

Abstract
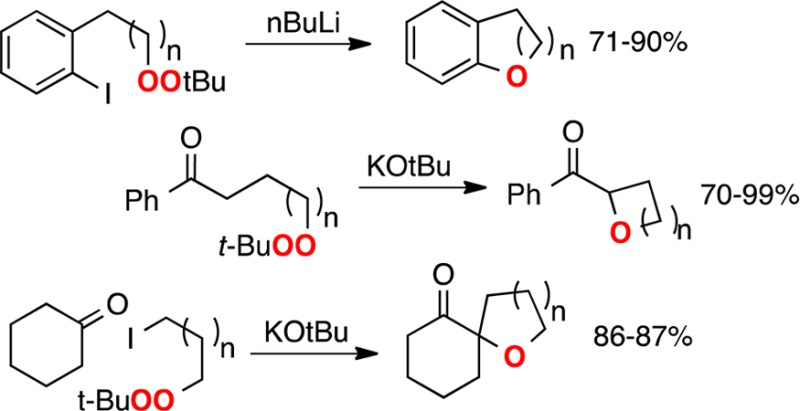
The intramolecular reaction of dialkyl peroxides with carbanions, generated via chemoselective metal-heteroatom exchange or deprotonation, provides a new approach to cyclic ethers. Applied in tandem with C–C bond formation, the strategy enables a one-step annelation to form oxaospirocycles.
Ethers, which comprise critical substructures in many bioactive molecules and natural products,1 are typically synthesized through attack of nucleophilic oxygen on an electrophilic carbon.1,2 The converse of this strategy, attack of a carbanion on electrophilic oxygen, has been investigated to only a limited extent for intermolecular reactions and is essentially unexplored for intramolecular reactions. We now demonstrate that chemoselective generation of carbanions in the presence of appropriately positioned O–O bonds of dialkyl peroxides allows the efficient introduction of cyclic ethers (Figure 1), including frameworks (for example, 15a or 19b, vide infra) challenging to approach via existing methodology.
Figure 1.
Overview of oxacycle synthesis via reactions of peroxides and carbanions.
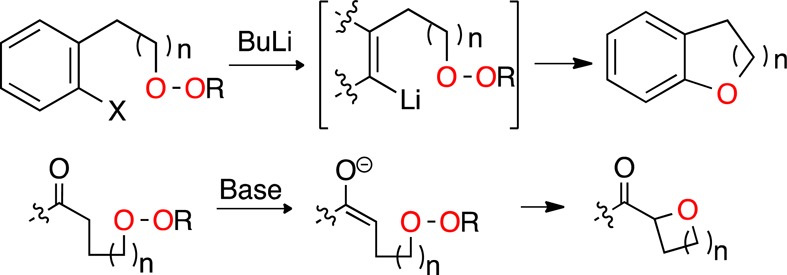
Previous reports have described the intermolecular reaction of simple organometallics with dialkyl peroxides,3 peresters,4 and endoperoxides.5 Bissilyl peroxides and lithiated hydroperoxides have been applied to oxygenation of lithiated arenes and alkenes.6,7 However, the only precedent for the corresponding intramolecular reactions of carbanions with peroxides is the 3-exo cyclization of short-lived enolate intermediates formed during nucleophilic epoxidation reactions.8
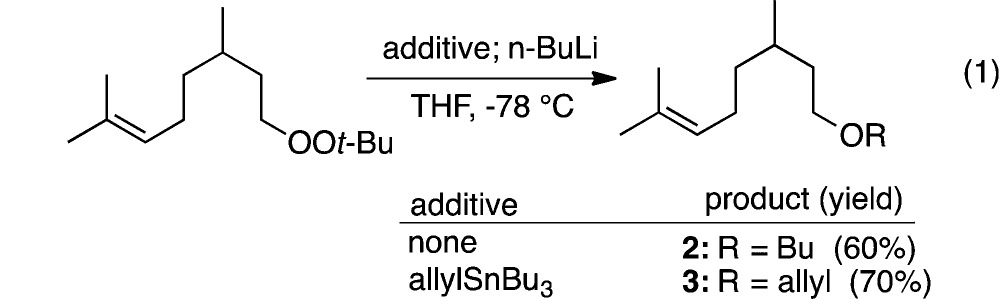
At the outset of these studies, we faced two major uncertainties: first, would it be possible to generate reactive carbanions in the presence of a peroxide; and, second, would the carbanion/peroxide pair undergo intramolecular C–O bond formation. The question of chemoselective generation of an organolithium nucleophile was initially investigated using a dialkyl peroxide (1) derived from reaction of t-butyl hydroperoxide with dihydrogeraniol methanesulfonate.9 Consistent with previous observations,3a reaction of 1 with n-BuLi proceeded readily to furnish butyl ether 2 (eq 1). Repeating this reaction in the presence of a slight excess of allyl tributylstannane resulted in predominant formation of the allyl ether (3), demonstrating that Li/heteroatom exchange proceeds much more rapidly than C–O bond formation.
 |
1 |
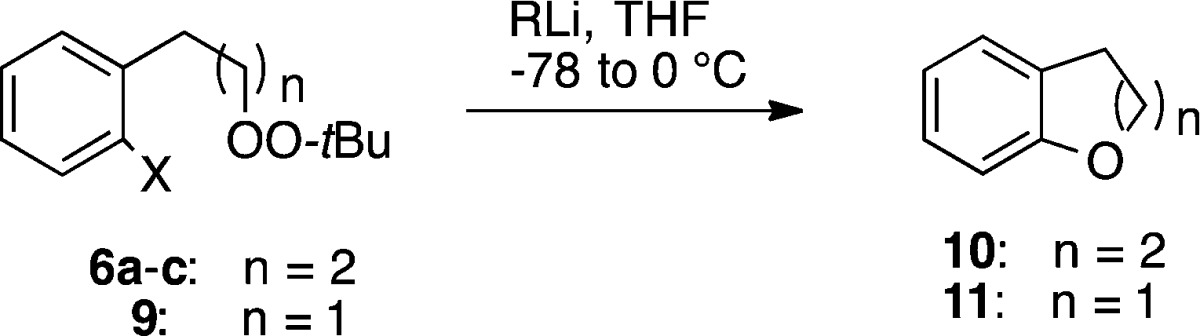
Confident of our ability to selectively generate a carbanion in the presence of a peroxide, we prepared a family of substrates (6a–c and 9) incorporating a dialkyl peroxide and a precursor aryllithium precursor (Scheme 1). The peroxide was either installed via displacement of a sulfonate (base) or iodide (Ag2O).10 The lower yield for phenethyl peroxide 9 reflects a tendency for Kornblum elimination in this skeleton.11
Scheme 1. Synthesis of Aryl Peroxide Substrates.

Table 1 illustrates the results of metal-heteroatom exchange for peroxides 6a–c and 9. Addition of either n-BuLi or PhLi to a–78 °C solution of 6a (iodide) or 6b (bromide), followed by warming of the reaction mixture to 0 °C, afforded good yields of dihydrobenzopyran 10. The corresponding reactions of the tributylstannyl arene (6c) proceeded in much lower yield (PhLi) or essentially not at all (BuLi). Lithiation of o-iodophenethyl peroxide 9 furnished dihydrobenzofuran in excellent yield.
Table 1. Lithiation/Cyclization.
| subst | n | X | prod | R | yield (%)a |
|---|---|---|---|---|---|
| 6a | 2 | I | 9 | n-Bu | 71 |
| 6a | 2 | I | 9 | Ph | 73 |
| 6b | 2 | Br | 9 | n-Bu | 71 |
| 6b | 2 | Br | 9 | Ph | 47 |
| 6c | 2 | SnBu3 | 9 | n-Bu | trace |
| 6c | 2 | SnBu3 | 9 | Ph | 19 |
| 9 | 1 | I | 10 | n-Bu | 90(58)b |
GC yields.
Isolated yield.
Repeating the lithiation of 6a and quenching the reaction with MeOH-d4 prior to warming resulted in formation of a monodeuterated peroxide (eq 2), demonstrating that lithiation occurs more rapidly and at lower temperature than intramolecular C–O bond formation.
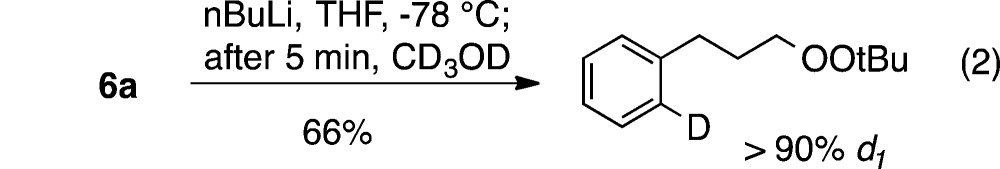 |
2 |
We next investigated corresponding cyclizations of enolate anions, prepared as illustrated in Scheme 2.
Scheme 2. Synthesis of Peroxyketones.

Conditions for cyclization were initially investigated for the 5-exo closure of peroxide 14b (Table 2). The reaction, which generates tert-butoxide anion as a byproduct, proceeded rapidly in THF in the presence of KOtBu or KH. Only traces of products were observed using LDA.
Table 2. Investigation of Cyclization Conditions.
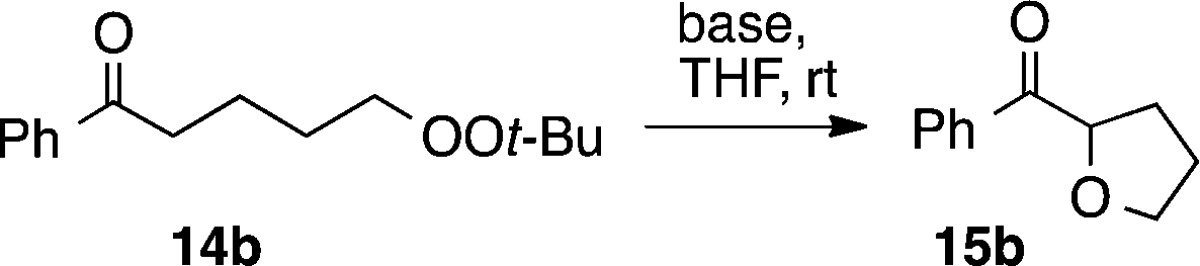
| base | equiv | isolated yield |
|---|---|---|
| KOt-Bu | 0.5 | 81% (99)a |
| KOt-Bu | 1.0 | 85% |
| KOt-Bu | 2.0b | 88%b |
| KH | 1.0 | 86% (99)a |
| KH | 2.0 | 89% |
| LDA | 1.0 | NR |
GC yields.
The use of greater amounts of KOtBu resulted in formation of products derived from peroxide decomposition.11
Application of the KOt-Bu conditions to the cyclization of 14a–c furnished good yields of the oxetane, tetrahydrofuran, and tetrahydropyran (Table 3). Peroxide 14d failed to undergo 7-exo closure to the oxepane, instead slowly undergoing decomposition.11
Table 3. Intramolecular C–O Formation.
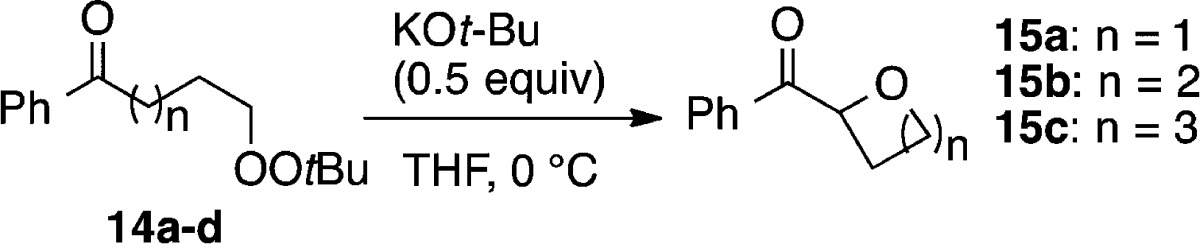
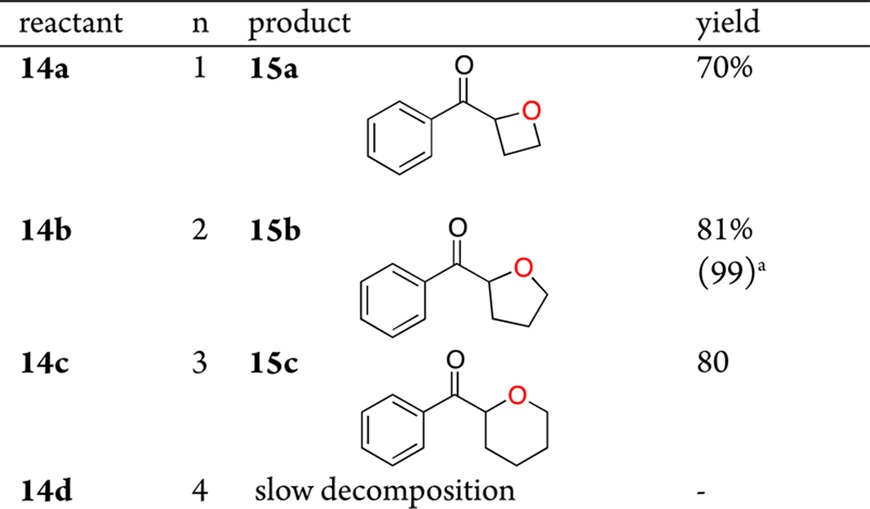
GC yield.
The transformation was not limited to aryl ketones, as evidenced by cyclization of peroxyhexanone 16 (eq 3); the volatile product was isolated after homologation with a Horner–Emmons reagent.
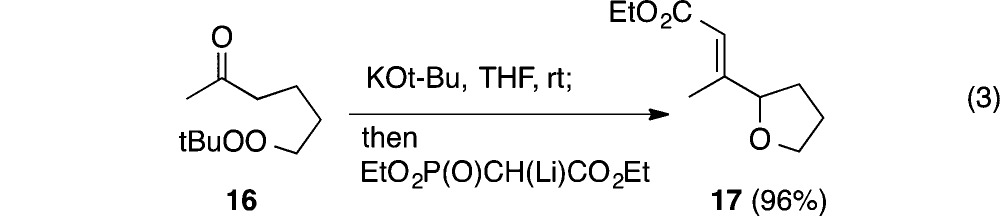 |
3 |
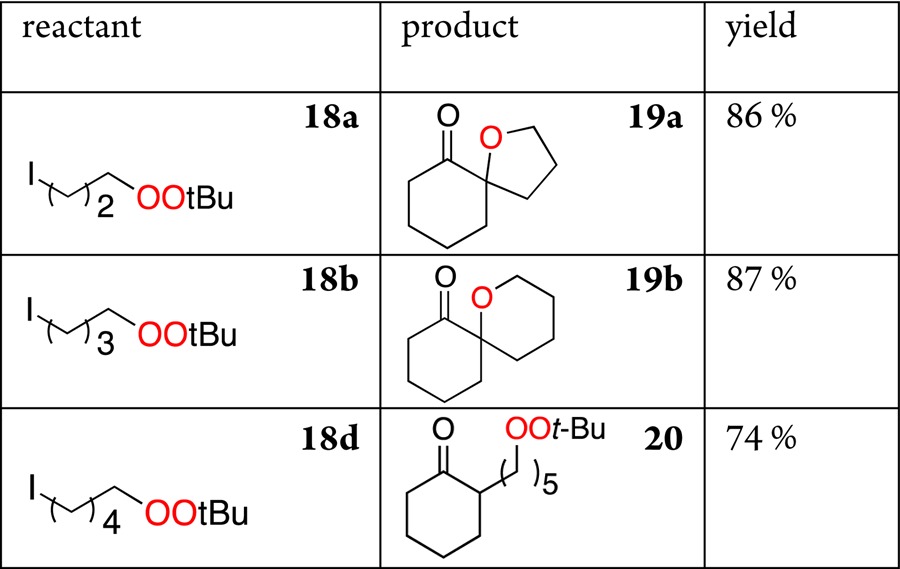
Finally, we investigated the reaction of enolates with a series of t-butyl iodoalkyl peroxides 18a–d, available in one step from the corresponding 1,n-dihalides (Table 4). The results demonstrate the ability to achieve, in one step, a high-yielding annelation of spirocyclic ethers onto ketone frameworks, opening the door to a class of spirocycles previously approachable mainly through cationic ring expansions.12 The isolation of the homologated peroxide (20) during attempted formation of a 7-membered ring provides strong evidence that the formation of the 5- and 6-membered spirocycle proceeds via initial formation of a C–C bond.
Table 4. Formation of Spirocyclic Ethers via Tandem C–C and C–O Bond Formation.
In conclusion, we have demonstrated the chemoselective generation of carbanions in the presence of dialkyl peroxides and the application of the resulting intermediates to establish new C–O bonds. This alternative to more traditional etherifications provides a new approach to synthesis of spirocyclic ethers, aryl ethers, and various oxacycles including oxetanes.
Acknowledgments
The authors wish to acknowledge Navid Rahmany for his contributions to synthesis of a cyclization precursor. Research was conducted with funding from the NSF (CHE 1057982) in facilities renovated with funding from NIH (RR016544).
Supporting Information Available
Experimental procedures, spectral listings, and selected 1H and 13C NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Teva Pharmaceuticals, Salt Lake City, UT.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Forsyth C. J., Ed. Science of Synthesis, v. 27: Ethers; George Thieme Verlag: Stuttgart, 2008. [Google Scholar]
- Hartwig J. F. Acc. Chem. Res. 1998, 31, 852–860. [Google Scholar]; Torraca K.; Huang X.; Parrish C.; Buchwald S. J. Am. Chem. Soc. 2001, 123, 10770–10771. [DOI] [PubMed] [Google Scholar]; Gowrisankar S.; Sergeev A. G.; Anbarasan P.; Spannenberg A.; Neumann H.; Beller M. J. Am. Chem. Soc. 2010, 132, 11592–11598. [DOI] [PubMed] [Google Scholar]; Winternheimer D. J.; Shade R. E.; Merlic C. A. Synthesis 2010, 2497–2511. [Google Scholar]; Naidu A. B.; Jaseer E. A.; Sekar G. J. Org. Chem. 2009, 74, 3675–3679. [DOI] [PubMed] [Google Scholar]; Larrosa I.; Romea P.; Urpí F. Tetrahedron 2008, 64, 2683–2723. [Google Scholar]
- Dialkyl peroxides:; a Baramki G. A.; Chang H. S.; Edward J. T. Can. J. Chem. 1962, 40, 441–445. [Google Scholar]; b Hou Y.; Meyers C. Y.; Akomeah M. J. Org. Chem. 2009, 74, 6362–6364. [DOI] [PubMed] [Google Scholar]
- Peresters:Lawesson S.-O.; Yang N. C. J. Am. Chem. Soc. 1959, 81, 4230–4233. [Google Scholar]
- Endoperoxides:; a Schwaebe M.; Little R. D. Tetrahedron Lett. 1996, 37, 6635–6638. [Google Scholar]; b Ziegert E.; Bräse S. Synlett 2006, 2119–2123. [Google Scholar]
- Taddei M.; Ricci A. Synthesis 1986, 633–635. [Google Scholar]; Dembech P.; Ricci A.; Seconi G.; Taddei M. Org. Synth. 1997, 74, 84. [Google Scholar]; Davis F. A.; Lai G. S.; Wei J. Tetrahedron Lett. 1988, 29, 4269–4272. [Google Scholar]
- Panek E. J.; Kaiser L. R.; Whitesides G. M. J. Am. Chem. Soc. 1977, 99, 3708–13. [Google Scholar]
- Meth-Cohn O.; Moore C.; Taljaard H. C. J. Chem. Soc., Perkin Trans. 1 1988, 2663–2674. [Google Scholar]; Porter M.; Skidmore J. Org. React. 2009, 74, 425–672. [Google Scholar]
- Kyasa S.; Puffer B. W.; Dussault P. H. J. Org. Chem. 2013, 78, 3452–3456and references within. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-S.; Nagai Y.; Ono K.; Begum K.; Wataya Y.; Hamada Y.; Tsuchiya K.; Masuyama A.; Nojima N.; McCullough K. J. J. Med. Chem. 2001, 44, 2357–2361. [DOI] [PubMed] [Google Scholar]; Bloodworth A. J.; Eggelte H. J. J. Chem. Soc., Perkin Trans. 1 1981, 3272–3278. [Google Scholar]
- The isolated byproduct resulted from intramolecular attack of the enolate on an aldehyde formed via E1CB fragmentation of the peroxid: Kornblum N.; DeLaMare H. E. J. Am. Chem. Soc. 1951, 73, 880–881. [Google Scholar]
- Rosenberg S.; Leino R. Synthesis 2009, 2651–2673. [Google Scholar]; Paquette L. A.; Negri J. T.; Rogers R. D. J. Org. Chem. 1992, 57, 3956–3965. [Google Scholar]; Zhang Q.-W.; Fan C.-A.; Zhang H.-J.; Tu Y.-Q.; Zhao Y.-M.; Gu P.; Chen Z.-M. Angew. Chem., Int. Ed. 2009, 48, 8572–8574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.