

Abstract

The direct decarboxylative arylation of α-amino acids has been achieved via visible light-mediated photoredox catalysis. This method offers rapid entry to prevalent benzylic amine architectures from an abundant biomass, specifically α-amino acid precursors. Significant substrate scope is observed with respect to both the amino acid and arene components.
The controlled excision of carbon dioxide from organic molecules, commonly known as decarboxylation, is a valuable transformation that is fundamental to biochemistry and broadly utilized in synthetic molecule construction. For example, in nature, enzymatically controlled regioselective decarboxylation allows the conversion of the abundant biomonomer glutamic acid to γ-aminobutyric acid (GABA), an important neurotransmitter in the mammalian central nervous system (eq 1).1 Synthetic chemists also rely on decarboxylative technologies as a means to remove extraneous carboxyl groups2 with protocols such as the Kolbe,3 Hunsdiecker,4 and Barton decarboxylations5 having long-proven value.
Given the worldwide abundance of biomass6 that incorporates carboxylate functionality (e.g., amino acids, α-hydroxy acids, fatty acids etc), there is a strong impetus to invent or discover chemical transformations that target carboxylic acids as chemoselective leaving groups for new and valuable C–C bond forming reactions. More specifically, the productive engagement of decarboxylation mechanisms into fragment-coupling bond constructions could enable the direct conversion of abundant, inexpensive substrates to complex, medicinally relevant pharmacophores. The seminal studies of Myers7 and Gooßen8 have demonstrated that classical organometallic catalytic coupling pathways (Heck, biaryl couplings) can be accessed via aryl and alkynyl carboxylic acids.9 Recently, we questioned whether Csp3–Csp2-bearing carboxylates (the carboxyl moiety most commonly found in biomass) might be rendered suitable for decarboxylative aryl couplings via the use of a novel photoredox-mediated radical–radical coupling pathway. Herein we describe the successful execution of these studies and demonstrate the direct conversion of broadly available α-amino acids or α-oxy acids with arene substrates to generate high-value benzylic amine or ether architectures using photoredox catalysis.
The benzylic amine is a central component of a wide range of medicinally relevant compounds, including pharmaceuticals, agrochemicals, and bioactive natural products.10 The development of improved methods for the synthesis of these important pharmacophores from readily available starting materials remains an important challenge in organic synthesis.11 A recent focus of our laboratory has been the development of visible light-mediated photoredox catalysis as a general strategy for achieving challenging bond constructions not readily accessible via classical two-electron pathways and without the need for harsh conditions or toxic reagents.12,13 Along these lines, we recently described a photoredox-catalyzed amine C–H arylation for the synthesis of benzylic amines via the coupling of tertiary amines with cyanoarenes.12 With the goal of further expanding the menu of amine α-functionalization capabilities, we anticipated that α-amino acid radical decarboxylation, coupled with in situ arylation, could provide rapid access to a wealth of complex primary and secondary benzylic amine pharmacophores using readily available biomass.
 |
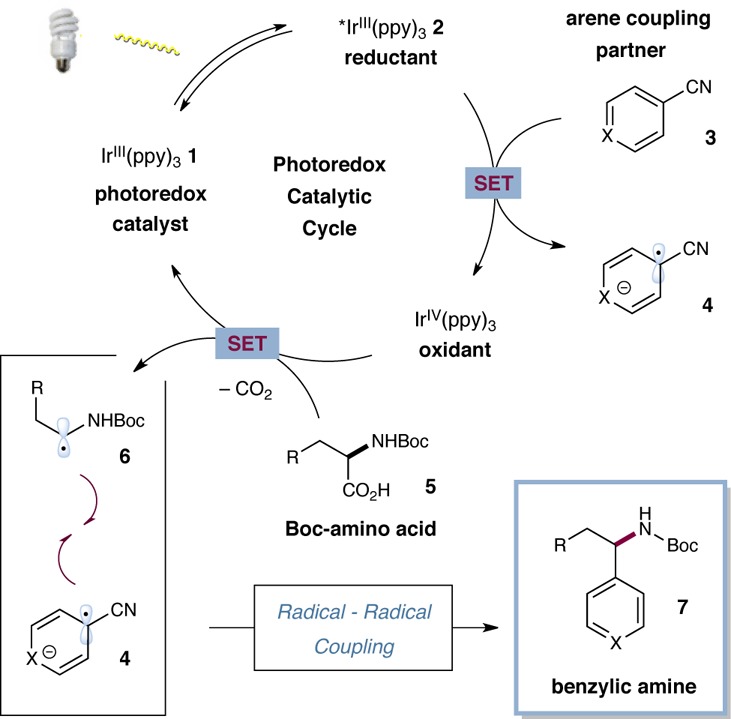
Design Plan. As outlined in eq 2, our general strategy envisioned photoredox-mediated single-electron oxidation of the carboxylate functionality of an α-amino acid, followed by loss of CO2, to render an α-carbamoyl radical. Radical–radical coupling of this species with the radical anion derived from an arene coupling partner should forge the benzylic C–C bond to provide, following aromatization via expulsion of cyanide, the high-value benzylic amine motif.
A detailed mechanism for the proposed photoredox decarboxylative aryl coupling is shown in Scheme 1. Irradiation of tris(2-phenylpyridinato-C2,N)iridium(III) [Ir(ppy)3] (1) with visible light produces a long-lived (1.9 μs) photoexcited state,14 *Ir(ppy)3 (2), which can be readily oxidized or reduced by an appropriate substrate quencher. Given that *Ir(ppy)3 (2) is a strong reductant (E1/2red [IrIV/*IrIII] = −1.73 V vs SCE),14 we presumed that facile reduction of 1,4-dicyanobenzene 3 (X = C–CN, E1/2red = −1.61 V vs SCE)15 via single electron transfer would generate the corresponding aryl radical anion 4.16 At this stage, oxidation of the α-amino acid substrate by IrIV(ppy)3 to generate the corresponding carboxyl radical species (with subsequent extrusion of CO2) would yield the requisite α-amino radical 6. A survey of relevant oxidation potentials reveals that this carboxylate oxidation step is endergonic as proposed (E1/2red [IrIV/IrIII] = +0.77 V vs SCE), (Boc-Pro-OCs+, Ered = +0.95 V vs SCE);17 however, we recognized that electronic adjustment of the photocatalyst ligands might render this decarboxylation thermodynamically feasible. At this stage we hoped that chemoselective heterocoupling of the persistent radical 4 with the transiently formed α-amino radical 6 would efficiently enable the requisite C–C fragment coupling, and subsequent loss of cyanide would deliver the benzylic amine 7.
Scheme 1. Proposed Catalytic Cycle for Decarboxylative Arylation.
Our initial investigations focused on the identification of a suitable photocatalyst capable of facilitating both single-electron reduction of the arene and single-electron oxidation of the α-amino acid (Table 1). We first examined the coupling of Boc-protected proline and 1,4-dicyanobenzene (1,4-DCB) at room temperature in the presence of K2HPO4, a 26W household fluorescent light bulb, and 2 mol% of Ir(ppy)3. While we were delighted to find that the proposed decarboxylative arylation coupling was feasible under these conditions, the observed reaction efficiency was poor (entry 1, 12% yield). As stated above, we recognized that electron transfer from Boc-prolinate to IrIV(ppy)3 (E1/2red [IrIV/IrIII]= +0.77 V vs SCE)14 could be thermodynamically challenging, and as such, we turned our attention to the identification of more strongly oxidizing photocatalysts. As expected, incorporation of a fluorine substituent onto the pyridine backbone of the photocatalyst provided a significant increase in efficiency. More specifically the Ir(p-F-ppy)3 and Ir(dFppy)3 photocatalysts (E1/2red [IrIV/IrIII] = +0.97 V and +1.13 V vs SCE in CH3CN, respectively)18 provided significantly enhanced yields of the desired α-amino arylation product (entries 2–3). It is important to note that highly oxidizing photocatalysts, such as Ir[dF(CF3)ppy]2(dtbbpy)PF6,19 were largely ineffective in this coupling protocol (entry 4), presumably due to their inability to participate in the arene reduction step of the catalytic cycle.
Table 1. Optimization of the Decarboxylative Arylation.

| entry | photocatalyst | base | % yielda |
|---|---|---|---|
| 1 | Ir(ppy)3 | K2HPO4 | 12 |
| 2 | Ir(p-F-ppy)3 | K2HPO4 | 58 |
| 3 | Ir(dFppy)3 | K2HPO4 | 54 |
| 4 | Ir[dF(CF3)ppy]2(dtbbpy) | K2HPO4 | trace |
| 5 | Ir[p-F(t-Bu)-ppy]3 | K2HPO4 | 73 |
| 6 | Ir[dF(t-Bu)-ppy]3 | K2HPO4 | 68 |
| 7 | Ir[p-F(t-Bu)-ppy]3 | CsF | 83 |
| 8b | Ir[p-F(t-Bu)-ppy]3 | CsF | 0 |
| 9 | none | CsF | 0 |
| 10 | Ir[p-F(t-Bu)-ppy]3 | none | trace |
Yield determined by 1H NMR using an internal standard.
Reaction performed in the absence of visible light.
A series of Ru-based catalysts were unsuccessful.
High-dielectric solvents are required (e.g., entry 7, DMSO = 83% yield, DMA = 74% yield, hexane = NR).
On the basis of these findings, we sought to design a photocatalyst system that would be more strongly reducing than Ir(p-F-ppy)3 (E1/2red [IrIV/*IrIII] = −1.60 V vs SCE) or Ir(dFppy)3 (E1/2red [IrIV/*IrIII] = −1.44 V vs SCE),18 yet at the same time could promote facile oxidation of the α-amino carboxylate moiety. We hypothesized that introduction of electron-donating tert-butyl groups into the pyridine ring of the ligand would serve to further enhance the reducing ability of the photocatalyst excited state while leaving the oxidation potential of the corresponding IrIV species largely unaffected. Indeed, we were pleased to find that two new photocatalysts, Ir[p-F(tBu)-ppy]3 (E1/2red [IrIV/*IrIII] = −1.67 V vs SCE)20 and Ir[dF(tBu)-ppy]3, exhibited a significant increase in overall efficiency to deliver the desired coupling adduct in 73% and 68% yield, respectively (entries 5 and 6). Further improvement in reaction yield was achieved through the use of CsF as the exogenous base, presumably due to increased solubility of the carboxylate species (entry 7). As expected, control experiments revealed that the photocatalyst, visible light, and the base were all essential components for the catalytic operation of this new coupling reaction (entries 8–10).
With optimal reaction conditions in hand, we next examined the scope of the amino acid component in this new decarboxylative arylation protocol. As illustrated in Table 2, this transformation generically proceeds at room temperature and in good to high yields with an extensive range of α-amino acids (products 8–20, 64–89% yield). These mild reaction conditions readily accommodate an array of useful functional groups, including esters, amides, carbamates, and ethers (products 13, 14, 18, 64–78% yield). Moreover, modification of the α-amino acid protecting group is well-tolerated between different carbamate systems (products 8 and 9, 75 and 82% yield, respectively). A wide range of acyclic α-amino acids incorporating aliphatic and aryl side chains are amenable to this decarboxylative coupling, furnishing benzylic amines in good to excellent yield (products 12–20, 64–89% yield). Remarkably, Boc-protected methionine, which is often prone to sulfide-oxidation, was a viable substrate, providing the corresponding arylation adduct in high yield (product 15, 81% yield). In addition, we have also demonstrated that unnatural α-amino acids, such as piperidine and morpholine-derived systems, are amenable to this decarboxylative arylation (products 10 and 11, 70 and 86% yield, respectively). Finally, and perhaps most notably, this transformation readily promotes C–C bond formation at sterically congested positions, an often underappreciated benefit of radical–radical couplings and their accompanying elongated transition-state bonds (product 19, 81% yield).21
Table 2. Decarboxylative Arylation: α-Amino Acid Scopea,b.

Performed using the optimized conditions from Table 1 (See Supporting Information).
Cited yields are of material isolated by column chromatography.
Unprotected indole Boc-Trp-OH resulted in lower efficiency (54% yield).
Determined by 1H NMR analysis to be 1.1:1 d.r.
We next evaluated the nature of the aryl coupling partner in this photoredox-mediated decarboxylative coupling. As shown in Table 3, a range of cyanoaromatics and cyano-heteroaromatics are able to participate in the requisite radical–radical coupling with good to excellent efficiency (entries 1–8, 52–85% yield). As detailed earlier, sterically encumbered substrates, such as 2,5-dimethylterephthalonitrile, are viable coupling partners (entry 1, 85% yield). Moreover, these mild reaction conditions allow a broad range of functional groups on the aryl ring (e.g., esters, lactones, phosphonate esters, and sulfones) to be readily accommodated (entries 3–6, 52–70% yield). With respect to pharmacophore production, it is important to note that this decarboxylative arylation is readily translatable to heteroaromatic group installation. For example, cyanopyridines serve as viable coupling partners to generate heterobenzylic amines in one step from amino acids (entries 7 and 8, 73–83% yield).
Table 3. Decarboxylative Arylation: Aromatic Scopea,b.


Cited yields are of material isolated by column chromatography. See Supporting Information for experimental details.
Regiomeric ratios (rr) determined by 1H NMR (major isomer is shown).
1.3:1 rr.
4:1 chemoselectivity for addition to ipso-CN vs SO2Ph position.
A defining aspect of this new decarboxylative functionalization protocol is its potential to provide direct access to a broad array of Csp3–Csp2 coupled arylation adducts from biomass-derived precursors. In this context, one particular challenge is the chemoselective decarboxylative functionalization of cyclic and acyclic α-oxygenated acids, moieties that are often found in the molecular skeletons of naturally occurring and biologically active substances. As shown in eq 3, exposing 2-tetrahydrofuroic acid to our optimized coupling conditions in the presence of 1,4-dicyanobenzene resulted in the desired arylation adduct in good yield, and notably, without oxidation to the corresponding furanyl species. Moreover, treatment of 2-methoxy-phenylacetic acid with 4-cyanopyridine in the presence of our photocatalyst and light led to the direct production of the corresponding heteroaryl ether with high levels of efficiency (eq 4).
 |
Acknowledgments
Financial support was provided by NIHGMS (R01 01 GM093213-01) and gifts from Merck and Amgen. Z.Z. is grateful for a postdoctoral fellowship from the Shanghai Institute of Organic Chemistry. We thank Dr. Yong Yan for his generous help with cyclic voltammetry experiments.
Supporting Information Available
Experimental procedures, structural proofs, and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lehninger A. L.; Nelson D. L.; Cox M. M.. Principles of Biochemistry; Worth Publishers: New York, 1993. [Google Scholar]; b Walsh C.Enzymatic Reaction Mechanisms; W. H. Freeman and Co.: New York, 1979. [Google Scholar]; c van Poelje P. D.; Snell E. E. Annu. Rev. Biochem. 1990, 59, 29. [DOI] [PubMed] [Google Scholar]
- a Vijh A. K.; Conway B. E. Chem. Rev. 1967, 67, 623. [Google Scholar]; b Johnson R. G.; Ingham R. K. Chem. Rev. 1956, 56, 219. [Google Scholar]; c Sheldon R. A.; Kochi J. K. Org. React. 1972, 19, 279. [Google Scholar]; d Barton D. H. R.; Crich D.; Motherwell W. B. Tetrahedron 1985, 41, 3901. [Google Scholar]
- Kolbe H. Liebigs Ann. Chem. 1849, 69, 257. [Google Scholar]
- Hunsdiecker H.; Hunsdiecker C. Chem. Ber. 1942, 75B, 291. [Google Scholar]
- Barton D. H. R.; Serebryakov E. P. Proc. Chem. Soc. 1962, 309. [Google Scholar]
- For examples of the use of biomass conversion to non-fuel, high-value chemical products see:Scott E.; Peter F.; Sanders J. Appl. Microbiol. Biotechnol. 2007, 75, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A. G.; Tanaka D.; Mannion M. R. J. Am. Chem. Soc. 2002, 124, 11250. [DOI] [PubMed] [Google Scholar]
- Gooßen L. J.; Deng G.; Levy L. M. Science 2006, 313, 662. [DOI] [PubMed] [Google Scholar]
- a Gooßen L. J.; Rodriguez N.; Gooßen K. Angew. Chem., Int. Ed. 2008, 47, 3100. [DOI] [PubMed] [Google Scholar]; b Cornella J.; Larrosa I. Synthesis 2012, 44, 653. [Google Scholar]; c Dzik W. I.; Lange P. P.; Gooßen L. J. Chem. Sci. 2012, 3, 2671. [Google Scholar]
- a Lawrence S. A.Amines: Synthesis, Properties, and Applications; Cambridge University Press: Cambridge, 2004. [Google Scholar]; b Lewis J. R. Nat. Prod. Rep. 2001, 95. [DOI] [PubMed] [Google Scholar]; c Lin N.-H.; Carrera G. M.; Anderson D. J. J. Med. Chem. 1994, 37, 3542. [DOI] [PubMed] [Google Scholar]
- a Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C. J. Am. Chem. Soc. 2006, 128, 3538. [DOI] [PubMed] [Google Scholar]; b Coldham I.; Leonori D. Org. Lett. 2008, 10, 3923. [DOI] [PubMed] [Google Scholar]; c Beng T. K.; Gawley R. E. Org. Lett. 2011, 13, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Seel S.; Thaler T.; Takatsu K.; Zhang C.; Zipse H.; Straub B. F.; Mayer P.; Knochel P. J. Am. Chem. Soc. 2011, 133, 4774. [DOI] [PubMed] [Google Scholar]
- a Nicewicz D.; MacMillan D. W. C. Science 2008, 322, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagib D. A.; Scott M. E.; MacMillan D. W. C. J. Am. Chem. Soc. 2009, 131, 10875. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shih H.-W.; Vander Wal M. N.; Grange R. L.; MacMillan D. W. C. J. Am. Chem. Soc. 2010, 132, 13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]; c Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. [DOI] [PubMed] [Google Scholar]; d Teply F. Collect. Czech. Chem. Commun. 2011, 76, 859. [Google Scholar]
- a Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F. Top. Curr. Chem. 2007, 281, 143. [Google Scholar]; b Dixon I. M.; Collin J.; Sauvage J.; Flamigni L.; Encinas S.; Barigelletti F. Chem. Soc. Rev. 2000, 29, 385. [Google Scholar]
- Mori Y.; Sakaguchi W.; Hayashi H. J. Phys. Chem. A 2000, 104, 4896. [Google Scholar]
- Studer A. Chem.—Eur. J. 2001, 7, 1159. [DOI] [PubMed] [Google Scholar]
- The reduction potential of Boc-Pro-OCs+ was measured in acetonitrile following the methods in; a Andrieux C. P.; Gonzalez F.; Saveant J. J. Electroanal. Chem. 2001, 498, 171. [Google Scholar]; b Galicia M.; Gonzalez F. J. J. Electrochem. Soc. 2002, 149, D46. [Google Scholar]
- a Dedeian K.; Djurovich P. I.; Garces F. O.; Carlson G.; Watts R. J. Inorg. Chem. 1991, 30, 1685. [Google Scholar]; b Tian N.; Lenkeit D.; Pelz S.; Fischer L. H.; Escudero D.; Schiewek R.; Klink D.; Schmitz O. J.; Gonzalez L.; Schaferling M.; Holder E. Eur. J. Inorg. Chem. 2010, 4875. [Google Scholar]; The excited-state potentials are typically calculated from ground-state potentials and zero–zero spectroscopic energy of the excited state, see:; c Balzani V.; Bolletta F.; Gandolfi M. T.; Maestri M. Top. Curr. Chem. 1978, 75, 1. [Google Scholar]
- Lowry M. S.; Goldsmith J. I.; Slinker J. D.; Rohl R.; Pascal R. A.; Malliaras G.; Bernhard S. Chem. Mater. 2005, 17, 5712. [Google Scholar]
- The reduction potential of Ir[p-F(tBu)-ppy]3 was determined by cyclic voltammetry. Stern–Volmer quenching studies revealed only 1,4-dicyanobenzene quenches the excited state of Ir[p-F(tBu)-ppy]3, lending support to our initial hypothesis in Scheme 1. See Supporting Information for details on luminescence quenching studies.
- a Damm W.; Giese B.; Hartung J.; Hasskerl T.; Houk K. N.; Hueter O.; Zipse H. J. Am. Chem. Soc. 1992, 114, 4067. [Google Scholar]; b Arnaud R.; Postlethwaite H.; Barone V. J. Phys. Chem. 1994, 98, 5913. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.