 38 mg of crude solid. Five milligrams of this was redissolved in 200 μL of DMSO, diluted with 200 μL of water, and purified by RP HPLC (Waters Symmetry 300 C4, 5 μm, 10 × 250 mm, 4 mL/min, 2–42% MeCN in H2O w/0.1% Formic Acid, over 60 min, retention time 52 min) to afford 1.5 mg of product, corresponding to an overall SPPS yield of 11% if the whole batch had been purified. LR ESI–MS: obsd average base peaks 868.79 [M + 5H]5+, 1085.75 [M + 4H]4+, 1447.23 [M + 3H]3+, corresponding to 4338.9 obsd average mass, calcd average mass 4339.9.
Glycosylation of Synthetic Peptide 10F2
10F2 peptide (0.6 mg, 0.14 μmol, 1 equiv) and Man9-azide (1.5 mg, 0.97 μmol, 7.0 equiv) were combined in a 0.5 mL Eppendorf tube by evaporation of stock solutions (tube A). A second tube was prepared, containing 9.8 μL (0.98 μmol, 3.0 equiv) of a 100 mM solution of THPTA ligand and 9.0 μL (0.90 μmol, 2.8 equiv) of a 100 mM solution of CuSO4 (tube B), and the tube was evaporated to dryness. Sodium ascorbate (3.0 mg, 15.2 μmol, 47 equiv) was placed in a third tube (tube C). The three tubes were placed in a two-neck pear (pointy-bottom) flask, and nitrogen atmosphere was established by cycles of vacuum and nitrogen refill. Under nitrogen efflux, 150 μL of DMSO (degassed by freeze–pump–thaw) was added to dissolve the peptide and sugar in tube A, and 75 μL of H2O (degassed by freeze–pump–thaw) was added to dissolve the contents of each of tubes B and C. The contents of tube B, and then tube C, were transferred by syringe to tube A. The resulting homogeneous mixture was allowed to react under nitrogen atmosphere for 20 h, at which time UPLC/MS analysis showed nearly complete conversion. The reaction was quenched by addition of TMEDA (1.5 μL, 3.22 μmol, 10 equiv.) and concentrated in vacuo. The residue was purified by RP-HPLC (same column and gradient method as for the unglycosylated 10F2 peptide, retention time 45 min) to afford pure glycopeptide 2. ESI–HRMS: obsd base peaks, 2058.0088 [M + 6H]6+, 2469.4028 [M + 5H]5+, 3086.7759 [M + 4H]4+, deconvoluted mass 12 334.962, calcd 12 334.980 ± 0.128.
Biotinylation of 10F2 Glycopeptide and Determination of 2G12 Binding by BLI (BioLayer Interferometry)
200 μg of 10F2 glycopeptide in 5.5 μL of water was treated with 6.5 mL of 50 mM TCEP·HCl/1 M Tris-HCl buffer, pH 7.8, under nitrogen, using the same inert gas setup employed in the click procedure. After 4.5 h, the reaction mixture was injected into HPLC (Waters Symmetry, 300 C4, 5 μm, 4.6 × 250 mm, 1 mL/min, 2–42% over 60 min, retention time 46.8 min).
The 2G12 binding of the resulting biotinylated glycopeptide 3 was determined using a BLItz instrument (Fortebio). Biotin-10F2 was loaded (120 s) onto a streptavidin biosensor as a 250 nM solution in buffer 1 (20 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgSO4, 0.20 mg/mL BSA, 0.02% Tween-20). The sensor was washed with buffer 1 for 60 s, after which time the net response due to loading was observed as 0.2 nm. The sensor was then equilibrated with buffer 2 (20 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgSO4, 2.0 mg/mL BSA, 0.1% v/v Tween-20) for 90 s. 2G12 (prepared in buffer 2) was associated at several concentrations (0.5, 1, 2, 4, 8, 16, 32 nM, in random order) for 600 s, followed by dissociation into blank buffer 2 for 600 s. After each 2G12 dissociation, the sensor was regenerated to remove remaining 2G12 by treatment with buffer 3 (10 mM glycine-HCl, pH 2.5) for 120 s, followed by 60 s of wash with buffer 1 and further washes to re-equilibrate the tip with buffer 2. Throughout the experiment, the shake rate was set at 1800 rpm. The use of buffer 2 (with high BSA) was important during association/dissociation to prevent nonspecific 2G12/streptavidin interactions, while buffer 1 (low BSA) was required during loading of the glycopeptide to the sensor surface. To further correct for residual nonspecific interactions, the data were referenced to a blank run using 0.5 nM 2G12 on a sensor containing no loaded peptide. The data were fit to a 1:1 binding model, yielding rate constants of kon = (11.1 ± 0.4) × 104 M–1 s–1 and koff = (1.51 ± 0.02) × 10–4 s–1, corresponding to a KD of 1.37 ± 0.02 nM.
Acknowledgments
I.J.K. gratefully acknowledges Brandeis University and the NIH (R01 AI090745). We are grateful to Polymun Scientific for a gift of mAb 2G12, to Prof. Bradley Pentelute for assistance with rapid flow solid-phase peptide synthesis, and to Prof. Jack Szostak for providing S.H. with the opportunity to learn mRNA display techniques in his laboratory.
Supporting Information Available
Supporting figures, tables, and note. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ 3B Pharmaceuticals GmbH, Magnusstrasse 11, 12489 Berlin, Germany.
The authors declare the following competing financial interest(s): A provisional patent application was filed on behalf of some of the authors.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Trkola A.; Purtscher M.; Muster T.; Ballaun C.; Buchacher A.; Sullivan N.; Srinivasan K.; Sodroski J.; Moore J. P.; Katinger H. J. Virol. 1996, 70, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Binley J. M.; Wrin T.; Korber B.; Zwick M. B.; Wang M.; Chappey C.; Stiegler G.; Kunert R.; Zolla-Pazner S.; Katinger H.; Petropoulos C. J.; Burton D. R. J. Virol. 2004, 78, 13232–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mascola J. R.; Stiegler G.; VanCott T. C.; Katinger H.; Carpenter C. B.; Hanson C. E.; Beary H.; Hayes D.; Frankel S. S.; Birx D. L.; Lewis M. G. Nat. Med. 2000, 6, 207–210. [DOI] [PubMed] [Google Scholar]; b Hessell A. J.; Poignard P.; Hunter M.; Hangartner L.; Tehrani D. M.; Bleeker W. K.; Parren P.; Marx P. A.; Burton D. R. Nat. Med. 2009, 15, 951–U155. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hessell A. J.; Rakasz E. G.; Poignard P.; Hangartner L.; Landucci G.; Forthal D. N.; Koff W. C.; Watkins D. I.; Burton D. R. PLoS Pathog. 2009, 5, e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Scanlan C. N.; Pantophlet R.; Wormald M. R.; Ollmann Saphire E.; Stanfield R.; Wilson I. A.; Katinger H.; Dwek R. A.; Rudd P. M.; Burton D. R. J. Virol. 2002, 76, 7306–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Calarese D. A.; Scanlan C. N.; Zwick M. B.; Deechongkit S.; Mimura Y.; Kunert R.; Zhu P.; Wormald M. R.; Stanfield R. L.; Roux K. H.; Kelly J. W.; Rudd P. M.; Dwek R. A.; Katinger H.; Burton D. R.; Wilson I. A. Science 2003, 300, 2065–2071. [DOI] [PubMed] [Google Scholar]; c Calarese D. A.; Lee H.-K.; Huang C.-Y.; Best M. D.; Astronomo R. D.; Stanfield R. L.; Katinger H.; Burton D. R.; Wong C.-H.; Wilson I. A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13372–13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan C. N.; Offer J.; Zitzmann N.; Dwek R. A. Nature 2007, 446, 1038–1045. [DOI] [PubMed] [Google Scholar]
- Ni J. H.; Song H. J.; Wang Y. D.; Stamatos N. M.; Wang L. X. Bioconjugate Chem. 2006, 17, 493–500. [DOI] [PubMed] [Google Scholar]
- Joyce J. G.; Krauss I. J.; Song H. C.; Opalka D. W.; Grimm K. M.; Nahas D. D.; Esser M. T.; Hrin R.; Feng M. Z.; Dudkin V. Y.; Chastain M.; Shiver J. W.; Danishefsky S. J. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 15684–15689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astronomo R. D.; Lee H. K.; Scanlan C. N.; Pantophlet R.; Huang C. Y.; Wilson I. A.; Blixt O.; Dwek R. A.; Wong C. H.; Burton D. R. J. Virol. 2008, 82, 6359–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astronomo R. D.; Kaltgrad E.; Udit A. K.; Wang S. K.; Doores K. J.; Huang C. Y.; Pantophlet R.; Paulson J. C.; Wong C. H.; Finn M. G.; Burton D. R. Chem. Biol. 2010, 17, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Luallen R. J.; Lin J. Q.; Fu H.; Cai K. K.; Agrawal C.; Mboudjeka I.; Lee F. H.; Montefiori D.; Smith D. F.; Doms R. W.; Geng Y. J. Virol. 2008, 82, 6447–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Luallen R. J.; Fu H.; Agrawal-Gamse C.; Mboudjeka I.; Huang W.; Lee F. H.; Wang L. X.; Doms R. W.; Geng Y. J. Virol. 2009, 83, 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Agrawal-Gamse C.; Luallen R. J.; Liu B.; Fu H.; Lee F.-H.; Geng Y.; Doms R. W. J. Virol. 2011, 85, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ciobanu M.; Huang K. T.; Daguer J. P.; Barluenga S.; Chaloin O.; Schaeffer E.; Mueller C. G.; Mitchell D. A.; Winssinger N. Chem. Commun. 2011, 47, 9321–9323. [DOI] [PubMed] [Google Scholar]; e Marradi M.; Di Gianvincenzo P.; Enriquez-Navas P. M.; Martinez-Avila O. M.; Chiodo F.; Yuste E.; Angulo J.; Penades S. J. Mol. Biol. 2011, 410, 798–810. [DOI] [PubMed] [Google Scholar]
- a MacPherson I. S.; Temme J. S.; Habeshian S.; Felczak K.; Pankiewicz K.; Hedstrom L.; Krauss I. J. Angew. Chem., Int. Ed. 2011, 50, 11238–11242. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Temme J. S.; Drzyzga M. G.; MacPherson I. S.; Krauss I. J. Chem.—Eur. J. 2013, 19, 17291–17295. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Temme J. S.; MacPherson I. S.; DeCourcey J. F.; Krauss I. J. J. Am. Chem. Soc. 2014, 136, 1726–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K.; Tsutsumi H.; Mihara H. Bioorg. Med. Chem. Lett. 2013, 23, 4940–4943. [DOI] [PubMed] [Google Scholar]
- Ng S.; Jafari M. R.; Matochko W. L.; Derda R. ACS Chem. Biol. 2012, 7, 1482–1487. [DOI] [PubMed] [Google Scholar]
- a Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]; b Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Roberts R. W.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Hest J. C. M.; Kiick K. L.; Tirrell D. A. J. Am. Chem. Soc. 2000, 122, 1282–1288. [Google Scholar]; b Tan Z.; Forster A. C.; Blacklow S. C.; Cornish V. W. J. Am. Chem. Soc. 2004, 126, 12752–12753. [DOI] [PubMed] [Google Scholar]
- a Shimizu Y.; Inoue A.; Tomari Y.; Suzuki T.; Yokogawa T.; Nishikawa K.; Ueda T. Nat. Biotechnol. 2001, 19, 751–755. [DOI] [PubMed] [Google Scholar]; b Josephson K.; Hartman M. C. T.; Szostak J. W. J. Am. Chem. Soc. 2005, 127, 11727–11735. [DOI] [PubMed] [Google Scholar]; c Shimizu Y.; Kanamori T.; Ueda T. Methods 2005, 36, 299–304. [DOI] [PubMed] [Google Scholar]; d Hartman M. C. T.; Josephson K.; Lin C.-W.; Szostak J. W. PLoS One 2007, 2, e972. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Guillen Schlippe Y. V.; Hartman M. C. T.; Josephson K.; Szostak J. W. J. Am. Chem. Soc. 2012, 134, 10469–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin H.; Walsh C. T. J. Am. Chem. Soc. 2004, 126, 13998–14003. [DOI] [PubMed] [Google Scholar]; b van Kasteren S. I.; Kramer H. B.; Jensen H. H.; Campbell S. J.; Kirkpatrick J.; Oldham N. J.; Anthony D. C.; Davis B. G. Nature 2007, 446, 1105–1109. [DOI] [PubMed] [Google Scholar]; c Huang W.; Groothuys S.; Heredia A.; Kuijpers B. H. M.; Rutjes F. P. J. T.; van Delft F. L.; Wang L.-X. ChemBioChem 2009, 10, 1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Artner L. M.; Merkel L.; Bohlke N.; Beceren-Braun F.; Weise C.; Dernedde J.; Budisa N.; Hackenberger C. P. R. Chem. Commun. 2012, 48, 522–524. [DOI] [PubMed] [Google Scholar]
- a Li H. G.; Wang L. X. Org. Biomol. Chem. 2003, 1, 3507–3513. [DOI] [PubMed] [Google Scholar]; b Li H. G.; Wang L. X. Org. Biomol. Chem. 2004, 2, 483–488. [DOI] [PubMed] [Google Scholar]; c Wang L. X.; Ni J. H.; Singh S.; Li H. G. Chem. Biol. 2004, 11, 127–134. [DOI] [PubMed] [Google Scholar]; d Krauss I. J.; Joyce J. G.; Finnefrock A. C.; Song H. C.; Dudkin V. Y.; Geng X.; Warren J. D.; Chastain M.; Shiver J. W.; Danishefsky S. J. J. Am. Chem. Soc. 2007, 129, 11042–11044. [DOI] [PubMed] [Google Scholar]; e Wang J.; Li H.; Zou G.; Wang L. X. Org. Biomol. Chem. 2007, 5, 1529–1540. [DOI] [PubMed] [Google Scholar]; f Wang S. K.; Liang P. H.; Astronomo R. D.; Hsu T. L.; Hsieh S. L.; Burton D. R.; Wong C. H. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 3690–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Gorska K.; Huang K.-T.; Chaloin O.; Winssinger N. Angew. Chem., Int. Ed. 2009, 48, 7695–7700. [DOI] [PubMed] [Google Scholar]; h Doores K. J.; Fulton Z.; Hong V.; Patel M. K.; Scanlan C. N.; Wormald M. R.; Finn M. G.; Burton D. R.; Wilson I. A.; Davis B. G. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17107–17112. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Clark B. E.; Auyeung K.; Fregolino E.; Parrilli M.; Lanzetta R.; De Castro C.; Pantophlet R. Chem. Biol. 2012, 19, 254–263. [DOI] [PubMed] [Google Scholar]
- Hoorelbeke B.; van Montfort T.; Xue J.; LiWang P. J.; Tanaka H.; Igarashi Y.; Van Dammef E. J. M.; Sanders R. W.; Balzarini J. FEBS Lett. 2013, 587, 860–866. [DOI] [PubMed] [Google Scholar]
- Abdiche Y.; Malashock D.; Pinkerton A.; Pons J. Anal. Biochem. 2008, 377, 209–217. [DOI] [PubMed] [Google Scholar]
- a Simon M. D.; Heider P. L.; Adamo A.; Vinogradov A. A.; Mong S. K.; Li X.; Berger T.; Policarpo R. L.; Zhang C.; Zou Y.; Liao X.; Spokoyny A. M.; Jensen K. F.; Pentelute B. L. ChemBioChem 2014, 15, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mong S. K.; Vinogradov A. A.; Simon M. D.; Pentelute B. L. ChemBioChem 2014, 15, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shimizu Y.; Ueda T. In Cell-Free Protein Production; Endo Y., Takai K., Ueda T., Eds.; Humana Press: Totowa, NJ, 2010; Methods Mol. Biol., Vol. 607, pp 11–21. [Google Scholar]; b Ma Z.; Hartman M. T. In Ribosome Display and Related Technologies; Douthwaite J. A., Jackson R. H., Eds.; Springer: New York, 2012; Methods Mol. Biol., Vol. 805, pp 367–390. [Google Scholar]
- Subtelny A. O.; Hartman M. C. T.; Szostak J. W. J. Am. Chem. Soc. 2008, 130, 6131–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi H.; Shimizu Y.; Ying B.-W.; Ueda T. Biochem. Biophys. Res. Commun. 2007, 352, 270–276. [DOI] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. G. Angew. Chem., Int. Ed. 2009, 48, 9879–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kurz M.; Gu K.; Lohse P. A. Nucleic Acids Res. 2000, 28, e83. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seelig B. Nat. Protoc. 2011, 6, 540–552. [DOI] [PubMed] [Google Scholar]
- Liu R. H.; Barrick J. E.; Szostak J. W.; Roberts R. W.. Rna-Ligand Interactions, Part B; Academic Press Inc.: San Diego, CA, 2000; Methods Enzymol., Vol. 318, pp 268–293. [Google Scholar]
- Chan W. C. W., Peter D.. Fmoc Solid Phase Peptide Synthesis; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.