

Abstract
OGlcNAcylation and phosphorylation are the major competing intracellular post-translational modifications of serine and threonine residues. The structural effects of both post-translational modifications on serine and threonine were examined within Baldwin model α-helical peptides (Ac-AKAAAAKAAAAKAAGY-NH2 or Ac-YGAKAAAAKAAAAKAA-NH2). At the N-terminus of an α-helix, both phosphorylation and OGlcNAcylation stabilized the α-helix relative to the free hydroxyls, with a larger induced structure for phosphorylation than for OGlcNAcylation, for the dianionic phosphate than for the monoanionic phosphate, and for modifications on threonine than for modifications on serine. Both phosphoserine and phosphothreonine resulted in peptides more α-helical than alanine at the N-terminus, with dianionic phosphothreonine the most α-helix-stabilizing residue here. In contrast, in the interior of the α-helix, both post-translational modifications were destabilizing with respect to the α-helix, with the greatest destabilization seen for threonine OGlcNAcylation at residue 5 and threonine phosphorylation at residue 10, with peptides containing either post-translational modification existing as random coils. At the C-terminus, both OGlcNAcylation and phosphorylation were destabilizing with respect to the α-helix, though the induced structural changes were less than in the interior of the α-helix. In general, the structural effects of modifications on threonine were greater than the effects on serine, because of both the lower α-helical propensity of Thr and the more defined induced structures upon modification of threonine than serine, suggesting threonine residues are particularly important loci for structural effects of post-translational modifications. The effects of serine and threonine post-translational modifications are analogous to the effects of proline on α-helices, with the effects of phosphothreonine being greater than those of proline throughout the α-helix. These results provide a basis for understanding the context-dependent structural effects of these competing protein post-translational modifications.
Phosphorylation and OGlcNAcylation are the major reversible intracellular post-translational modifications of serine and threonine residues. Phosphorylation in humans is controlled by more than 650 protein kinases and protein phosphatases, which provide exquisite control over site-specific phosphorylation at numerous sites in proteins, with the majority of intracellular proteins subject to phosphorylation.1 In contrast, in humans, OGlcNAcylation (β-O-GlcNAc modification of serine or threonine, via addition of β-d-N-acetylglucosamine) is controlled by only one OGlcNAc transferase (OGT), with three isoforms in humans, which adds OGlcNAc, and only one OGlcNAcase (OGA), which removes OGlcNAc (Figure 1).2,3 Despite this more limited set of enzymes for the addition and removal of β-O-GlcNAc, hundreds of intracellular proteins have been positively identified as being modified by OGlcNAcylation, with OGlcNAc cycling on serine and threonine residues on time scales similar to those of phosphorylation. OGlcNAcylation is responsive to energy homeostasis and nucleotide, amino acid, and lipid availability, with sugar transfer via UDP-OGlcNAc, providing responsiveness to metabolism, stress, and nutrient access.2 Interestingly, OGlcNAcylation often occurs at sites of phosphorylation, with the effects of OGlcNAcylation sometimes opposing those of phosphorylation, and sometimes having effects that are similar to those of phosphorylation.4,5 Competing phosphorylation and OGlcNAcylation appear to be particularly important in transcription, including modifications of the RNA polymerase II C-terminal domain, c-myc, CREB, C/EBP, and the estrogen receptor, among other examples.3,6−11
Figure 1.
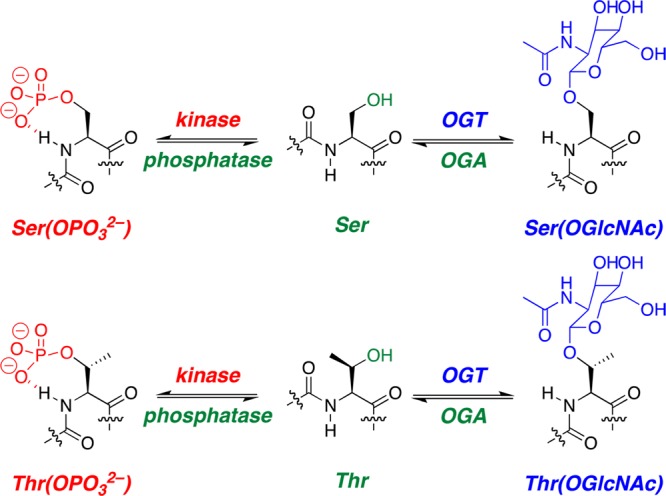
Phosphorylation and OGlcNAcylation are dynamic intracellular post-translational modifications of serine and threonine residues of proteins. OGT denotes O-GlcNAc transferase and OGA O-GlcNAcase.
To provide a structural basis for understanding some of the effects of phosphorylation versus OGlcNAcylation, we examine herein the effects of phosphorylation versus OGlcNAcylation within the α-helix, the most common protein secondary structure. The effects of phosphorylation on α-helical structure have been examined in a number of contexts, including model α-helical peptides, coiled coil peptides, and designed peptides.12−19 The effects of phosphorylation on structure have also been examined in random coil, polyproline II helix (PPII), and β-hairpin contexts, as well as within globular proteins.20−33
In model α-helices, Doig found that phosphoserine (pSer) stabilized the α-helix when at or near the N-terminus (N-cap or first three residues of the α-helix) and destabilized the α-helix when in the interior.14 Doig also found that phosphoserine could be stabilizing in the interior of an α-helix when phosphoserine was located with an i, i + 4 relationship with respect to a lysine residue.16 No examination of the effects of threonine phosphorylation, or of modification at the C-terminus of the α-helix, was conducted in these critical initial studies. In a model peptide and in a designed four-helix bundle, De Grado found that an Arg-X-pSer sequence was highly stabilizing at the N-terminus of the α-helix, with phosphorylation at all four sites of the tetramer resulting in a significant stabilization of the helical bundle relative to the nonphosphorylated bundle.15 In contrast, Vinson found substantial destabilization of a coiled coil upon phosphorylation at an interior helical position, with greater destabilization due to phosphothreonine (pThr) than phosphoserine.12 Interestingly, when the internal serine was placed within the structural context of multiple arginine residues, serine phosphorylation was found to be stabilizing to the coiled coil, although notably phosphothreonine here was still destabilizing.13 No structural basis for the differential effects of serine phosphorylation versus threonine phosphorylation was provided beyond the α-helical propensity of threonine being lower than that of serine. These effects in model peptides, with phosphorylation generally stabilizing at the N-terminus and generally destabilizing in the interior of an α-helix unless phosphoserine is in the vicinity of positively charged residues, are consistent with data for several native proteins subject to phosphorylation.8,34−36
In contrast, relatively few data exist on the effects of OGlcNAcylation on structure, or on the direct comparison of the effects of phosphorylation and OGlcNAcylation. In the RNA polymerase II C-terminal domain repeat (SYSPTSP), phosphorylation of serine residues induces binding to the Pin1 WW domain as a polyproline helix, while OGlcNAcylation of Thr induces a β-turn conformation.37−39 In the disordered N-terminus of the estrogen receptor, OGlcNAcylation also induces a β-turn formation, whereas phosphorylation of the same residue opposes the turn conformation.8 Within the loop region between two α-helices of a designed α-helical hairpin, Chan found that both phosphorylation and OGlcNAcylation reduced the rate of fibril formation, even though neither modification appreciably affected the structure of the soluble protein or its Tm.40 Waters has demonstrated in model β-hairpin peptides that phosphorylation disrupts β-hairpin formation, whereas OGlcNAcylation at a different residue stabilizes the β-hairpin, both via native effects of the post-translational modifications on the β conformation and via either repulsive (phosphorylation) or attractive (OGlcNAc) interactions with a cross-strand tryptophan residue.24−26,41 In the only examination of the effects of phosphorylation and glycosylation on α-helicity, Koksch, Hackenberger, and co-workers found that phosphorylation and glycosylation (though here β-galactose, not β-OGlcNAc; others have found substantial structural differences between modification with a sugar and the N-acetyl-amino sugar analogue42) were destabilizing in the solvent-exposed central sequence of a coiled coil, although a single O-galactose could be incorporated with no effect on α-helicity.19
Recently, we examined the effects of phosphorylation and OGlcNAcylation on the structure of peptides from the proline-rich domain of the protein tau, in which hyperphosphorylation is associated with formation of the neurofibrillary tangles (NFT) observed in Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE), and other tauopathies, while OGlcNAcylation is protective against NFT formation.7,21,29,43,44 In that study, we found that, within proline-rich sequences, phosphorylation induces polyproline helix while OGlcNAcylation opposes polyproline helix, with the structural effects of threonine phosphorylation being greater than those of serine phosphorylation and the structural effects of phosphorylation being greater than those of OGlcNAcylation.21,29 Particular conformational restriction was observed at dianionic phosphothreonine residues, with highly restricted 3JαN coupling constants (mean 3JαN = 3.5 Hz across eight phosphothreonine residues in six peptides; 3JαN can be corresponded to the ϕ backbone torsion angle through a parametrized Karplus relationship45) and evidence of a stable hydrogen bond between phosphothreonine and its own amide hydrogen. Interestingly, in that study, one peptide, tau196–209, exhibited nascent α-helical structure. In that peptide, which contains PGSPG(S/T) repeats observed as α-helix nucleation sites in the Protein Data Bank (PDB), we found that both phosphorylation and OGlcNAcylation had similar structural effects, with both post-translational modifications opposing α-helix formation. These results suggest a context dependence of the structural effects of phosphorylation and OGlcNAcylation, consistent with the observation that at some sites these modifications have opposing functional effects whereas at other sites these modifications have similar functional effects. Therefore, we sought to conduct a rigorous examination of the effects of phosphorylation and OGlcNAcylation of serine and threonine on α-helicity, within a well-controlled model peptide system, as a function of helical position, to provide a structural basis for understanding functional effects of the major intracellular serine and threonine post-translational modifications.
Results
A series of peptides was synthesized on the basis of model Baldwin α-helical peptides, Ac-(YG)AKAAAAKAAAAKAA(GY)-NH2 (Figure 2), with the Tyr added for concentration determination and located at the N-terminus (for peptides with C-terminal modifications) or C-terminus (all other peptides) as appropriate to avoid interactions with the sites of modification. Serine and threonine residues were introduced at the N-terminal amino acid (residue 1) (Ac-XKAAAAKAAAAKAAGY-NH2), at an internal amino acid (residue 5) (Ac-AKAAXAKAAAAKAAGY-NH2), and at the C-terminal amino acid (residue 14, counting from the first Ala to employ consistent numbering across all peptides) (Ac-YGAKAAAAKAAAAKAX-NH2) to examine the effects of post-translational modifications at the N-terminus, in the interior, and at the C-terminus of an α-helix (Figure 2). All positions were chosen to avoid potential i, i + 4 interactions with lysine residues.16 Peptides with serine, phosphoserine, threonine, and phosphothreonine residues were synthesized via trityl-protected serine and threonine, followed by trityl deprotection, phosphitylation, oxidation, and peptide cleavage/deprotection, yielding the peptides with site-specifically phosphorylated amino acids.17,21 OGlcNAcylated peptides were synthesized using the protected Fmoc-Ser/Thr-(Ac3OGlcNAc)-OH, synthesized via a modification of the method of Arsequell, followed by initial peptide purification, O-deacetylation, and final peptide purification, yielding peptides site-specifically incorporating a single Ser or Thr OGlcNAc.29,46,47 All peptides were examined by circular dichroism at 0.5 °C (Figures 3–12 and Table 1). Peptides were also examined by nuclear magnetic resonance (NMR) to determine residue-specific structural changes (Tables 2 and 3 and Supporting Information).48,49
Figure 2.

Sequences of peptides examined in this study. Top: the alanine-rich model α-helical peptide. Middle: peptides with modifications at the N-terminal residue (residue 1), at a central residue (residue 5), and at the C-terminal residue (residue 14; numbering based on other peptides, using the first Ala as residue 1, excluding the N-terminal YG) of a model α-helix. GY or YG sequences were added for concentration determination. The peptide with the C-terminal modifications employed the YG at the N-terminus to avoid potential interaction of the post-translational modification with the tyrosine. Bottom: peptides examined for the effects of phosphorylation at residues 2 and 10. Peptides were also synthesized with proline in place of serine or threonine at residues 1, 2, 5, 10, and 14.
Figure 3.
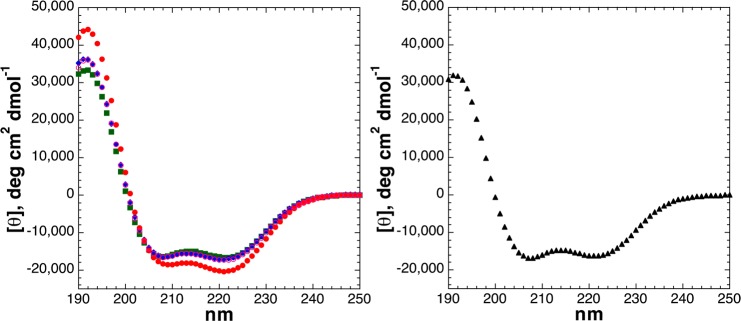
CD spectra of Ac-SKAAAAKAAAAKAAGY-NH2 peptides with serine modifications [unmodified Ser (free hydroxyl), SerOGlcNAc, SerOPO3H– (pH 4), and SerOPO32– (pH 8)] (left) and with Ser replaced with Ala (right): unmodified Ser (green squares), dianionic phosphoserine (pH 8) (red circles), monoanionic phosphoserine (pH 4) (open magenta circles), SerOGlcNAc (blue diamonds), and Ala (black triangles). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C. Data are the average of at least three independent trials. Data were background corrected but were not smoothed. Individual CD spectra of all peptides, with error bars shown, are given in the Supporting Information.
Figure 12.

Comparison of the effects of threonine post-translational modifications as a function of α-helical position: (a) threonine, (b) phosphothreonine (pH 8), and (c) Thr(OGlcNAc). Red denotes modifications at residue 1, orange those at residue 2, green those at residue 5, cyan those at residue 10, and blue those at residue 14. Each plot in this figure includes the CD data of a series of isomeric peptides (N1, N5, N10, and N14).
Table 1. Summary of CD Data for All Peptidesa.
| peptide | [θ]222 | [θ]208 | [θ]190 | [θ]222/[θ]208 | –[θ]190/[θ]208 | % helix |
|---|---|---|---|---|---|---|
| Ac-SerKAAAAKAAAAKAAGY-NH2 | –16621 | –16434 | 32243 | 1.01 | 1.96 | 49.7 |
| Ac-Ser(OGlcNAc)KAAAAKAAAAKAAGY-NH2 | –16944 | –16358 | 35272 | 1.04 | 2.16 | 50.6 |
| Ac-Ser(OPO3H–)KAAAAKAAAAKAAGY-NH2 | –17030 | –16635 | 33901 | 1.02 | 2.04 | 50.8 |
| Ac-Ser(OPO32–)KAAAAKAAAAKAAGY-NH2 | –19993 | –18384 | 42092 | 1.09 | 2.29 | 58.6 |
| Ac-ThrKAAAAKAAAAKAAGY-NH2 | –14863 | –15423 | 27161 | 0.96 | 1.76 | 45.1 |
| Ac-Thr(OGlcNAc)KAAAAKAAAAKAAGY-NH2 | –17267 | –17120 | 32808 | 1.01 | 1.92 | 51.4 |
| Ac-Thr(OPO3H–)KAAAAKAAAAKAAGY-NH2 | –16661 | –15997 | 33161 | 1.04 | 2.07 | 49.8 |
| Ac-Thr(OPO32–)KAAAAKAAAAKAAGY-NH2 | –20495 | –18290 | 43519 | 1.12 | 2.38 | 60.0 |
| Ac-ASerAAAAKAAAAKAAGY-NH2 | –16650 | –16246 | 32311 | 1.02 | 1.99 | 49.8 |
| Ac-ASer(OPO3H–)AAAAKAAAAKAAGY-NH2 | –19142 | –17756 | 39137 | 1.08 | 2.20 | 56.4 |
| Ac-ASer(OPO32–)AAAAKAAAAKAAGY-NH2 | –22401 | –19376 | 47754 | 1.16 | 2.46 | 65.0 |
| Ac-AThrAAAAKAAAAKAAGY-NH2 | –15248 | –15328 | 29238 | 0.99 | 1.91 | 46.1 |
| Ac-AThr(OPO3H–)AAAAKAAAAKAAGY-NH2 | –19076 | –17383 | 34079 | 1.10 | 1.96 | 56.2 |
| Ac-AThr(OPO32–)AAAAKAAAAKAAGY-NH2 | –23977 | –20776 | 50682 | 1.15 | 2.44 | 69.2 |
| Ac-AKAASerAKAAAAKAAGY-NH2 | –9489 | –12001 | 13623 | 0.79 | 1.14 | 30.9 |
| Ac-AKAASer(OGlcNAc)AKAAAAKAAGY-NH2 | –7362 | –10510 | 9108 | 0.70 | 0.87 | 25.3 |
| Ac-AKAASer(OPO3H–)AKAAAAKAAGY-NH2 | –5233 | –8782 | 3170 | 0.60 | 0.36 | 19.6 |
| Ac-AKAASer(OPO32–)AKAAAAKAAGY-NH2 | –5019 | –9115 | 25 | 0.55 | 0.00 | 19.1 |
| Ac-AKAAThrAKAAAAKAAGY-NH2 | –4593 | –8386 | 727 | 0.55 | 0.09 | 17.9 |
| Ac-AKAAThr(OGlcNAc)AKAAAAKAAGY-NH2 | –145 | –6835 | –14035 | 0.02 | –2.05 | 6.2 |
| Ac-AKAAThr(OPO3H–)AKAAAAKAAGY-NH2 | –1868 | –6555 | –5647 | 0.28 | –0.86 | 10.7 |
| Ac-AKAAThr(OPO32–)AKAAAAKAAGY-NH2 | –2742 | –8292 | –6777 | 0.33 | –0.82 | 13.0 |
| Ac-AKAAAAKAASerAKAAGY-NH2 | –7883 | –11530 | 8113 | 0.68 | 0.70 | 26.6 |
| Ac-AKAAAAKAASer(OPO3H–)AKAAGY-NH2 | –2936 | –8145 | –5752 | 0.36 | –0.71 | 13.6 |
| Ac-AKAAAAKAASer(OPO32–)AKAAGY-NH2 | –431 | –6789 | –14930 | 0.06 | –2.20 | 6.9 |
| Ac-AKAAAAKAAThrAKAAGY-NH2 | –6020 | –10003 | 3732 | 0.60 | 0.37 | 21.7 |
| Ac-AKAAAAKAAThr(OPO3H–)AKAAGY-NH2 | 382 | –5396 | –13528 | –0.07 | –2.51 | 4.8 |
| Ac-AKAAAAKAAThr(OPO32–)AKAAGY-NH2 | 1967 | –5542 | –18077 | –0.35 | –3.26 | 0.6 |
| Ac-YGAKAAAAKAAAAKASer-NH2 | –11375 | –13477 | 20103 | 0.84 | 1.49 | 35.9 |
| Ac-YGAKAAAAKAAAAKASer(OGlcNAc)-NH2 | –8461 | –11840 | 12910 | 0.71 | 1.09 | 28.2 |
| Ac-YGAKAAAAKAAAAKASer(OPO3H–)-NH2 | –5630 | –9603 | 4445 | 0.59 | 0.46 | 20.7 |
| Ac-YGAKAAAAKAAAAKASer(OPO32–)-NH2 | –3734 | –8814 | –2015 | 0.42 | –0.23 | 15.7 |
| Ac-YGAKAAAAKAAAAKAThr-NH2 | –9190 | –12024 | 13630 | 0.76 | 1.13 | 30.1 |
| Ac-YGAKAAAAKAAAAKAThr(OGlcNAc)-NH2 | –6685 | –11064 | 5746 | 0.60 | 0.52 | 23.5 |
| Ac-YGAKAAAAKAAAAKAThr(OPO3H–)-NH2 | –4984 | –9420 | 1437 | 0.53 | 0.15 | 19.0 |
| Ac-YGAKAAAAKAAAAKAThr(OPO32–)-NH2 | –3417 | –8524 | –2248 | 0.40 | –0.26 | 14.8 |
| Ac-AKAAAAKAAAAKAAGY-NH2 | –16325 | –16775 | 30780 | 0.97 | 1.83 | 48.9 |
| Ac-ProKAAAAKAAAAKAAGY-NH2 | –12026 | –13891 | 16191 | 0.87 | 1.17 | 37.6 |
| Ac-AProAAAAKAAAAKAAGY-NH2 | –10462 | –11395 | 11687 | 0.92 | 1.03 | 33.4 |
| Ac-AKAAProAKAAAAKAAGY-NH2 | 1147 | –7230 | –16913 | –0.16 | –2.34 | 2.8 |
| Ac-AKAAAAKAAProAKAAGY-NH2 | 1261 | –7756 | –17185 | –0.16 | –2.22 | 2.5 |
| Ac-YGAKAAAAKAAAAKAPro-NH2 | –5527 | –10737 | 3261 | 0.51 | 0.30 | 20.4 |
CD data were collected at 0.5 °C in water with 10 mM phosphate [pH 4 (OPO3H– peptides), pH 8 (OPO32– peptides), or pH 7 (all other peptides)] and 25 mM KF. The percent α-helix was calculated by the method of Baldwin,48,49 where % helix = 100[([θ]222 – [θ]C)/([θ]H – [θ]C)], where [θ]C is the mean residue ellipticity at 222 nm of 100% random coil, which equals 2220 – 53T, [θ]H is the mean residue ellipticity at 222 nm of 100% α-helix, which equals (−44000 + 250T)/(1 – 3/n), where T is the temperature in degrees Celsius (0.5) and n is the number of residues (16).
Table 2. Summary of Key Ser/Thr NMR Data for All Peptidesa.
| peptide | δ (HN) | 3JαN | δ (Hα) | δ (H-acetyl) |
|---|---|---|---|---|
| Ac-SerKAAAAKAAAAKAAGY-NH2 | 8.58 | 5.1 | 4.34 | 2.10 |
| Ac-Ser(OGlcNAc)KAAAAKAAAAKAAGY-NH2 | 8.62 | 3.3 | 4.33 | 2.13, 2.07 |
| Ac-Ser(OPO3H–)KAAAAKAAAAKAAGY-NH2 | 8.91 | 4.4 | 4.40 | 2.13 |
| Ac-Ser(OPO32–)KAAAAKAAAAKAAGY-NH2 | 9.41 | 3.6 | 4.29 | 2.15 |
| Ac-ThrKAAAAKAAAAKAAGY-NH2 | 8.40 | 6.2 | 4.25 | 2.12 |
| Ac-Thr(OGlcNAc)KAAAAKAAAAKAAGY-NH2 | 8.34 | 5.1 | 4.18 | 2.15, 2.08 |
| Ac-Thr(OPO3H–)KAAAAKAAAAKAAGY-NH2 | 8.80 | 4.8 | 4.23 | 2.16 |
| Ac-Thr(OPO32–)KAAAAKAAAAKAAGY-NH2 | 9.57 | 3.5 | 4.04 | 2.16 |
| Ac-ASerAAAAKAAAAKAAGY-NH2 | 8.62 | 5.7 | 4.41 | 2.07 |
| Ac-ASer(OPO3H–)AAAAKAAAAKAAGY-NH2 | 8.97 | 5.1 | 4.51 | 2.09 |
| Ac-ASer(OPO32–)AAAAKAAAAKAAGY-NH2 | 9.57 | 4.3 | 4.41 | 2.13 |
| Ac-AThrAAAAKAAAAKAAGY-NH2 | 8.63 | 4.7 | 4.26 | 2.06 |
| Ac-AThr(OPO3H–)AAAAKAAAAKAAGY-NH2 | 9.08 | ndb | 4.16 | 2.11 |
| Ac-AThr(OPO32–)AAAAKAAAAKAAGY-NH2 | ||||
| 278 K | 10.24 | 3.7 | 4.02 | 2.15 |
| 285 K | 10.15 | 4.0 | 4.02 | 2.14 |
| 298 K | 9.94 | 4.0 | 4.04 | 2.13 |
| 305 K | 9.81 | 4.9 | 4.05 | 2.12 |
| Ac-AKAASerAKAAAAKAAGY-NH2 | 8.44 | ndc | 4.37 | 2.05 |
| Ac-AKAASer(OGlcNAc)AKAAAAKAAGY-NH2 | 8.45 | ndc | 4.46 | 2.04, 2.02 |
| Ac-AKAASer(OPO3H–)AKAAAAKAAGY-NH2 | 8.71 | 5.7 | 4.51 | 2.03 |
| Ac-AKAASer(OPO32–)AKAAAAKAAGY-NH2 | 9.06 | 4.5 | 4.44 | 2.03 |
| Ac-AKAAThrAKAAAAKAAGY-NH2 | 8.29 | ndc | 4.23 | 2.04 |
| Ac-AKAAThr(OGlcNAc)AKAAAAKAAGY-NH2 | 8.16 | ndc | 4.34 | 2.06, 2.01 |
| Ac-AKAAThr(OPO3H–)AKAAAAKAAGY-NH2 | 8.57 | ndc | 4.36 | 2.02 |
| Ac-AKAAThr(OPO32–)AKAAAAKAAGY-NH2 | 9.50 | 4.1 | 4.11 | 2.01 |
| Ac-AKAAAAKAASerAKAAGY-NH2 | 8.30 | ndc | 4.37 | 2.06 |
| Ac-AKAAAAKAASer(OPO3H–)AKAAGY-NH2 | 8.54 | ndc | 4.50 | 2.03 |
| Ac-AKAAAAKAASer(OPO32–)AKAAGY-NH2 | 9.01 | 3.3 | 4.42 | 2.03 |
| Ac-AKAAAAKAAThrAKAAGY-NH2 | 8.19 | 6.8 | 4.25 | 2.06 |
| Ac-AKAAAAKAAThr(OPO3H–)AKAAGY-NH2 | 8.44 | ndc | 4.40 | 2.02 |
| Ac-AKAAAAKAAThr(OPO32–)AKAAGY-NH2 | 9.41 | 4.5 | 4.09 | 2.03 |
| Ac-YGAKAAAAKAAAAKASer-NH2 | 8.22 | ndc | 4.37 | 1.99 |
| Ac-YGAKAAAAKAAAAKASer(OGlcNAc)-NH2 | 8.30 | ndc | 4.48 | 2.03, 1.98 |
| Ac-YGAKAAAAKAAAAKASer(OPO3H–)-NH2 | 8.57 | 6.8 | 4.50 | 1.97 |
| Ac-YGAKAAAAKAAAAKASer(OPO32–)-NH2 | 9.02 | 6.3 | 4.42 | 1.98 |
| Ac-YGAKAAAAKAAAAKAThr-NH2 | 8.18 | 7.9 | 4.29 | 1.98 |
| Ac-YGAKAAAAKAAAAKAThr(OGlcNAc)-NH2 | 8.18 | 8.0 | 4.42 | 2.05, 1.98 |
| Ac-YGAKAAAAKAAAAKAThr(OPO3H–)-NH2 | 8.42 | 7.9 | 4.39 | 1.98 |
| Ac-YGAKAAAAKAAAAKAThr(OPO32–)-NH2 | 8.93 | 6.2 | 4.26 | 1.99 |
Chemical shifts (δ) [amide (HN) and Hα] and backbone coupling constants (3JαN, which corresponds to the ϕ torsion angle) of serine or threonine resonances and acetyl (N-terminal and OGlcNAc) resonances for all Ser- and Thr-containing peptides. Experiments were conducted at 278 K (unless otherwise indicated) in 5 mM phosphate buffer with 25 mM NaCl at pH 7.2 (dianionic phosphorylated peptides) or pH 4 (other peptides). Complete NMR data are given in the Supporting Information.
Not determined due to broadening.
Not determined due to spectral overlap.
Table 3. 31P NMR-Derived Coupling Constants for Phosphothreonine-Containing Peptidesa.
| peptide | 3JPHβ |
|---|---|
| Ac-Thr(OPO32–)KAAAAKAAAAKAAGY-NH2 | |
| 278 K | 8.6 |
| 298 K | 7.9 |
| Ac-AThr(OPO32–)AAAAKAAAAKAAGY-NH2 | |
| 278 K (30% TFE) | 9.6 |
| 278 K | 9.5 |
| 298 K | 9.0 |
| 310 K | 8.3 |
| 323 K | 7.4 |
| Ac-AKAAThr(OPO32–)AKAAAAKAAGY-NH2 | |
| 278 K | 8.9 |
| 298 K | 8.8 |
| Ac-AKAAAAKAAThr(OPO32–)AKAAGY-NH2 | |
| 278 K | 8.5 |
| 298 K | 8.4 |
| 310 K | 8.3 |
| 323 K | 8.3 |
| 338 K | 8.1 |
| Ac-YGAKAAAAKAAAAKAThr(OPO32–)-NH2 | |
| 278 K | 7.8 |
| 298 K | 7.8 |
Experiments were conducted in 5 mM phosphate buffer (pH 8.0) with 25 mM NaCl in D2O (unless otherwise indicated). The peptide Ac-ASer(OPO32–)AAAAKAAAAKAAGY-NH2 exhibited a 3JPHβ of 7.4 Hz (with identical coupling to both diastereotopic serine Hβ protons) at 278 K and a 3JPHβ of 6.7 Hz at 298 K (Figure S93 of the Supporting Information).
Peptides with an N-terminal serine residue all exhibited α-helicity comparable to or greater than that of alanine (Figure 3), consistent with the known ability of serine to function as an N-cap via hydrogen bonding of the serine lone pair electrons with unsatisfied amide hydrogens.14,50−57 Notably, phosphoserine and serine-O-GlcNAc have multiple hydrogen bond acceptor lone pairs, suggesting their capability to function similarly. Phosphoserine may also contribute to stability at the N-terminus of an α-helix via favorable helix dipole interactions.14,15,58,59 Interestingly, Ser-OGlcNAc exhibited α-helicity comparable to that of Ser but had a substantially more α-helical 3JαN coupling constant, which correlates with the ϕ torsion angle (here, 3JαN = 3.3 Hz for SerOGlcNAc, corresponding to ϕ = −53°, compared to 3JαN = 5.1 Hz, average ϕ = −68°, for Ser, as expected for a peptide that is 50% α-helical and 50% random coil), suggesting substantial conformational restriction is involved in the N-terminal helix-stabilizing effects of Ser-OGlcNAc. The magnitude of this coupling constant in Ser-OGlcNAc strongly suggests a conformationally restricting interaction that is present in both the α-helical and random coil states.
As has been previously observed, a dianionic N-terminal phosphoserine is more α-helix-stabilizing than the monoanionic phosphoserine (typical pKa of ∼5.8).14 The increased α-helicity for the phosphoserine dianion could be due to a more favorable helix–dipole interaction, better oxygen hydrogen bond acceptors in N-capping due to a greater negative charge per oxygen, and/or greater conformational restriction of dianionic phosphoserine than serine or monoanionic phosphoserine. NMR of the phosphoserine residue (Table 2 and Supporting Information) indicates a highly downfield-shifted phosphoserine amide proton chemical shift, which has previously been observed by us and others for dianionic phosphoserine and phosphothreonine, and is consistent with a hydrogen bonding interaction of phosphoserine with its own amide hydrogen.20,23,29,36,60−65 The dianionic phosphoserine also exhibited greater ϕ conformational restriction than the monoanionic phosphoserine [3JαN = 3.6 Hz (average ϕ = −56°) and 4.4 Hz (average ϕ = −62°), respectively]. We have also observed in polyproline helices, which lack both secondary structure interactions with hydrogen bond donors and an appreciable helix dipole, that the dianionic phosphorylated residues induce a greater structural change than monoanionic phosphates, both by CD and by NMR.21,29 These data suggest a role for conformational restriction due to phosphorylation, in addition to the electrostatic effects and hydrogen bonding effects of phosphorylation, in inducing structural change.
The peptide with an N-terminal threonine residue was less α-helical than the peptide with Ser or Ala at the N-terminus, consistent with previous data on α-helix propensities in general and at the N-terminus in particular (Figures 3 and 4 and Table 1).52,56,57,66−69 OGlcNAcylation of threonine resulted in a significant increase in α-helicity, generating a peptide with α-helicity comparable to that of the Ala peptide. As was observed for serine, threonine phosphorylation resulted in an increase in α-helicity, with greater α-helicity for the dianionic state than the monoanionic state. For both OGlcNAcylation and phosphorylation, a higher level of induced structure and a greater overall structural change were observed for threonine modification than for serine modification: of all peptides in the series with N-terminal modifications, the least α-helical peptide contained unmodified threonine while the most α-helical peptide contained dianionic phosphothreonine. NMR confirmed CD data (Table 2 and Supporting Information): unmodified threonine was more disordered than serine, whereas phosphothreonine was highly ordered, with an overall change in ϕ (3JαN = 3.5 Hz for ThrOPO32–, corresponding to an average ϕ = −55°, compared to 3JαN = 6.2 Hz for Thr, corresponding to an average ϕ = −76°; overall Δ3JαN = 2.7 Hz) larger than the change for Ser (Δ3JαN = 1.5 Hz).45 Dianionic phosphothreonine also induced greater amide chemical shift dispersion than any other peptide with N-terminal Ser and/or Thr residues. Interestingly, while the peptide with ThrOGlcNAc had greater induced α-helicity than that with SerOGlcNAc by CD, greater ϕ main chain conformational restriction was observed for SerOGlcNAc by NMR, suggesting additional conformational restriction induced by the sugar.
Figure 4.

CD spectra of Ac-TKAAAAKAAAAKAAGY-NH2 peptides with threonine modifications [unmodified Thr (free hydroxyl), ThrOGlcNAc, ThrOPO3H– (pH 4), and ThrOPO32– (pH 8)]: unmodified Thr (green squares), dianionic phosphothreonine (pH 8) (red circles), monoanionic phosphothreonine (pH 4) (open magenta circles), and ThrOGlcNAc (blue diamonds). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
Doig observed that the largest structural effects of serine phosphorylation were at the second residue of α-helical peptides.14 We found that the effects of serine and threonine phosphorylation were also magnified at this position, with dianionic phosphothreonine at residue 2 generating the most α-helical peptide in this study (Figure 5). By NMR, both phosphopeptides also exhibited highly restricted backbone conformations at the phosphorylated residue [3JαN values of 4.3 Hz (pSer) and 3.7 Hz (pThr) as dianionic phosphates] compared to those of the nonphosphorylated peptides (Table 2). The phosphothreonine amide hydrogen exhibited a particularly downfield chemical shift (δ = 10.24 ppm, compared to δ = 8.63 ppm for nonphosphorylated Thr) as well as exchange dynamics (including reduced peak magnitude) that were different from those of any amide in this study, consistent with an especially favorable interaction with this amide in this peptide. In addition, NMR data indicated particular χ1 conformational restriction for phosphothreonine in this peptide, with a 3JHαHβ of 10.4 Hz, which corresponds70,71 to phosphothreonine adopting almost exclusively a χ1 = −60° torsion angle (g– or m)72 (Figure S25 of the Supporting Information). Dianionic phosphothreonine at residue 2 also induced two of the four most downfield resonances of alanine amide protons (δ = 8.97 and 8.72 ppm) of any peptides in this study, with an alanine in the peptides with phosphothreonine at residue 5 (δ = 8.82 ppm) and at residue 10 (δ = 8.80 ppm) being the others (see the Supporting Information for complete NMR data), suggestive of strong local stabilizing interactions induced by phosphothreonine. These structural effects correlated with α-helical structure: with increased temperature, which resulted in reduced α-helical content [CD (Figure S16 of the Supporting Information)], the phosphothreonine amide chemical shift became more upfield and the 3JαN increased, as was observed for other, less α-helical peptides in this study.
Figure 5.

CD spectra of Ac-A(S/T)AAAAKAAAAKAAGY-NH2 peptides with serine (left) or threonine (right) modifications [unmodified Ser/Thr (free hydroxyl), Ser/ThrOPO3H– (pH 4), and Ser/ThrOPO32– (pH 8)]: unmodified Ser/Thr (green squares), dianionic phosphoserine/phosphothreonine (pH 8) (red circles), and monoanionic phosphoserine/phosphothreonine (pH 4) (open magenta circles). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
The effects of serine post-translational modifications were examined in the interior of the α-helix at residue 5 (Figure 6). As described previously by Vinson and by Doig,12,14 phosphorylation of serine here was significantly destabilizing with respect to α-helicity, with the destabilizing effect of phosphorylation being larger than the stabilizing effect observed at the N-terminus. Interestingly, despite the lower α-helicity observed by CD, the dianionic phosphoserine by NMR still exhibited an α-helical 3JαN (4.5 Hz) and a more restricted 3JαN than monoanionic phosphoserine (5.7 Hz), suggestive of a strong local stabilizing interaction. Serine OGlcNAcylation also destabilized the α-helix, though to an extent smaller than that observed for phosphorylation.
Figure 6.
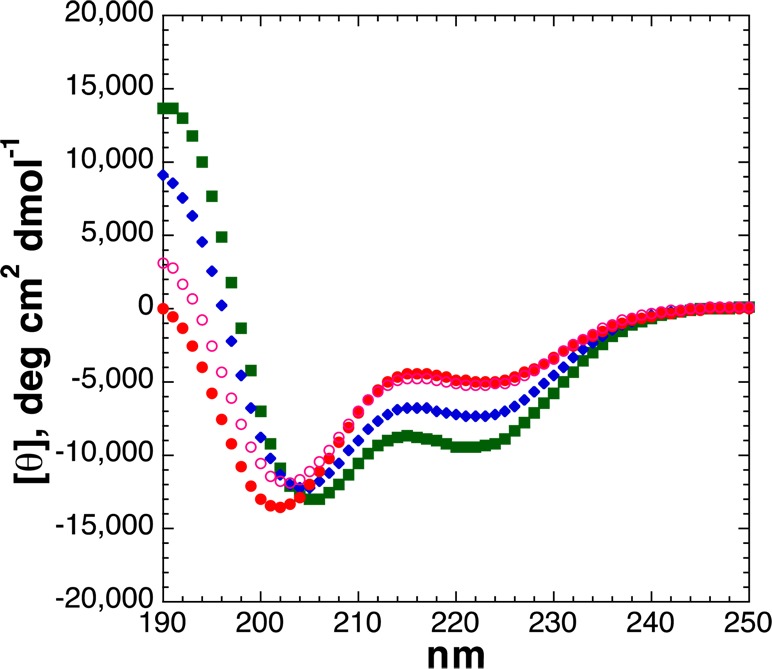
CD spectra of Ac-AKAASAKAAAAKAAGY-NH2 peptides with serine modifications [unmodified Ser (free hydroxyl), SerOGlcNAc, SerOPO3H– (pH 4), and SerOPO32– (pH 8)]: unmodified Ser (green squares), dianionic phosphoserine (pH 8) (red circles), monoanionic phosphoserine (pH 4) (open magenta circles), and SerOGlcNAc (blue diamonds). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
Threonine is more α-helix-destabilizing than serine. Both residues are helix-destabilizing because of the combination of side chain hydrogen bond donors and acceptors capable of multiple hydrogen bonds with the main chain, with Thr being additionally destabilizing because of the greater steric demand of a β-branched amino acid.66,67,73,74 Thus, as expected, the peptide with an interior threonine (residue 5) was substantially less α-helical (Figure 7) than the peptide with serine (Figure 6), and both peptides were less α-helical than the peptide with alanine here (Figure 3 and Table 1). Both post-translational modifications exacerbated the low α-helical propensity of threonine. In this case, in contrast to all previous results and all other results herein, the dianionic phosphothreonine was less structurally modifying (here, less destabilizing to the α-helix) than the monoanionic phosphothreonine. This reduced α-helical disruption could be due to the combination of α-helix-destabilizing effects of phosphothreonine being partially counteracted by the ability of phosphothreonine to more effectively nucleate the shorter C-terminal α-helix (e.g., in residues 5–14, with strong α-helix nucleation by phosphothreonine at the N-terminus of the short α-helix, as seen above). Consistent with this interpretation, by NMR, phosphothreonine at residue 5 exhibited a quite downfield amide chemical shift (δ = 9.50 ppm) and a small 3JαN of 4.1 Hz consistent with α-helix. Alternatively, the increased α-helicity of dianionic versus monoanionic phosphothreonine here could potentially be due to a favorable interaction with the i – 3 lysine residue, though this interaction is expected to contribute minimally and was not apparently significant for phosphoserine at this position.75 Here, the most disruptive post-translational modification was Thr OGlcNAcylation, which rendered the peptide almost fully random coil. ThrOGlcNAc is a highly sterically demanding amino acid, which in general leads to a bias against α-helix and polyproline helix and a greater preference for more extended conformations, as has been observed for other sterically demanding amino acids such as tert-leucine, fluorinated amino acids, and non-Aib α,α-dialkylglycines.21,27,29,76−80
Figure 7.
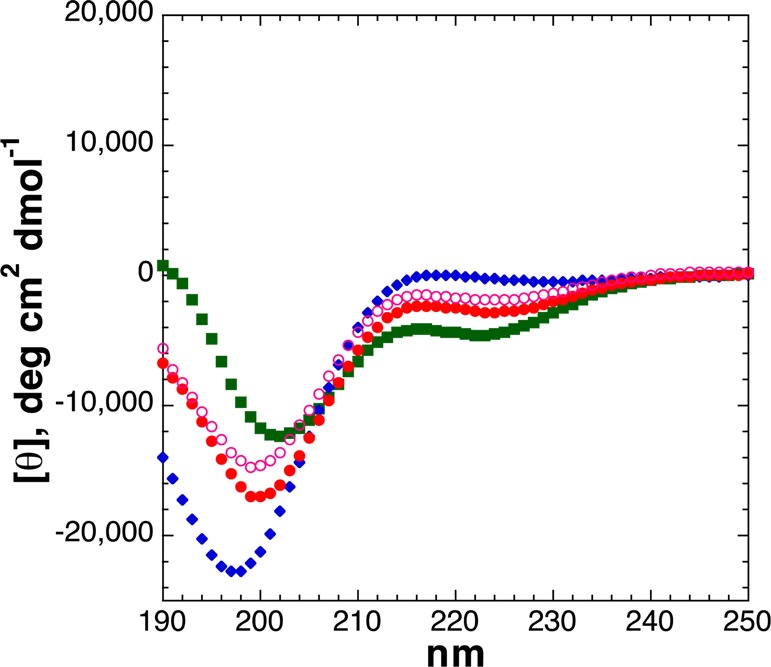
CD spectra of Ac-AKAATAKAAAAKAAGY-NH2 peptides with threonine modifications [unmodified Thr (free hydroxyl), ThrOGlcNAc, ThrOPO3H– (pH 4), and ThrOPO32– (pH 8)]: unmodified Thr (green squares), dianionic phosphothreonine (pH 8) (red circles), monoanionic phosphothreonine (pH 4) (open magenta circles), and ThrOGlcNAc (blue diamonds). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
To examine the basis for the unexpected reduced structural effects of dianionic versus monoanionic phosphothreonine at residue 5 of the α-helix, peptides were synthesized with threonine or phosphothreonine at residue 10 of the α-helix (Figures 2 and 8). Peptides were also synthesized with serine or phosphoserine here, to determine whether there is an inherent effect of α-helical position on the structural effects of phosphorylation (Figure 9). The unmodified Thr-containing peptides (Thr at residue 5 or 10) had similar α-helicities, indicating that there is no inherent difference in the effects of threonine at different interior residues of the α-helix, as expected on the basis of previous data on helix propensities as a function of α-helical position.81,82 In contrast, at residue 10, phosphothreonine was highly disruptive to the α-helix, with the dianionic phosphothreonine inducing a complete random coil CD spectrum of the peptide, and the monoanionic phosphothreonine also inducing disruption of the α-helix greater than that observed by any modification at residue 5 of the α-helix. Similarly, phosphoserine was also highly disruptive to the α-helix at residue 10, with a greater disruption of the α-helix by phosphoserine at residue 10 than at residue 5 and with a greater disruption as dianionic phosphoserine than as monoanionic phosphoserine. Interestingly, by NMR, phosphothreonine and phosphoserine still exhibited significantly downfield chemical shifts (δ = 9.41 and 9.01 ppm, respectively) and small 3JαN values (4.5 and 3.3 Hz, respectively) indicative of restricted ϕ (corresponding to average ϕ values of −63° and −53°, respectively),45 consistent with α-helix formation but also observed in proline-rich peptides adopting a polyproline helix.21,29 In addition, the peptide with dianionic phosphothreonine also exhibited the greatest chemical shift dispersion of the alanine amide protons (σ = 0.43 ppm, compared to σ = 0.32 ppm for the peptide with dianionic phosphothreonine at residue 2 and σ = 0.08–0.19 ppm for all other Thr-containing peptides), with two significantly upfield alanine amide resonances (δ = 8.80 and 8.61 ppm), suggestive of substantial local structure around phosphothreonine despite an overall random coil structure. These data are consistent with the interpretation that the structural effects of threonine phosphorylation at residue 5 are a balance of disruption of α-helical structure of the residues N-terminal to phosphothreonine via a phosphate–amide interaction, versus a significant induction of a shorter α-helix for the C-terminal residues (e.g., residues 5–14, as was observed at position 1 or 2 of an α-helix). In contrast, at residue 10, the effects of phosphorylation on α-helical induction are inherently minimal because only four residues are C-terminal (one α-helical turn; in general, very short α-helices are inherently unstable),83 while exhibiting a strong anti-α-helix signal due to the phosphate–amide interaction disrupting the hydrogen bonding pattern necessary for α-helix propagation. More generally, these data indicate that phosphorylation in the interior of an α-helix is more disruptive when the phosphorylation site is closer to the C-terminus. Most broadly, these data indicate a particularly strong disruption of the α-helix by phosphothreonine when in the interior of an α-helix, comparable to the effects of proline within α-helices.66,84−86
Figure 8.
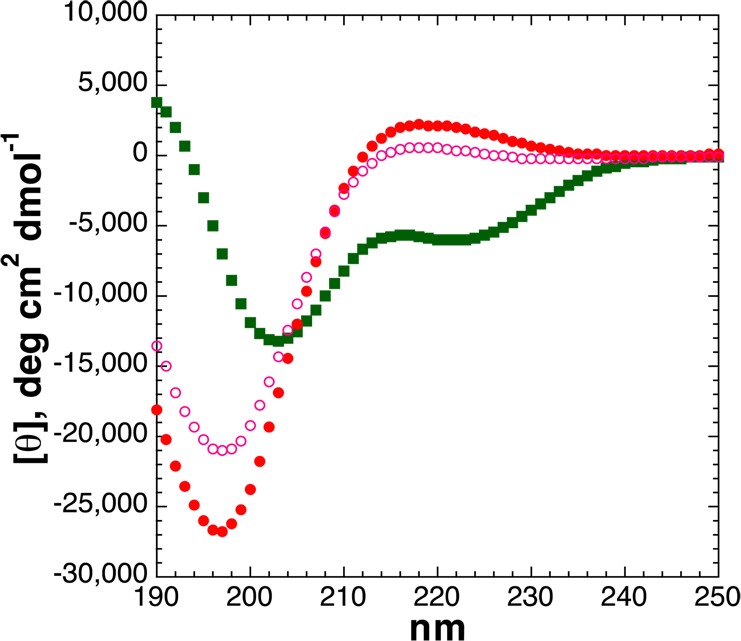
CD spectra of Ac-AKAAAAKAATAKAAGY-NH2 peptides with threonine modifications [unmodified Thr (free hydroxyl), ThrOPO3H– (pH 4), and ThrOPO32– (pH 8)]: unmodified Thr (green squares), dianionic phosphothreonine (pH 8) (red circles), and monoanionic phosphothreonine (pH 4) (open magenta circles). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
Figure 9.

CD spectra of Ac-AKAAAAKAASAKAAGY-NH2 peptides with serine modifications [unmodified Ser (free hydroxyl), SerOPO3H– (pH 4), and SerOPO32– (pH 8)]: unmodified Ser (green squares), dianionic phosphoserine (pH 8) (red circles), and monoanionic phosphoserine (pH 4) (open magenta circles). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
The effects of serine and threonine post-translational modifications were examined at the C-terminus of the α-helix (Figure 10). There has been no prior measurement of the effects of phosphorylation at the C-terminus of an α-helix, although Doig noted that molecular dynamics simulations, which can underestimate hydrogen bonding and n → π* noncovalent interactions and overestimate electrostatic interactions, suggested strong disruption of the α-helix.14 Serine and threonine exhibit enhanced frequencies and α-helical propensities at the C-termini of α-helices because of their ability to function effectively as C-caps via their hydrogen bond donor capabilities in bonding to exposed amide carbonyls.51,53,55,57,87−91 In contrast, in α-helices, the helix macrodipole is stabilized by positively charged residues at the C-terminus and destabilized here by negatively charged residues.58,59,88 The data indicate that both serine and threonine exhibit enhanced α-helical propensity at the C-terminus relative to interior positions, with somewhat greater α-helicity for serine than for threonine, as expected. In contrast, both OGlcNAcylation and phosphorylation disrupted the α-helix at the C-terminus of α-helices, with greater α-helical disruption upon phosphorylation than OGlcNAcylation. Interestingly, by NMR, dianionic phosphoserine and phosphothreonine exhibited substantially larger 3JαN values (6.3 and 6.2 Hz, respectively) and more upfield amide chemical shifts (Table 2) than in any other phosphopeptides in this study or in proline-rich peptides examined previously, suggesting a weakening of the phosphate–amide interaction, potentially due to either an unfavorable α-helix macrodipole or the importance of amide hydrogen bonding to the i – 4 carbonyl in α-helices, which competes with the amide–phosphate interaction. Interestingly, the C-terminal carboxamide chemical shifts for the peptides with ThrOGlcNAc (δ = 7.50 and 7.39 ppm) and ThrOPO32– (δ = 7.89 and 7.25 ppm) were divergent from those of any other peptides in this study [7.63–7.70 and 7.22–7.25 ppm (7.30–7.35 ppm for C-terminal modifications)] (Tables S30 and S40 of the Supporting Information). Notably, while both post-translational modifications were disruptive at the interior and at the C-terminus of α-helices, for threonine the effects of phosphorylation and OGlcNAcylation were observed to be greater at the interior of α-helices than at the C-terminus, whereas for serine the effects were comparable in both locations (Figures 11 and 12). These differences may potentially be due to the less favorable phosphate–amide interactions for phosphoserine than for phosphothreonine (e.g., as observed in smaller downfield amide chemical shifts for phosphoserine in all peptides).
Figure 10.
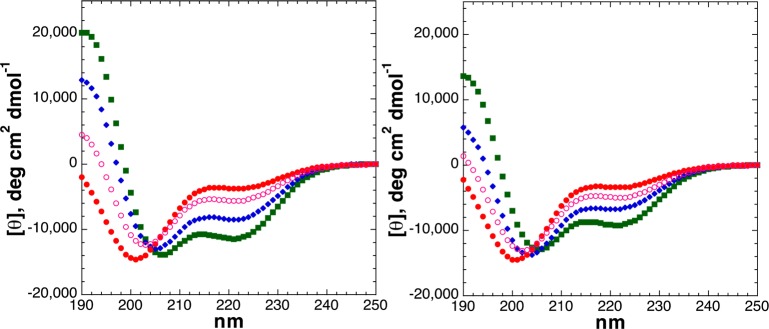
CD spectra of Ac-YGAKAAAAKAAAAKA(S/T)-NH2 peptides with serine (left) or threonine (right) modifications [unmodified Ser/Thr (free hydroxyl), Ser/ThrOPO3H– (pH 4), and Ser/ThrOPO32– (pH 8)]: unmodified Ser/Thr (green squares), dianionic phosphoserine/phosphothreonine (pH 8) (red circles), and monoanionic phosphoserine/phosphothreonine (pH 4) (open magenta circles). CD experiments were conducted in water with 10 mM phosphate (pH 7 or as indicated) and 25 mM KF at 0.5 °C.
Figure 11.
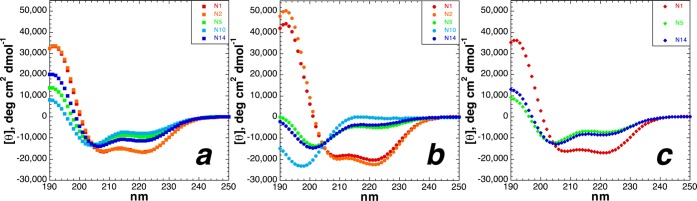
Comparison of the effects of serine post-translational modification as a function of α-helical position: (a) serine, (b) phosphoserine (pH 8), and (c) Ser(OGlcNAc). Red denotes modifications at residue 1, orange those at residue 2, green those at residue 5, cyan those at residue 10, and blue those at residue 14. Each plot in this figure includes the CD data of a series of isomeric peptides (N1, N5, N10, and N14).
Interestingly, the effects of post-translational modifications described above are qualitatively similar to those that are expected for proline residues. N-Terminal proline residues nucleate the α-helix via conformational restriction and effective presentation of the carbonyl hydrogen bond acceptors for i + 4 amide protons. Internal proline residues strongly disrupt α-helices, and C-terminal proline residues serve as α-helix stop signals, as a result of proline preventing propagation of the hydrogen bonding pattern of an α-helix due to the absence of an amide hydrogen, as well as a steric clash between proline and the prior amino acid when both are in α-helical conformations.50,86,87,92−94 To directly compare the effects of serine and threonine post-translational modifications with those of proline, peptides were synthesized with proline at residues 1, 2, 5, 10, and 14 of the model peptides (Figure 2). As expected, peptides with proline at the N-terminus (residue 1 or 2) exhibited substantial α-helix, though the level was lower than that observed for peptides with N-terminal serine or threonine derivatives (Figure 13a).56,95 In contrast, internal proline residues (position 5 or 10) strongly disrupted the α-helix, resulting in a random coil CD signature, similar to those observed for Thr(OGlcNAc) at residue 5 or for phosphothreonine or phosphoserine at residue 10 (Figure 13b). C-Terminal proline residues modestly disrupted α-helicity, in a manner similar to the effects of phosphoserine or phosphothreonine at these positions (Figure 13c). Overall, proline substantially modulated α-helicity in this series of peptides as a function of position, as expected (Figure 13d).
Figure 13.

Comparison of the effects of threonine modification with proline modification on α-helicity as a function of α-helical position: (a) proline (black triangles) and dianionic phosphothreonine (red circles) at the N-terminus [residue 1 (filled symbols) and residue 2 (open symbols)] of an α-helix, (b) proline at residues 5 (filled triangles) and 10 (open triangles) of an α-helix, ThrOGlcNAc at residue 5 (filled diamonds) of an α-helix, and dianionic phosphothreonine at residues 5 (filled circles) and 10 (open circles) of an α-helix, and (c) proline (black triangles), threonine (green squares), Thr(OGlcNAc) (blue diamonds), and dianionic phosphothreonine (red circles) at the C-terminus of an α-helix. (d) Comparison of circular dichroism spectra of peptides containing proline at all α-helical positions: red for proline at residue 1, orange for proline at residue 2, green for proline at residue 5, cyan for proline at residue 10, and blue for proline at residue 14.
Phosphorylation of threonine at residue 2 and at residue 10 produced the greatest structural effects of any peptides in this study: phosphothreonine at residue 2 yielded the most α-helical peptide herein, while phosphothreonine at residue 10 yielded the peptide with the least α-helical content, with the peptide exhibiting a complete random coil signature. Peptides with threonine or phosphothreonine at residue 2 or 10 were examined further by heteronuclear NMR spectroscopy, using 1H–13C HSQC experiments, to identify the residue-specific effects of modification in these peptides (Figure 14; expanded spectra in Figures S90 and S91 of the Supporting Information and Tables S23 and S24 of the Supporting Information). By chemical shift index analysis, upfield changes in the Hα chemical shift, downfield changes in the Cα chemical shift, and upfield changes in the Cβ chemical shift are indicative of increased α-helical content at a given residue.96,97 These experiments indicated that the structural effects observed by circular dichroism were consistent with structural changes throughout the peptides, with phosphorylation making alanine and lysine residues more α-helical (i.e., southeastern movement of resonances in Figure 14a) in the peptide with phosphothreonine at residue 2 but making alanine and lysine residues less α-helical upon phosphorylation at residue 10 (i.e., northwestern movement of resonances in Figure 14b). The mean chemical shift of alanine Cα resonances was 51.39 ppm for the peptide with phosphothreonine at residue 2 (Δδ = +0.65 ppm upon phosphorylation), versus 49.33 ppm for the peptide with phosphothreonine at residue 10 (Δδ = −0.74 ppm upon phosphorylation), indicating significant overall differences in induced structure upon phosphorylation as a function of location within the α-helix (Table S45 of the Supporting Information). Of additional note is the difference in phosphorylation-induced changes in the Hα and 13Cα chemical shifts of threonine, which exhibited 0.24 ppm upfield and 3.41 ppm downfield changes, respectively, upon phosphorylation at residue 2, but 0.16 ppm upfield and 0.61 ppm downfield changes, respectively, upon phosphorylation at residue 10. Overall, the phosphothreonine 13Cα chemical shift at residue 2 was 2.89 ppm downfield of that at residue 10, despite almost no difference (0.09 ppm) in the 13Cα threonine chemical shift in the nonphosphorylated peptides, consistent with the large difference in α-helicity of the two phosphorylated peptides. The effects of phosphorylation on the Hβ, Cβ, and Cγ Thr resonances were also substantially dependent on structural context, as were the effects on Hα, Hβ, and Cβ resonances of Ala and Lys residues [see the Supporting Information for quantitative comparisons of NMR data for threonine-containing peptides (Tables S34–S46) and serine-containing peptides (Tables S25–S33)].
Figure 14.
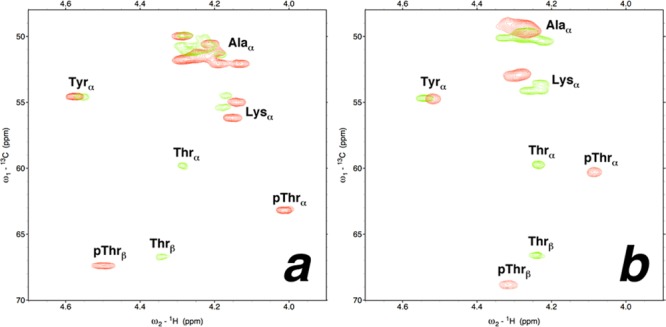
1H–13C HSQC spectra (Hα–Cα region) of peptides with nonphosphorylated (green) and phosphorylated (red) threonine. Experiments were conducted at 278 K in D2O containing 5 mM phosphate (pH 4 for nonphosphorylated peptides and pH 8 for phosphorylated peptides) and 25 mM NaCl. Full spectra are given in the Supporting Information. (a) Peptides with threonine or phosphothreonine at residue 2. (b) Peptides with threonine or phosphothreonine at residue 10.
The effects of serine/threonine post-translational modifications on α-helicity could also be read out across all serine/threonine-containing peptides via analysis of the chemical shift of the N-terminal acetyl group, which reports on the structure at the N-terminus of the peptide (Table 2 and Tables S29 and S39 of the Supporting Information). The most α-helical peptides exhibited the most downfield acetyl chemical shifts, with N-terminal phosphorylation inducing further downfield shifts. In contrast, the least α-helical peptides exhibited more upfield acetyl chemical shifts.
To confirm the reversible structural effects of phosphorylation, the peptides with phosphothreonine at residue 2 and phosphothreonine at residue 10 were incubated with Antarctic phosphatase, a nonspecific phosphatase. The peptide with phosphothreonine at residue 2 exhibited a reduced α-helical content as a function of time upon exposure to phosphatase, while the peptide with phosphothreonine at residue 10 exhibited a change from a random coil signature to α-helix upon desphosphorylation by phosphatase, in a manner consistent with the effects observed in isolated peptides (Figures S17–S20 of the Supporting Information). In general, phosphatases act preferentially on random coil structures. Interestingly, despite the random coil overall structure of the peptide with phosphothreonine at residue 10, this peptide exhibited relatively slower dephosphorylation, consistent with local structure around phosphothreonine potentially sterically reducing access to the substrate. Collectively, these results emphasize that reversible phosphorylation may function as a trigger to induce or disrupt α-helix, depending on the position of the phosphorylation site within an α-helix.
We have previously identified that phosphothreonine may adopt a highly conformationally restricted structure, in which the Cβ–Hβ bond is, surprisingly, in an eclipsed or near-eclipsed conformation with the Oγ–P bond.29 On the basis of a parametrized Karplus relationship for P–H three-bond coupling constants, the expected 3JHβP for an eclipsing interaction is 10.6 Hz.98 All phosphothreonine-containing peptides were analyzed by 31P NMR (Table 3). Peptides with greater α-helicity or conditions that strongly promote α-helix formation [lower temperature and/or addition of trifluoroethanol (TFE)] were observed to have larger (more conformationally restricted) 3JHβP values, close to that expected for an eclipsed C–H/O–P bond (maximum observed 3JHβP of 9.6 Hz), while peptides or conditions (higher temperature) that exhibited lower α-helical content by CD were observed to have smaller 3JHβP values, consistent with more disordered conformations (minimum observed 3JHβP of 7.8 Hz, which still represents only a 30° average deviation from an eclipsed conformation). Notably, even under conditions where the peptide is disordered, the 3JHβP for phosphothreonine residues was larger than has been normally observed for phosphoserine residues (typically 3JHβP ∼ 6 Hz for each of the diastereotopic β protons), as has been seen previously for phosphothreonine in other peptides and proteins, suggestive of inherently substantially greater conformational restriction at the individual amino acid side chain level for phosphothreonine than phosphoserine.99−103
Discussion
We have described the direct comparison of the effects of two competing intracellular post-translational modifications, phosphorylation and OGlcNAcylation, on the stability of the α-helix, the most common secondary structure in proteins, examining separately the effects of serine and threonine modification and the effects of modifications at the N-terminus, at internal positions, and at the C-terminus of the α-helix. Within α-helices, we found the effects of phosphorylation and OGlcNAcylation to be qualitatively similar, though differing in magnitude, with effects of phosphorylation in most cases being greater than those of OGlcNAcylation. At the N-terminus of the α-helix, both post-translational modifications were stabilizing, with phosphorylation in the more physiologically important dianionic state the most stabilizing. In addition, we found greater induced α-helicity with phosphothreonine than phosphoserine. Notably, all post-translational modifications at the N-terminus (residue 1 or 2) generated peptides that were more α-helical than peptides with alanine, the most helix-stabilizing canonical residue. In contrast, at a position in the interior of an α-helix, both post-translational modifications were destabilizing, with particular α-helix destabilization for post-translationally modified threonine residues. Both threonine OGlcNAcylation and threonine phosphorylation were capable of nearly complete disruption of the α-helix, in a manner dependent on position in the α-helix, with phosphothreonine effects representing a balance of strong helix-inducing effects at the N-terminus of an α-helix and strong helix-disrupting effects in the interior of an α-helix. Phosphorylation was more disruptive to the α-helix at a more C-terminal interior position (residue 10) than at a more N-terminal interior position (residue 5) of the α-helix. The data on phosphorylation at the N-terminus and interior of α-helices are consistent with previous data for phosphorylation in model peptides and in coiled coils, although larger effects were observed in coiled coils, potentially because of the roles of tertiary structure, additional side chain–side chain interactions, and multiple modifications (two to four phosphorylated residues) in these contexts.12,14,15 Most previous studies examined only serine phosphorylation, although Vinson observed severe effects of threonine phosphorylation on coiled coil stability.12
This work represents the first direct comparison of the effects of phosphorylation versus OGlcNAcylation within the context of an α-helix. These data suggest α-helices as one context in which phosphorylation and OGlcNAcylation may have similar structural and, potentially, functional effects, the latter of which are observed in some cases of these post-translational modifications.4,5 Both post-translational modifications are stabilizing at the N-terminus of an α-helix but are destabilizing at the interior or at the C-terminus of an α-helix, effects that enhance the native α-helix propensities of serine and threonine. The contrasting effects of these post-translational modifications at the N-terminus (induce α-helix) versus the interior or C-terminus (disrupt α-helix) of an α-helix suggest that both OGlcNAcylation and phosphorylation enhance α-helical start and stop signals in proteins, defining the boundaries of α-helical structure and recognition epitopes.104,105 Given the general observation of OGlcNAcylation and phosphorylation in transcriptional regulatory elements and the broad importance of α-helical recognition epitopes in transcription, these data suggest the possibility of direct structural effects of both post-translational modifications on transcription, via both induction and disruption of recognition α-helices.5,8,9,106−114
The results herein in α-helices stand in contrast to the results of our previous investigation of the effects of phosphorylation and OGlcNAcylation on the structure of the proline-rich domain of tau and of proline-rich model peptides. In these proline-rich contexts, the post-translational modifications had opposing structural effects, with phosphorylation inducing polyproline helix formation and OGlcNAcylation confirming the native biases of serine and threonine and opposing polyproline helix.21,27,29 The effects of OGlcNAcylation in disfavoring formation of both the polyproline helix and the α-helix at the interior of an α-helix, and with greater induced effects observed in the more compact structure of an α-helix compared to the somewhat more extended polyproline helix, are consistent with the effects of OGlcNAcylation being significantly steric in nature. The role of the OGlcNAc in stabilizing the N-terminus of an α-helix is less clear. The effect could be due to overall conformational restriction, in the presence of reduced steric restraints at the N-terminus of the α-helix. Alternatively, the sugar notably adds several additional hydrogen bond acceptors, which could potentially function in helix capping. In contrast to OGlcNAcylation, the effects of phosphorylation appear to be primarily mediated through the interaction of the phosphate with the amide backbone (see below), with additional effects due to α-helix macrodipole interactions (for anions, favorable at the N-terminus and unfavorable at the C-terminus). In total, these data emphasize the importance of structural context when considering the effects of protein post-translational modifications and provide a basis for understanding situations in which phosphorylation and OGlcNAcylation may be opposing (e.g., yin-yang) versus complementary in their effects, both of which have been described in numerous biological contexts.3,5,7,10,115,116
We previously observed in proline-rich peptides that the structural effects of threonine modification were greater than those of serine modification, with a highly conformationally restricted structure observed for phosphothreonine, including restriction of ϕ to a conformation compatible with either α-helix or polyproline helix (mean 3JαN = 3.5 Hz for dianionic pThr, versus mean 3JαN = 5.4 Hz for dianionic pSer) and evidence of a phosphate–amide side chain–main chain hydrogen bond, via highly downfield amide H chemical shifts (mean δ = 9.63 ppm for pThr, and mean δ = 8.99 ppm for pSer) and slow amide exchange at the phosphorylated residues at pH 8, even at high salt concentrations (1 M NaCl) and elevated temperatures.21,29,117,118 Both phosphoserine and phosphothreonine exhibited conformations distinct from expected random coil values (3JαN ∼ 6–8 Hz) observed for nonphosphorylated residues in multiple proline-rich peptide contexts, and in particular, phosphothreonine and phosphoserine had greater conformational restriction than the standard phosphomimic Glu (Glu 3JαN = 5.8–6.3 Hz). Notably, the side chain–main chain hydrogen bonding previously observed, by us and others, would be highly disruptive within α-helices.23,117−124 Vinson previously observed in coiled coil peptides that threonine phosphorylation was particularly structurally disruptive to α-helices, compared to serine phosphorylation.12,13 Similarly, Hilser observed larger effects of threonine over serine phosphorylation in model polyproline helix-mediated protein–protein interactions.28 In addition, Corzana et al. have observed greater conformational restriction in ThrOGlcNAc over SerOGlcNAc amino acids, as well as more generally in glycosylated threonine over glycosylated serine amino acids.28,42,125−127 The data herein are consistent with all of these observations, with structural effects of threonine phosphorylation and threonine OGlcNAcylation observed herein greater than those of the same post-translational modifications on serine. In particular, the significant conformational restriction at phosphothreonine, combined with larger induced structural effects seen by CD and by NMR, suggests a special role for phosphothreonine residues in protein structure. Collectively, these data from multiple structural contexts suggest threonine residues as potential hot spots in structural modulation via protein post-translational modifications compared to serine residues, with larger induced effects for threonine modification than serine modification.128,129
Notably, the effects of serine and (particularly) threonine modification by OGlcNAcylation and (particularly) phosphorylation are similar to those observed for proline residues on α-helices. Proline is an α-helix inducer at the N-terminus of α-helices, because of the conformational restriction of its ϕ torsion angle to one similar to that in α-helices and its ready presentation of its carbonyl as a hydrogen bond acceptor to interact with i + 4 amide hydrogens and thus nucleate the first turn of the α-helix.55−57,95,130−133 In contrast, proline is widely recognized as being highly disruptive to α-helices in their central residues, primarily due to proline’s inability to propagate hydrogen bonding patterns of α-helices because of the absence of an amide hydrogen bond donor.50,53,66,67,69,84,93,134,135 Interestingly, the kinks induced in α-helices with central proline residues can cause these exposed hydrogen bonding groups to be potent sites for protein–protein interactions in membranes.85,86,136−138 Proline residues are also destabilizing at the C-terminus of α-helices, again because of the absence of a hydrogen bond donor, as well as a preference for a non-α-helical conformation in the pre-proline residue, which can cause fraying and distortions of the last turn of the α-helix or alternatively adoption of nonhelical conformations at proline.50,51,55,82,87,92,93,135,139 The effects of proline disruption of α-helices are reduced at the C-terminus compared to in the interior of the α-helix, with greater interior effects closer to the C-terminus than to the N-terminus, as was observed here for phosphorylation and OGlcNAcylation.138 We previously observed that phosphothreonine is a particularly conformationally restricted amino acid, capable of adopting a structure similar to that of proline via two noncovalent interactions: (1) side chain cyclization via side chain–main chain phosphate–amide hydrogen bonding and (2) an n → π* interaction between consecutive carbonyls.29 Because of its dramatic structural effects, proline is widely recognized as a start and stop signal for α-helices.50,87,92−94 Notably, the structural effects of threonine phosphorylation on α-helix stability were found herein to be greater than those of proline, and the effects of threonine OGlcNAcylation were found to be comparable to those of proline (Figures 12b,c and 13). The data herein suggest phosphorylation and OGlcNAcylation can function like proline, but as inducible start and stop signals in α-helices, with induction of the α-helix at the N-terminus and disruption of the α-helix in the interior and at the C-terminus of α-helices.
At the N-terminus of peptides, both phosphoserine and phosphothreonine strongly promoted α-helix, with phosphorylation inducing α-helicity greater than that observed in peptides containing alanine. In addition, greater induced α-helicity was observed for phosphothreonine at residue 2 than at residue 1. Analysis of phosphoserine and phosphothreonine residues in α-helices in high-resolution structures in the PDB (structures at ≤2.4 Å, 90% sequence identity cutoff) indicated a strong preference for phosphoserine and phosphothreonine to be at or near the helical N-terminus when they were present in α-helices (pSer: 1h4x, 2fwn, 3mk1, 3ql6, 2w5w, 2bik, 3f3z, 1mki, and 3qic; pThr: 3ot9, 4iza, 2ga3, 2jfl, 2w8d, 2wtv, and 3u02; PDB entry 1r0z was the only example among these structures in which an α-helical phosphorylated residue was outside the first turn of the α-helix). In a majority of these examples, the phosphorylated residue was the first α-helical residue. These structures revealed a common structural motif in which multiple noncovalent interactions centered on phosphorylation lead to stabilization of the α-helix (Figure 15).140−142 The phosphate interacts with its own amide hydrogen (residue i) via hydrogen bonding. The i – 1 carbonyl, conjugated to the phosphoresidue amide, engages in an n → π* interaction with the i (phosphorylated) residue carbonyl (an interaction expected to be strengthened due to the amide–phosphate hydrogen bond, generating a better donor carbonyl group at the i – 1 residue, as is done by thioamide donors, and a better hydrogen bond acceptor at the i residue due to the n → π* interaction143,144). The i – 1 and i carbonyls hydrogen bond with the i + 3 and i + 4 amide hydrogens. In addition, in some cases (e.g., PDB entry 3ql6), the i – 1 carbonyl also interacts with the i + 1 amide in a bidentate hydrogen bond and the i – 2 carbonyl engages in a hydrogen bond with the i + 2 amide hydrogen. In these structures, the phosphate group interacts with only its own amide hydrogen and does not directly interact with the subsequent amides. One exception is PDB entry 2wtv, which contains two consecutive phosphothreonine residues, one at the N-cap position (which exhibits i – 1···i + 1 and i – 1···i + 2 phosphate–amide hydrogen bonds) and one at the N-terminus of the α-helix (which interacts with its own amide hydrogen but does not interact with other helical residues).
Figure 15.

Structures of α-helices with (a and b) phosphoserine [(a) PDB entry 3ql6,141 bovine lactoperoxidase, 1.70 Å resolution, residues 196–205; (b) PDB entry 1h4x,140Bacillus subtilis sporulation factor SpoIIAB, 1.16 Å resolution, residues 56–71] and (c) phosphothreonine (right, pdb 2w8d,142B. subtilis lipoteichoic acid synthase, 2.35 Å resolution, residues 295–305) residues at the N-terminus of the α-helix. The lactoperoxidase structure (a) also includes an Asn-linked β-OGlcNAc two residues C-terminal to the α-helix (residue 205, with residues 204 and 205 in extended conformations). Hydrogen bonds are denoted with black dashed lines; n → π* interactions (here, Oi–1–Ci=Oi distances of 2.85–2.90 Å and Oi–1–Ci=Oi angles of 88–115°) are denoted with magenta dashed lines.
These structures suggest a general mode by which phosphorylation stabilizes α-helices when it occurs at the N-terminus: in addition to a favorable interaction of the dianionic phosphate with the α-helix macrodipole,58,59 an interaction that would be maximized by the phosphate being located directly adjacent to the peptide backbone, phosphorylation promotes a strong phosphate–amide hydrogen bond with its own amide hydrogen; this entropically favorable intraresidue phosphate–amide interaction structurally organizes the prior residue (e.g., as in pre-proline effects) and aligns both the i – 1 and i carbonyls via n → π* interactions (favorable in α-helices)145−148 to effectively hydrogen bond with the i + 3 and i + 4 amide hydrogens. This interaction also may promote interactions of the i – 1 and/or i – 2 carbonyl with the i + 1 and/or i + 2 amide hydrogens, potentially providing hydrogen bond acceptors for all α-helical amide groups and suggesting a plausible structural basis for the observation of the α-helicity for phosphorylated residues at the N2 position being greater than that at the N1 position of alanine-rich peptides. This observation also correlates with observed downfield N-terminal acetyl chemical shifts for N1 and N2 phosphopeptides compared to other phosphopeptides (Table 2). The strength of this network of noncovalent interactions also explains the observation that the peptide with the N5 phosphothreonine is more α-helical as a dianion than a monoanion, because of strong helix-inducing effects (e.g., organizing residue 4, and possibly residue 3) at the N-terminus of the shorter α-helix in this case counteracting the strong helix-destabilizing effects of phosphothreonine in the interior of an α-helix.
Serine/threonine phosphorylation is the most prominent reversible intracellular post-translational modification, with phosphorylation observed on a majority of proteins involved in signal transduction and transcription. OGlcNAcylation has also been observed on numerous intracellular proteins that are important in signal transduction and transcription, often on sites that have also been identified as phosphorylation sites. Because of a reduced number of tools for the examination of OGlcNAcylation compared to the number for phosphorylation, including in particular a limited repertoire of antibodies, the absence of convenient radiolabeling methods, and more challenging synthetic methods, the effects of OGlcNAcylation on protein structure and function are substantially less well understood.11,149−151 Even greater challenges exist for the more complex glycosylation events observed extracellularly, which are employed in cell–cell communication and cell recognition and are modulated in several diseases, including cancers, inflammatory diseases, and bacterial and viral infections.152−157 This work examined the effects of β-OGlcNAc modification on α-helical peptides. Notably, these effects are expected to be general for OGlcNAcylation but are not expected to be general for all serine/threonine glycosylation events nor for N-linked glycosylation.19,158−167 In particular, the effects of β-OGlcNAc modification might be quite different from the effects of modification with α anomers of sugars, which is common in extracellular proteins. Indeed, in a limited number of examples, including antifreeze proteins and the prion protein, dramatic structural differences were observed between modification of Ser/Thr residues with α anomers versus β anomers of sugars.42,168,169 Data to date suggest that β anomers exhibit substantial steric effects near the backbone, similar to β-branched and aromatic amino acids, whereas α anomers are more compatible with compact protein conformations.
The data herein, combined with those previously observed for the effects of phosphorylation on structure in proline-rich peptides,21,27,29 suggest a general mode for effects of serine/threonine phosphorylation on structure. Previously, we found that phosphorylation induced polyproline helix and resulted in particular ϕ conformational restriction (3JαN = 3.0–5.5 Hz, corresponding to ϕ = −50° to −70°), with a value of ϕ consistent with both polyproline helix and α-helix, as well as strong evidence of a phosphoserine/phosphothreonine side chain–main chain hydrogen bond and suggestion of a potential n → π* interaction between the n – 1 carbonyl (conjugated to the pSer/pThr amide) and the carbonyl of the phosphoresidue, an interaction consistent with either polyproline helix or α-helix.145,147,148,170 Phosphorylation, particularly on threonine, has strong conformational effects, inducing a compact value of ϕ (which can be observed as a small 3JαN) and interaction with its own amide hydrogen (which can be observed as downfield phosphoserine/phosphothreonine amide chemical shifts and reduced rates of amide exchange at pH 7–8, even at elevated temperatures or high salt concentrations). Downfield amide chemical shifts on phosphorylation have also been observed by others, within both disordered and ordered peptides, although usually these are not investigated in a pH range of 7–8 that is necessary to ensure dianionic phospho-residues (typical pKa values of 5.5–6.0), because of expected (though not observed by us) increases in amide exchange rates decreasing sensitivity in the NMR experiment. The effects of phosphorylation on structure observed herein and previously indicate that side chain–main chain phosphate–amide hydrogen bonding could be the primary context for the interpretation of the local structural effects of serine/threonine phosphorylation, with conformational changes that could be favorable or unfavorable with respect to secondary structure in a manner readily rationalized on the basis of these interactions. These results also suggest that serine/threonine phosphorylation should be highly disruptive to β-sheet formation.24−26 Notably, tyrosine phosphorylation is not expected to allow local interaction with the protein backbone and thus is not expected to exhibit structural effects similar to those observed for serine/threonine phosphorylation.
Conclusion
The results herein comprise a systematic investigation of the structural effects of phosphorylation versus modification with β-d-O-GlcNAc (OGlcNAcylation) on the stability of α-helices via modification on serine versus threonine hydroxyls, and at the N-terminus, internal, or C-terminal α-helical positions. Phosphorylation and OGlcNAcylation are the major intracellular post-translational modifications of serine and threonine, occurring via regulated enzymatic responses to a multitude of intracellular and extracellular signals and resulting in diverse downstream intracellular responses. The results herein, in combination with our previous results on the effects of phosphorylation and OGlcNAcylation in disordered regions of proteins, should provide a broad context for interpreting structural and potentially functional effects of post-translational modifications of serine and threonine in intracellular proteins.
Acknowledgments
We thank Anil Pandey for experimental assistance.
Supporting Information Available
Synthesis of protected Fmoc-OGlcNAcylated amino acids, peptide synthesis and characterization data, CD data for all peptides, experimental procedures, and full NMR spectroscopy data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
We thank the National Institutes of Health (GM93225) and the University of Delaware for funding.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Hart G. W.; Housley M. P.; Slawson C. (2007) Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022. [DOI] [PubMed] [Google Scholar]
- Hu P.; Shimoji S.; Hart G. W. (2010) Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. 584, 2526–2538. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Gucek M.; Hart G. W. (2008) Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A. 105, 13793–13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart G. W.; Slawson C.; Ramirez-Correa G.; Lagerlof O. (2011) Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu. Rev. Biochem. 80, 825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer F. I.; Hart G. W. (2001) Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry 40, 7845–7852. [DOI] [PubMed] [Google Scholar]
- Liu F.; Iqbal K.; Grundke-Iqbal I.; Hart G. W.; Gong C. X. (2004) O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 10804–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. X.; Du J. T.; Zhou L. X.; Liu X. H.; Zhao Y. F.; Nakanishi H.; Li Y. M. (2006) Alternative O-GlcNAcylation/O-phosphorylation of Ser(16) induce different conformational disturbances to the N terminus of murine estrogen receptor β. Chem. Biol. 13, 937–944. [DOI] [PubMed] [Google Scholar]
- Slawson C.; Hart G. W. (2011) O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 11, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrant M. K.; Rho H. S.; Xie Z.; Jiang Y. L.; Gross C.; Culhane J. C.; Yan G.; Qian J.; Ichikawa Y.; Matsuoka T.; Zachara N.; Etzkorn F. A.; Hart G. W.; Jeong J. S.; Blackshaw S.; Zhu H.; Cole P. A. (2012) Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat. Chem. Biol. 8, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach J. E.; Clark P. M.; Mason D. E.; Neve R. L.; Peters E. C.; Hsieh-Wilson L. C. (2012) Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat. Chem. Biol. 8, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilak L.; Moitra J.; Krylov D.; Vinson C. (1997) Phosphorylation destabilizes α-helices. Nat. Struct. Biol. 4, 112–114. [DOI] [PubMed] [Google Scholar]
- Szilak L.; Moitra J.; Vinson C. (1997) Design of a leucine zipper coiled coil stabilized 1.4 kcal mol–1 by phosphorylation of a serine in the e position. Protein Sci. 6, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew C. D.; Warwicker J.; Jones G. R.; Doig A. J. (2002) Effect of phosphorylation on α-helix stability as a function of position. Biochemistry 41, 1897–1905. [DOI] [PubMed] [Google Scholar]
- Signarvic R. S.; DeGrado W. F. (2003) De Novo Design of a Molecular Switch: Phosphorylation-Dependent Association of Designed Peptides. J. Mol. Biol. 334, 1–12. [DOI] [PubMed] [Google Scholar]
- Errington N.; Doig A. J. (2005) A phosphoserine-lysine salt bridge with an α-helical peptide, the strongest α-helix side-chain interaction measured to date. Biochemistry 44, 7553–7558. [DOI] [PubMed] [Google Scholar]
- Balakrishnan S.; Zondlo N. J. (2006) Design of a Protein Kinase-Inducible Domain. J. Am. Chem. Soc. 128, 5590–5591. [DOI] [PubMed] [Google Scholar]
- Zondlo S. C.; Gao F.; Zondlo N. J. (2010) Design of an Encodable Tyrosine Kinase-Inducible Domain: Detection of Tyrosine Kinase Activity by Terbium Luminescence. J. Am. Chem. Soc. 132, 5619–5621. [DOI] [PubMed] [Google Scholar]
- Broncel M.; Falenski J. A.; Wagner S. C.; Hackenberger C. P. R.; Koksch B. (2010) How Post-Translational Modifications Influence Amyloid Formation: A Systematic Study of Phosphorylation and Glycosylation in Model Peptides. Chem.—Eur. J. 16, 7881–7888. [DOI] [PubMed] [Google Scholar]
- Tholey A.; Lindemann A.; Kinzel V.; Reed J. (1999) Direct effects of phosphorylation on the preferred backbone conformation of peptides: A nuclear magnetic resonance study. Biophys. J. 76, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielska A. A.; Zondlo N. J. (2006) Hyperphosphorylation of tau induces local polyproline II helix. Biochemistry 45, 5527–5537. [DOI] [PubMed] [Google Scholar]
- Meng H. Y.; Thomas K. M.; Lee A. E.; Zondlo N. J. (2006) Effects of i and i+3 Residue Identity on Cis-Trans Isomerism of the Aromatici+1-Prolyli+2 Amide Bond: Implications for Type VI β-turn Formation. Biopolymers 84, 192–204. [DOI] [PubMed] [Google Scholar]
- Lee K. K.; Joo C.; Yang S.; Han H.; Cho M. (2007) Phosphorylation effect on the GSSS peptide conformation in water: Infrared, vibrational circular dichroism, and circular dichroism experiments and comparisons with molecular dynamics simulations. J. Chem. Phys. 126, 235102. [DOI] [PubMed] [Google Scholar]
- Riemen A. J.; Waters M. L. (2009) Controlling Peptide Folding with Repulsive Interactions between Phosphorylated Amino Acids and Tryptophan. J. Am. Chem. Soc. 131, 14081–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemen A. J.; Waters M. L. (2010) Dueling Post-Translational Modifications Trigger Folding and Unfolding of a β-Hairpin Peptide. J. Am. Chem. Soc. 132, 9007–9013. [DOI] [PubMed] [Google Scholar]
- Riemen A. J.; Waters M. L. (2010) Positional effects of phosphoserine on β-hairpin stability. Org. Biomol. Chem. 8, 5411–5417. [DOI] [PubMed] [Google Scholar]
- Brown A. M.; Zondlo N. J. (2012) A Propensity Scale for Type II Polyproline Helices (PPII): Aromatic Amino Acids in Proline-Rich Sequences Strongly Disfavor PPII Due to Proline-Aromatic Interactions. Biochemistry 51, 5041–5051. [DOI] [PubMed] [Google Scholar]
- Elam W. A.; Schrank T. P.; Campagnolo A. J.; Hilser V. J. (2013) Evolutionary conservation of the polyproline II conformation surrounding intrinsically disordered phosphorylation sites. Protein Sci. 22, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister M. A.; Pandey A. K.; Bielska A. A.; Zondlo N. J. (2014) OGlcNAcylation and Phosphorylation Have Opposing Structural Effects in tau: Phosphothreonine Induces Particular Conformational Order. J. Am. Chem. Soc. 136, 3803–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. N.; Lewis R. J. (2001) Structural basis for control by phosphorylation. Chem. Rev. 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- Huse M.; Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Steichen J. M.; Iyer G. H.; Li S.; Saldanha S. A.; Deal M. S.; Woods V. L.; Taylor S. S. (2010) Global Consequences of Activation Loop Phosphorylation on Protein Kinase A. J. Biol. Chem. 285, 3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen J. M.; Kuchinskas M.; Keshwani M. M.; Yang J.; Adams J. A.; Taylor S. S. (2012) Structural Basis for the Regulation of Protein Kinase A by Activation Loop Phosphorylation. J. Biol. Chem. 287, 14672–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan I.; Perez-Alvarado G. C.; Parker D.; Dyson H. J.; Montminy M.; Wright P. E. (1997) Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator-coactivator interactions. Cell 91, 741–752. [DOI] [PubMed] [Google Scholar]
- Parker D.; Jhala U. S.; Radhakrishnan I.; Yaffe M. B.; Reyes C.; Shulman A. I.; Cantley L. C.; Wright P. E.; Montminy M. (1998) Analysis of an activator:coactivator complex reveals an essential role for secondary structure in transcriptional activation. Mol. Cell 2, 353–359. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I.; Pérez-Alvarado G. C.; Dyson H. J.; Wright P. E. (1998) Conformational preferences in Ser133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS Lett. 430, 317–322. [DOI] [PubMed] [Google Scholar]
- Simanek E. E.; Huang D. H.; Pasternack L.; Machajewski T. D.; Seitz O.; Millar D. S.; Dyson H. J.; Wong C. H. (1998) Glycosylation of threonine of the repeating unit of RNA polymerase II with β-linked N-acetylglucosame leads to a turnlike structure. J. Am. Chem. Soc. 120, 11567–11575. [Google Scholar]
- Verdecia M. A.; Bowman M. E.; Lu K. P.; Hunter T.; Noel J. P. (2000) Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 7, 639–643. [DOI] [PubMed] [Google Scholar]
- Noble C. G.; Hollingworth D.; Martin S. R.; Ennis-Adeniran V.; Smerdon S. J.; Kelly G.; Taylor I. A.; Ramos A. (2005) Key features of the interaction between Pcf11 CID and RNA polymerase II CTD. Nat. Struct. Mol. Biol. 12, 144–151. [DOI] [PubMed] [Google Scholar]
- Liang F.-C.; Chen R. P.-Y.; Lin C.-C.; Huang K.-T.; Chan S. I. (2006) Tuning the conformation properties of a peptide by glycosylation and phosphorylation. Biochem. Biophys. Res. Commun. 342, 482–488. [DOI] [PubMed] [Google Scholar]
- Laughrey Z. R.; Kiehna S. E.; Riemen A. J.; Waters M. L. (2008) Carbohydrate-π Interactions: What Are They Worth?. J. Am. Chem. Soc. 130, 14625–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana Y.; Fletcher G. L.; Fujitani N.; Tsuda S.; Monde K.; Nishimura S. I. (2004) Antifreeze glycoproteins: Elucidation of the structural motifs that are essential for antifreeze activity. Angew. Chem., Int. Ed. 43, 856–862. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Zhang L.; Li X. J.; Run X. Q.; Liang Z. H.; Li Y.; Liu Y.; Lee M. H.; Grundke-Iqbal I.; Iqbal K.; Vocadlo D. J.; Liu F.; Gong C. X. (2012) Differential Effects of an O-GlcNAcase Inhibitor on Tau Phosphorylation. PLoS One 7, e35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa S. A.; Shan X. Y.; Macauley M. S.; Clark T.; Skorobogatko Y.; Vosseller K.; Vocadlo D. J. (2012) Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399. [DOI] [PubMed] [Google Scholar]
- Vuister G. W.; Bax A. (1993) Quantitative J Correlation: A New Approach for Measuring Homonuclear Three-Bond JHNHα Coupling Constants in 15N-enriched Proteins. J. Am. Chem. Soc. 115, 7772–7777. [Google Scholar]
- Arsequell G.; Krippner L.; Dwek R. A.; Wong S. Y. C. (1994) Building blocks for solid-phase glycopeptide synthesis: 2-Acetamido-2-deoxy-β-d-glycosides of FmocSerOH and FmocThrOH. Chem. Commun. 2383–2384. [Google Scholar]
- Sjölin P.; Elofsson M.; Kihlberg J. (1996) Removal of Acyl Protective Groups from Glycopeptides: Base Does Not Epimerize Peptide Stereocenters, and β-Elimination Is Slow. J. Org. Chem. 61, 560–565. [DOI] [PubMed] [Google Scholar]
- Luo P. Z.; Baldwin R. L. (1997) Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 36, 8413–8421. [DOI] [PubMed] [Google Scholar]
- Rohl C. A.; Baldwin R. L. (1997) Comparison of NH Exchange and Circular Dichroism as Techniques for Measuring the Parameters of the Helix-Coil Transition in Peptides. Biochemistry 36, 8435–8442. [DOI] [PubMed] [Google Scholar]
- Richardson J. S.; Richardson D. C. (1988) Amino-Acid Preferences for Specific Locations at the Ends of α-Helices. Science 240, 1648–1652. [DOI] [PubMed] [Google Scholar]
- Serrano L.; Fersht A. R. (1989) Capping and α-Helix Stability. Nature 342, 296–299. [DOI] [PubMed] [Google Scholar]
- Lyu P. C.; Zhou H. X.; Jelveh N.; Wemmer D. E.; Kallenbach N. R. (1992) Position-dependent stabilizing effects in α-helices: N-terminal capping in synthetic model peptides. J. Am. Chem. Soc. 114, 6560–6562. [Google Scholar]
- Chakrabartty A.; Doig A. J.; Baldwin R. L. (1993) Helix Capping Propensities in Peptides Parallel Those in Proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 11332–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig A. J.; Chakrabartty A.; Klingler T. M.; Baldwin R. L. (1994) Determination of Free-Energies of N-Capping in α-Helices by Modification of the Lifson-Roig Helix-Coil Theory to Include N-Capping and C-Capping. Biochemistry 33, 3396–3403. [DOI] [PubMed] [Google Scholar]
- Doig A. J.; Baldwin R. L. (1995) N- and C-Capping Preferences for All 20 Amino-Acids in α-Helical Peptides. Protein Sci. 4, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran D. A. E.; Penel S.; Doig A. J. (2001) Effect of the N1 residue on the stability of the α-helix for all 20 amino acids. Protein Sci. 10, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora R.; Rose G. D. (1998) Helix capping. Protein Sci. 7, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol W. G. J.; Vanduijnen P. T.; Berendsen H. J. C. (1978) α-Helix Dipole and Properties of Proteins. Nature 273, 443–446. [DOI] [PubMed] [Google Scholar]
- Shoemaker K. R.; Kim P. S.; York E. J.; Stewart J. M.; Baldwin R. L. (1987) Tests of the Helix Dipole Model for Stabilization of α-Helices. Nature 326, 563–567. [DOI] [PubMed] [Google Scholar]
- Bienkiewicz E. A.; Lumb K. J. (1999) Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 15, 203–206. [DOI] [PubMed] [Google Scholar]
- Landrieu I.; Lacosse L.; Leroy A.; Wieruszeski J. M.; Trivelli X.; Sillen A.; Sibille N.; Schwalbe H.; Saxena K.; Langer T.; Lippens G. (2006) NMR analysis of a Tau phosphorylation pattern. J. Am. Chem. Soc. 128, 3575–3583. [DOI] [PubMed] [Google Scholar]
- Lippens G.; Landrieu I.; Hanoulle X. (2008) Studying posttranslational modifications by in-cell NMR. Chem. Biol. 15, 311–312. [DOI] [PubMed] [Google Scholar]
- Lippens G.; Amniai L.; Wieruszeski J. M.; Sillen A.; Leroy A.; Landrieu I. (2012) Towards understanding the phosphorylation code of tau. Biochem. Soc. Trans. 40, 698–703. [DOI] [PubMed] [Google Scholar]
- Sibille N.; Huvent I.; Fauquant C.; Verdegem D.; Amniai L.; Leroy A.; Wieruszeski J. M.; Lippens G.; Landrieu I. (2012) Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered tau protein. Proteins 80, 454–462. [DOI] [PubMed] [Google Scholar]
- Theillet F. X.; Smet-Nocca C.; Liokatis S.; Thongwichian R.; Kosten J.; Yoon M. K.; Kriwacki R. W.; Landrieu I.; Lippens G.; Selenko P. (2012) Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR 54, 217–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil K. T.; Degrado W. F. (1990) A Thermodynamic Scale for the Helix-Forming Tendencies of the Commonly Occurring Amino-Acids. Science 250, 646–651. [DOI] [PubMed] [Google Scholar]
- Lyu P. C.; Liff M. I.; Marky L. A.; Kallenbach N. R. (1990) Side chain contributions to the stability of α-helical structure in peptides. Science 250, 669–673. [DOI] [PubMed] [Google Scholar]
- Lyu P. C.; Wemmer D. E.; Zhou H. X.; Pinker R. J.; Kallenbach N. R. (1993) Capping Interactions in Isolated α-Helices: Position-Dependent Substitution Effects and Structure of a Serine-Capped Peptide Helix. Biochemistry 32, 421–425. [DOI] [PubMed] [Google Scholar]
- Blaber M.; Zhang X. J.; Matthews B. W. (1993) Structural Basis of Amino-Acid α-Helix Propensity. Science 260, 1637–1640. [DOI] [PubMed] [Google Scholar]
- Pérez C.; Lühr F.; Rüterjans H.; Schmidt J. M. (2001) Self-Consistent Karplus Parametrization of 3J Couplings Depending on the Polypeptide Side-Chain Torsion χ1. J. Am. Chem. Soc. 123, 7081–7093. [DOI] [PubMed] [Google Scholar]
- Schmidt J. M. (2007) Asymmetric Karplus Curves for the Protein Side-Chain 3J couplings. J. Biomol. NMR 37, 287–301. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Word J. M.; Richardson J. S.; Richardson D. C. (2000) The Penultimate Rotamer Library. Proteins 40, 389–408. [PubMed] [Google Scholar]
- Creamer T. P.; Rose G. D. (1992) Side-Chain Entropy Opposes α-Helix Formation but Rationalizes Experimentally Determined Helix-Forming Propensities. Proc. Natl. Acad. Sci. U.S.A. 89, 5937–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaber M.; Zhang X. J.; Lindstrom J. D.; Pepiot S. D.; Baase W. A.; Matthews B. W. (1994) Determination of α-Helix Propensity within the Context of a Folded Protein: Sites 44 and 131 in Bacteriophage-T4 Lysozyme. J. Mol. Biol. 235, 600–624. [DOI] [PubMed] [Google Scholar]
- Liehr S.; Chenault H. K. (1999) A comparison of the α-helix forming propensities and hydrogen bonding properties of serine phosphate and α-amino-γ-phosphonobutyric acid. Bioorg. Med. Chem. Lett. 9, 2759–2762. [DOI] [PubMed] [Google Scholar]
- Lyu P. C.; Sherman J. C.; Chen A.; Kallenbach N. R. (1991) α-Helix Stabilization by Natural and Unnatural Amino-Acids with Alkyl Side-Chains. Proc. Natl. Acad. Sci. U.S.A. 88, 5317–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formaggio F.; Baldini C.; Moretto V.; Crisma M.; Kaptein B.; Broxterman Q. B.; Toniolo C. (2005) Preferred conformations of peptides containing tert-leucine, a sterically demanding, lipophilic α-amino acid with a quaternary side-chain C-β atom. Chem.—Eur. J. 11, 2395–2404. [DOI] [PubMed] [Google Scholar]
- Chiu H.-P.; Suzuki Y.; Gullickson D.; Ahmad R.; Kokona B.; Fairman R.; Cheng R. P. (2006) Helix Propensity of Highly Fluorinated Amino Acids. J. Am. Chem. Soc. 128, 15556–15557. [DOI] [PubMed] [Google Scholar]
- Chiu H.-P.; Kokona B.; Fairman R.; Cheng R. P. (2009) Effect of Highly Fluorinated Amino Acids on Protein Stability at a Solvent-Exposed Position on an Internal Strand of Protein G B1 Domain. J. Am. Chem. Soc. 131, 13192–13192. [DOI] [PubMed] [Google Scholar]
- Toniolo C.; Crisma M.; Formaggio F.; Peggion C. (2001) Control of peptide conformation by the Thorpe-Ingold effect (C(α)-tetrasubstitution). Biopolymers 60, 396–419. [DOI] [PubMed] [Google Scholar]
- Engel D. E.; DeGrado W. F. (2004) Amino acid propensities are position-dependent throughout the length of α-helices. J. Mol. Biol. 337, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd N. E.; Hoang H. N.; Abbenante G.; Fairlie D. P. (2005) Single turn peptide α helices with exceptional stability in water. J. Am. Chem. Soc. 127, 2974–2983. [DOI] [PubMed] [Google Scholar]
- Nilsson I.; Saaf A.; Whitley P.; Gafvelin G.; Waller C.; von Heijne G. (1998) Proline-induced disruption of a transmembrane α-helix in its natural environment. J. Mol. Biol. 284, 1165–1175. [DOI] [PubMed] [Google Scholar]
- Cordes F. S.; Bright J. N.; Sansom M. S. (2002) Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323, 951–960. [DOI] [PubMed] [Google Scholar]
- Barlow D. J.; Thornton J. M. (1988) Helix Geometry in Proteins. J. Mol. Biol. 201, 601–619. [DOI] [PubMed] [Google Scholar]
- Presta L. G.; Rose G. D. (1988) Helix Signals in Proteins. Science 240, 1632–1641. [DOI] [PubMed] [Google Scholar]
- Schneider J. P.; DeGrado W. F. (1998) The design of efficient α-helical C-capping auxiliaries. J. Am. Chem. Soc. 120, 2764–2767. [Google Scholar]
- Kallenbach N. R.; Gong Y. X. (1999) C-terminal capping motifs in model helical peptides. Bioorg. Med. Chem. 7, 143–151. [DOI] [PubMed] [Google Scholar]
- Serrano L.; Sancho J.; Hirshberg M.; Fersht A. R. (1992) α-Helix Stability in Proteins. 1. Empirical Correlations Concerning Substitution of Side-Chains at the N and C-Caps and the Replacement of Alanine by Glycine or Serine at Solvent-Exposed Surfaces. J. Mol. Biol. 227, 544–559. [DOI] [PubMed] [Google Scholar]
- Petukhov M.; Uegaki K.; Yumoto N.; Serrano L. (2002) Amino acid intrinsic α-helical propensities III: Positional dependence at several positions of the C terminus. Protein Sci. 11, 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P. R.; Flory P. J. (1968) Confromational energies and configurational statistics of copolypeptides containing l-proline. J. Mol. Biol. 34, 105–120. [DOI] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. (1991) Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K.; Nagarajaram H. A.; Ramakrishnan C.; Balaram P. (1998) Stereochemical punctuation marks in protein structures: Glycine and proline containing helix stop signals. J. Mol. Biol. 275, 917–932. [DOI] [PubMed] [Google Scholar]
- Cochran D. A. E.; Doig A. J. (2001) Effect of the N2 residue on the stability of the α-helix for all 20 amino acids. Protein Sci. 10, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D.; Richards F. M. (1992) The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) The C-13 Chemical Shift Index: A Simple Method for the Identification of Protein Secondary Structure Using C-13 Chemical-Shift Data. J. Biomol. NMR 4, 171–180. [DOI] [PubMed] [Google Scholar]
- Lankhorst P. P.; Haasnoot C. A. G.; Erkelens C.; Altona C. (1984) Nucleic Acid Constituents. 36. C-13 NMR in Conformational-Analysis of Nucleic-Acid Fragments. 2. A Reparametrization of the Karplus Equation for Vicinal NMR Coupling-Constants in CCOP and HCOP Fragments. J. Biomol. Struct. Dyn. 1, 1387–1405. [DOI] [PubMed] [Google Scholar]
- Ho C.; Magnuson J. A.; Wilson J. B.; Magnuson N. S.; Kurland R. J. (1969) Phosphorus Nuclear Magnetic Resonance Studies of Phosphoproteins and Phosphorylated Molecules. 2. Chemical Nature of Phosphorus Atoms in αs-Casein-B and Phosvitin. Biochemistry 8, 2074–2082. [DOI] [PubMed] [Google Scholar]
- Vogel H. J.; Bridger W. A. (1982) P-31 Nuclear Magnetic-Resonance Studies of the 2 Phosphoserine Residues of Hen Egg-White Ovalbumin. Biochemistry 21, 5825–5831. [DOI] [PubMed] [Google Scholar]
- Brauer M.; Sykes B. D. (1984) P-31 Nuclear Magnetic-Resonance Studies of Phosphorylated Proteins. Methods Enzymol. 107, 36–81. [DOI] [PubMed] [Google Scholar]
- Matheis G.; Whitaker J. R. (1984) P-31 NMR Chemical-Shifts of Phosphate Covalently Bound to Proteins. Int. J. Biochem. 16, 867–873. [DOI] [PubMed] [Google Scholar]
- McIntosh L. P.; Kang H. S.; Okon M.; Nelson M. L.; Graves B. J.; Brutscher B. (2009) Detection and assignment of phosphoserine and phosphothreonine residues by 13C-31P spin-echo difference NMR spectroscopy. J. Biomol. NMR 43, 31–37. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. X.; Lyu P. C.; Wemmer D. E.; Kallenbach N. R. (1994) α-Helix Capping in Synthetic Model Peptides by Reciprocal Side-Chain Main-Chain Interactions: Evidence for an N-Terminal Capping Box. Proteins 18, 1–7. [DOI] [PubMed] [Google Scholar]
- Lu M.; Shu W.; Ji H.; Spek E.; Wang L. Y.; Kallenbach N. R. (1999) Helix capping in the GCN4 leucine zipper. J. Mol. Biol. 288, 743–752. [DOI] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. (2010) Systematic Analysis of Helical Protein Interfaces Reveals Targets for Synthetic Inhibitors. ACS Chem. Biol. 5, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarracino D. A.; Bullock B. N.; Arora P. S. (2011) Protein-Protein Interactions in Transcription: A Fertile Ground for Helix Mimetics. Biopolymers 95, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Perumal N. B.; Oldfield C. J.; Su E. W.; Uversky V. N.; Dunker A. K. (2006) Intrinsic Disorder in Transcription Factors. Biochemistry 45, 6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbur J. R.; Tabor A. D.; Cheng X. D.; Wang H.; Uesugi M.; Lozano G.; Zhang W. (2002) MDM-2 binding and TAFII31 recruitment is regulated by hydrogen bond disruption between the p53 residues Thr18 and Asp21. Oncogene 21, 7100–7113. [DOI] [PubMed] [Google Scholar]
- Iakoucheva L. M.; Radivojac P.; Brown C. J.; O’Connor T. R.; Sikes J. G.; Obradovic Z.; Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlummer S.; Vetter R.; Kuder N.; Henkel A.; Chen Y. X.; Li Y. M.; Kuhlmann J.; Waldmann H. (2006) Influence of serine O-glycosylation or O-phoshorylation close to the vJun nuclear localisation sequence on nuclear import. ChemBioChem 7, 88–97. [DOI] [PubMed] [Google Scholar]
- Schon O.; Friedler A.; Bycroft M.; Freund S. M. V.; Fersht A. R. (2002) Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 323, 491–501. [DOI] [PubMed] [Google Scholar]
- Teufel D. P.; Bycroft M.; Fersht A. R. (2009) Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 28, 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. W.; Ferreon J. C.; Ferreon A. C. M.; Arai M.; Wright P. E. (2010) Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 19290–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. J.; Bullen J. W.; Hart G. W. (2008) Cross-talk between GlcNAcylation and phosphorylation: Roles in insulin resistance and glucose toxicity. Am. J. Physiol. 295, E17–E28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidan Q.; Hart G. W. (2010) The intersections between O-GlcNAcylation and phosphorylation: Implications for multiple signaling pathways. J. Cell Sci. 123, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.-T.; Li Y.-M.; Wei W.; Wu G.-S.; Zhao Y.-F.; Kanazawa K.; Nemoto T.; Nakanishi H. (2005) Low-barrier hydrogen bond between phosphate and the amide group in phosphopeptide. J. Am. Chem. Soc. 127, 16350–16351. [DOI] [PubMed] [Google Scholar]
- Wong S. E.; Bernacki K.; Jacobson M. (2005) Competition between intramolecular hydrogen bonds and solvation in phosphorylated peptides: Simulations with explicit and implicit solvent. J. Phys. Chem. B 109, 5249–5258. [DOI] [PubMed] [Google Scholar]
- Vijayakumar M.; Qian H.; Zhou H. X. (1999) Hydrogen bonds between short polar side chains and peptide backbone: Prevalence in proteins and effects on helix-forming propensities. Proteins 34, 497–507. [PubMed] [Google Scholar]
- Eswar N.; Ramakrishnan C. (2000) Deterministic features of side-chain main-chain hydrogen bonds in globular protein structures. Protein Eng. 13, 227–238. [DOI] [PubMed] [Google Scholar]
- Ramelot T. A.; Nicholson L. K. (2001) Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J. Mol. Biol. 307, 871–884. [DOI] [PubMed] [Google Scholar]
- Groban E. S.; Narayanan A.; Jacobson M. P. (2006) Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput. Biol. 2, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-K.; Kitn E.; Joo C.; Song J.; Han H.; Cho M. (2008) Site-selective Intramolecular Hydrogen-Bonding Interactions in Phosphorylated Serine and Threonine Dipeptides. J. Phys. Chem. B 112, 16782–16787. [DOI] [PubMed] [Google Scholar]
- Kim S. Y.; Jung Y.; Hwang G. S.; Han H.; Cho M. (2011) Phosphorylation alters backbone conformational preferences of serine and threonine peptides. Proteins 79, 3155–3165. [DOI] [PubMed] [Google Scholar]
- Corzana F.; Busto J. H.; Jimenez-Oses G.; de Luis M. G.; Asensio J. L.; Jimenez-Barbero J.; Peregrina J. M.; Avenoza A. (2007) Serine versus threonine glycosylation: The methyl group causes a drastic alteration on the carbohydrate orientation and on the surrounding water shell. J. Am. Chem. Soc. 129, 9458–9467. [DOI] [PubMed] [Google Scholar]
- Fernandez-Tejada A.; Corzana F.; Busto J. H.; Jimenez-Oses G.; Jimenez-Barbero J.; Avenoza A.; Peregrina J. M. (2009) Insights into the Geometrical Features Underlying β-O-GlcNAc Glycosylation: Water Pockets Drastically Modulate the Interactions between the Carbohydrate and the Peptide Backbone. Chem.—Eur. J. 15, 7297–7301. [DOI] [PubMed] [Google Scholar]
- Corzana F.; Fernandez-Tejada A.; Busto J. H.; Joshi G.; Davis A. P.; Jimenez-Barbero J.; Avenoza A.; Peregrina J. M. (2011) Molecular Recognition of β-O-GlcNAc Glycopeptides by a Lectin-Like Receptor: Binding Modulation by the Underlying Ser or Thr Amino Acids. ChemBioChem 12, 110–117. [DOI] [PubMed] [Google Scholar]
- Clackson T.; Wells J. A. (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267, 383–386. [DOI] [PubMed] [Google Scholar]
- Thanos C. D.; Randal M.; Wells J. A. (2003) Potent small-molecule binding to a dynamic hot spot on IL-2. J. Am. Chem. Soc. 125, 15280–15281. [DOI] [PubMed] [Google Scholar]
- Kemp D. S.; Curran T. P.; Boyd J. G.; Allen T. J. (1991) Studies of N-Terminal Templates for α-Helix Formation: Synthesis and Conformational-Analysis of Peptide Conjugates of (2S,5S,8S,11S)-1-Acetyl-1,4-diaza-3-keto-5-carboxy-10-thiatricyclo[2.8.1 0.0(4,8)]tridecane (Ac-Hel1-Oh). J. Org. Chem. 56, 6683–6697. [Google Scholar]
- Kemp D. S.; Curran T. P.; Davis W. M.; Boyd J. G.; Muendel C. (1991) Studies of N-Terminal Templates for α-Helix Formation: Synthesis and Conformational-Analysis of (2S,5S,8S,11S)-1-Acetyl-1,4-diaza-3-keto-5-carboxy-10-thiatricyclo[2.8.1 0.0(4,8)]tridecane (Ac-Hel1-Oh). J. Org. Chem. 56, 6672–6682. [Google Scholar]
- Kemp D. S.; Allen T. J.; Oslick S. L. (1995) The Energetics of Helix Formation by Short Templated Peptides in Aqueous Solution. 1. Characterization of the Reporting Helical Template Ac-Hel1. J. Am. Chem. Soc. 117, 6641–6657. [Google Scholar]
- Viguera A. R.; Serrano L. (1999) Stable proline box motif at the N-terminal end of α-helices. Protein Sci. 8, 1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiersen H.; Rees A. R. (2001) The hunchback and its neighbours: Proline as an environmental modulator. Trends Biochem. Sci. 26, 679–684. [DOI] [PubMed] [Google Scholar]
- Senes A.; Engel D. E.; DeGrado W. F. (2004) Folding of helical membrane proteins: The role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 14, 465–479. [DOI] [PubMed] [Google Scholar]
- Li S. C.; Goto N. K.; Williams K. A.; Deber C. M. (1996) α-Helical, but not β-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. U.S.A. 93, 6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P.; Chakrabarti S. (1998) C–H···O hydrogen bond involving proline residues in α-helices. J. Mol. Biol. 284, 867–873. [DOI] [PubMed] [Google Scholar]
- Orzaez M.; Salgado J.; Gimenez-Giner A.; Perez-Paya E.; Mingarro I. (2004) Influence of proline residues in transmembrane helix packing. J. Mol. Biol. 335, 631–640. [DOI] [PubMed] [Google Scholar]
- Zondlo S. C.; Lee A. E.; Zondlo N. J. (2006) Determinants of Specificity of MDM2 for the Activation Domains of p53 and p65: Proline27 Disrupts the MDM2-binding motif of p53. Biochemistry 45, 11945–11957. [DOI] [PubMed] [Google Scholar]
- Seavers P. R.; Lewis R. J.; Brannigan J. A.; Verschueren K. H. G.; Murshudov G. N.; Wilkinson A. J. (2001) Structure of the Bacillus cell fate determinant SpollAA in phosphorylated and unphosphorylated forms. Structure 9, 605–614. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Singh N.; Sharma S.; Singh S. B.; Kaur P.; Bhushan A.; Srinivasan A.; Singh T. P. (2008) Crystal structure of lactoperoxidase at 2.4 angstrom resolution. J. Mol. Biol. 376, 1060–1075. [DOI] [PubMed] [Google Scholar]
- Schirner K.; Marles-Wright J.; Lewis R. J.; Errington J. (2009) Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 28, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; VanVeller B.; Guzei I. A.; Raines R. T. (2013) n→π* Interactions of Amides and Thioamides: Implications for Protein Stability. J. Am. Chem. Soc. 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Newberry R. W.; VanVeller B.; Raines R. T.; Woolfson D. N. (2013) Interplay of Hydrogen Bonds and n→π* Interactions in Proteins. J. Am. Chem. Soc. 135, 18682–18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderaker M. P.; Raines R. T. (2003) An electronic effect on protein structure. Protein Sci. 12, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges J. A.; Raines R. T. (2006) Energetics of an n→π* interaction that impacts protein structure. Org. Lett. 8, 4695–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. (2010) n→π* interactions in proteins. Nat. Chem. Biol. 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Raines R. T. (2011) Signature of n→π* interactions in α-helices. Protein Sci. 20, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo L. D.; Krishnamoorthy L.; Mahal L. K. (2006) A Cellular FRET-Based Sensor for β-O-GlcNAc, a Dynamic Carbohydrate Modification Involved in Signaling. J. Am. Chem. Soc. 128, 14768–14769. [DOI] [PubMed] [Google Scholar]
- Rexach J. E.; Clark P. M.; Hsieh-Wilson L. C. (2008) Chemical approaches to understanding O-GlcNAc glycosylation in the brain. Nat. Chem. Biol. 4, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo L. D.; Froemming J. A.; Mahal L. K. (2011) Targeted in Vivo O-GlcNAc Sensors Reveal Discrete Compartment-specific Dynamics during Signal Transduction. J. Biol. Chem. 286, 6650–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt M. R.; Bertozzi C. R. (2005) Synthetic glycopeptides and glycoproteins as tools for biology. Chem. Soc. Rev. 34, 58–68. [DOI] [PubMed] [Google Scholar]
- Dube D. H.; Bertozzi C. R. (2005) Glycans in cancer and inflammation: Potential for therapeutics and diagnostics. Nat. Rev. Drug Discovery 4, 477–488. [DOI] [PubMed] [Google Scholar]
- Gamblin D. P.; Scanlan E. M.; Davis B. G. (2009) Glycoprotein Synthesis: An Update. Chem. Rev. 109, 131–163. [DOI] [PubMed] [Google Scholar]
- Kiessling L. L.; Splain R. A. (2010) Chemical Approaches to Glycobiology. Annu. Rev. Biochem. 79, 619–653. [DOI] [PubMed] [Google Scholar]
- Wang P.; Dong S.; Brailsford J. A.; Iyer K.; Townsend S. D.; Zhang Q.; Hendrickson R. C.; Shieh J.; Moore M. A. S.; Danishefsky S. J. (2012) At Last: Erythropoietin as a Single Glycoform. Angew. Chem., Int. Ed. 51, 11576–11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes G. C. L.; Castagner B.; Seeberger P. H. (2009) Combined Approaches to the Synthesis and Study of Glycoproteins. ACS Chem. Biol. 4, 703–713. [DOI] [PubMed] [Google Scholar]
- O’Connor S. E.; Imperiali B. (1997) Conformational switching by asparagine-linked glycosylation. J. Am. Chem. Soc. 119, 2295–2296. [Google Scholar]
- Imperiali B.; O’Connor S. E. (1999) Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Struct. Biol. 3, 643–649. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Powers D. L.; Powers E. T.; Kelly J. W. (2011) Glycosylation of the enhanced aromatic sequon is similarly stabilizing in three distinct reverse turn contexts. Proc. Natl. Acad. Sci. U.S.A. 108, 14127–14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culyba E. K.; Price J. L.; Hanson S. R.; Dhar A.; Wong C. H.; Gruebele M.; Powers E. T.; Kelly J. W. (2011) Protein Native State State Stabilization by Placing Aromatic Side Chains in N-Glycosylated Reverse Turns. Science 331, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C. R.; Maiti B.; Noid W. G. (2012) Specific and Nonspecific Effects of Glycosylation. J. Am. Chem. Soc. 134, 8184–8193. [DOI] [PubMed] [Google Scholar]
- McManus A. M.; Otvos L. Jr.; Hoffmann R.; Craik D. J. (1999) Conformational Studies by NMR of the Antimicrobial Peptide, Drosocin, and Its Non-Glycosylated Derivative: Effects of Glycosylation on Solution Conformation. Biochemistry 38, 705–714. [DOI] [PubMed] [Google Scholar]
- Seitz O. (2000) Glycopeptide synthesis and the effects of glycosylation on protein structure and activity. ChemBioChem 1, 215–246. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Shental-Bechor D.; Dhar A.; Turner M. J.; Powers E. T.; Gruebele M.; Levy Y.; Kelly J. W. (2010) Context-Dependent Effects of Asparagine Glycosylation on Pin WW Folding Kinetics and Thermodynamics. J. Am. Chem. Soc. 132, 15359–15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam R. Y.; Rowley C. N.; Petrov I.; Zhang T. Y.; Afagh N. A.; Woo T. K.; Ben R. N. (2009) Solution Conformation of C-Linked Antifreeze Glycoprotein Analogues and Modulation of Ice Recrystallization. J. Am. Chem. Soc. 131, 15745–15753. [DOI] [PubMed] [Google Scholar]
- Culyba E. K.; Price J. L.; Hanson S. R.; Dhar A.; Wong C. H.; Gruebele M.; Powers E. T.; Kelly J. W. (2011) Protein Native-State Stabilization by Placing Aromatic Side Chains in N-Glycosylated Reverse Turns. Science 331, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-Y.; Lin C.-C.; Chang Y.-T.; Lin S.-C.; Chan S. I. (2002) One O-linked sugar can affect the coil-to-β structural transition of the prion peptide. Proc. Natl. Acad. Sci. U.S.A. 99, 13633–13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcilius L.; Santhakumar G.; Stone R. S.; Capicciotti C. J.; Joseph S.; Matthews J. M.; Ben R. N.; Payne R. J. (2013) Synthesis of peptides and glycopeptides with polyproline II helical topology as potential antifreeze molecules. Bioorg. Med. Chem. 21, 3569–3581. [DOI] [PubMed] [Google Scholar]
- Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. (2001) Conformational Stability of Collagen Relies on a Stereoelectronic Effect. J. Am. Chem. Soc. 123, 777–778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.