

Abstract
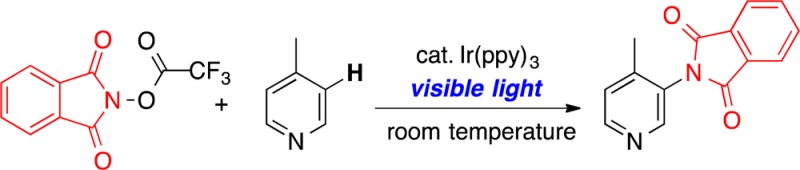
This paper reports a room temperature visible light photocatalyzed method for the C–H amination of arenes and heteroarenes. A key enabling advance in this work is the design of N-acyloxyphthalimides as precursors to nitrogen-based radical intermediates for these transformations. A broad substrate scope is presented, including the selective meta-amination of pyridine derivatives. A radical aromatic substitution mechanism is proposed.
Aromatic and heteroaromatic amines are widely prevalent in pharmaceuticals,1 agrochemicals,2 and organic materials.3 These molecules are most commonly prepared by nucleophilic aromatic substitution,4 transition-metal-catalyzed cross-coupling,5 or reduction of nitroarenes.6 While all of these methods have proven valuable for a variety of applications, they suffer from the disadvantage of requiring prefunctionalized starting materials.
Over the past decade, there has been increasing effort to circumvent the requirement for prefunctionalization by achieving the direct C–H amination of aromatic substrates.7 The vast majority of synthetically useful arene C–H amination methods involve either ligand-directed8−11 or intramolecular12,13 C–N bond formation. In contrast, intermolecular C–H amination of simple arenes/heteroarenes remains significantly more challenging.14−19 Current methods suffer from significant limitations, including the requirement for high temperatures,14b,14c,15,19 UV photolysis,18,19 and/or superstoichiometric quantities of expensive oxidants.14−16 Additionally, some reported systems require specialized catalyst scaffolds.15 Finally, competitive formation of C–H oxygenation15 or C–H bromination18 side products is observed in several cases.
We sought to develop a new photocatalytic method for the intermolecular C–H amination of arenes and heteroarenes that would address many of the challenges described above. Our approach to this problem was inspired by the work of Skell, who demonstrated that UV photolysis of N-bromophthalimide releases PhthN• and Br•, which can participate in competing C–H amination and C–H bromination of the benzene solvent (Scheme 1).18,20 We hypothesized that visible light photocatalysis21,22 could be used to form analogous nitrogen-based radical intermediates under much milder conditions from readily available starting materials (Scheme 2).23 We anticipated that this could render these reactions synthetically useful by (1) precluding the concomitant formation of Br•, (2) affording enhanced yields of the desired aryl amines, and (3) enabling an expansion of the (currently extremely narrow) substrate scope.
Scheme 1. Benzene C–H Amination via UV Photolysis of N-Bromophthalimide.

Scheme 2. (a) Known and (b) Proposed Reductive Fragmentation Pathways for N-Acyloxyphthalimides.
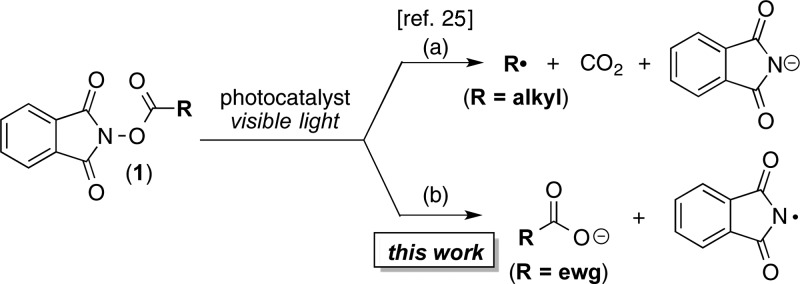
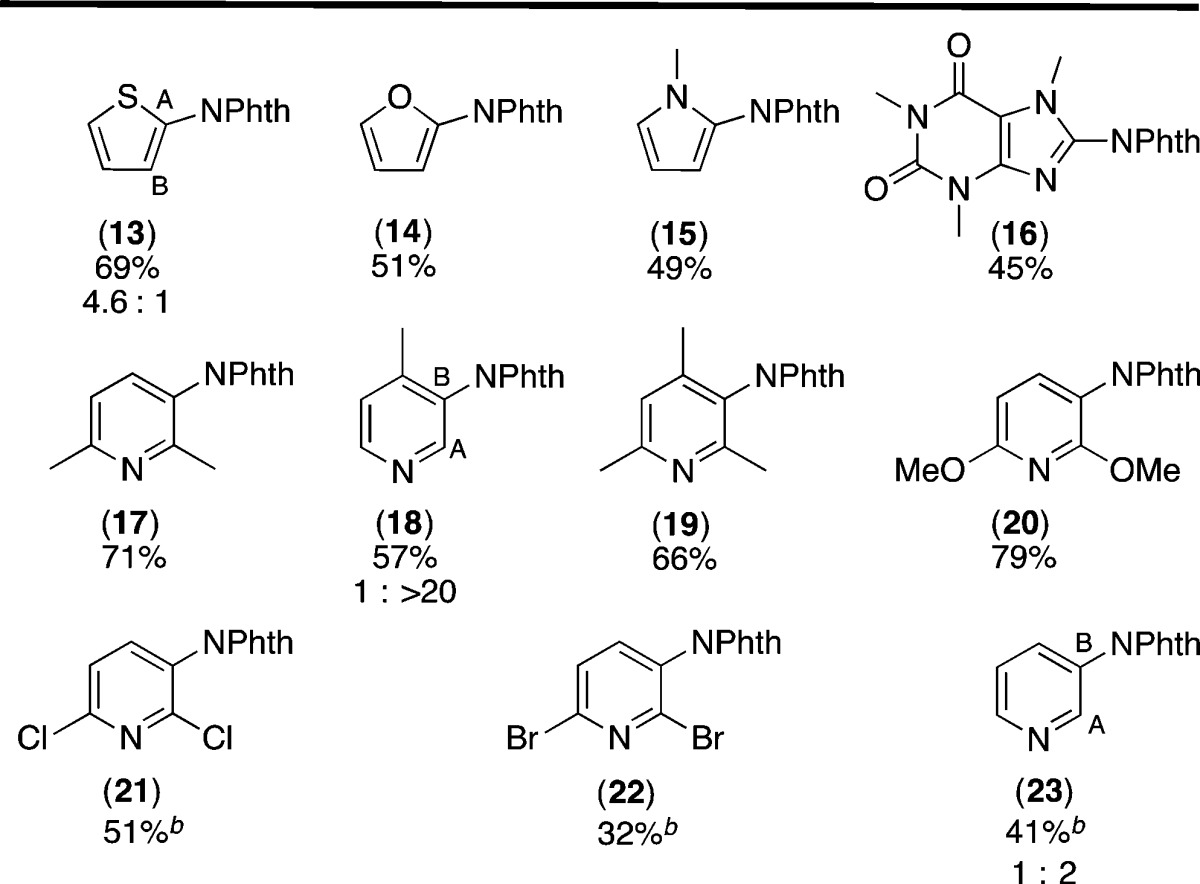
Our initial efforts toward this goal focused on the use of N-acyloxyphthalimides (1) as aminating reagents. These compounds can be readily synthesized from N-hydroxyphthalimide and carboxylic acid derivatives.24 In addition, several reports have shown that they undergo visible light photocatalyzed reduction.25 With R = alkyl, this reduction results in decarboxylative fragmentation to release CO2, R•, and phthalimide anion (Scheme 2a). However, we hypothesized that the use of more electron-withdrawing R substituents would enhance the leaving group ability of the carboxylate anion, thereby favoring fragmentation to release RCO2– and PhthN• (Scheme 2b). The PhthN• could then participate in C–H amination of diverse substrates.
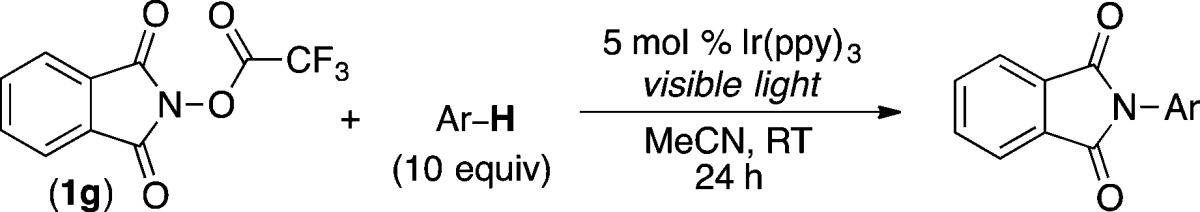
To test this hypothesis, we examined the C–H amination of benzene with a series of electronically different N-acyloxyphthalimides in the presence of 5 mol % of commercially available Ir(ppy)326 and visible light. As shown in Table 1, entry 1, acyloxyphthalimide 1a (with R = CH3) did not react to afford detectable quantities of C–H amination product 2. This is consistent with literature reports showing that alkyl-substituted acyloxyphthalimides undergo selective decarboxylative fragmentation via pathway a (Scheme 2).25 However, as predicted, increasing the electron-withdrawing character of the R substituent resulted in the formation of product 2 in modest to excellent yield (entries 2–7). In particular, trifluoromethylacyloxyphthalimide (1g), which is available in a single step from commercial N-hydroxyphthalimide and trifluoroacetic anhydride, afforded 81% yield of 2.27
Table 1. Visible Light Photocatalyzed C–H Amination of Benzene with N-Acyloxyphthalimide Derivatives.
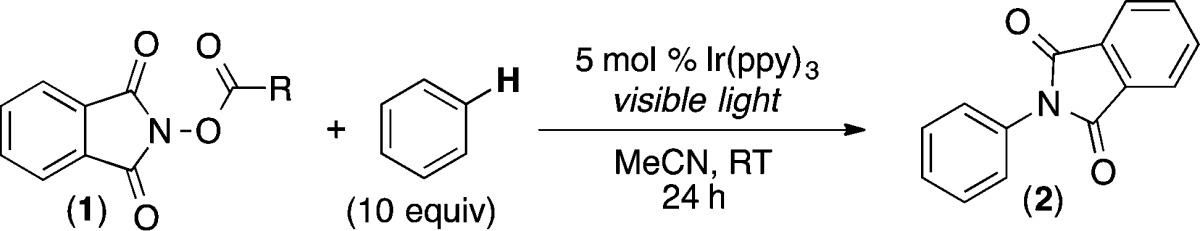
| entry | R | yielda |
|---|---|---|
| 1 | CH3 (1a) | ndb |
| 2 | p-MeOPh (1b) | 7% |
| 3 | Ph (1c) | 8% |
| 4 | p-CF3Ph (1d) | 20% |
| 5 | C6F6 (1e) | 53% |
| 6 | CH2CF3 (1f) | 62% |
| 7 | CF3 (1g) | 81% |
General conditions: 1 (1 equiv), Ir(ppy)3 (5 mol %), benzene (10 equiv), MeCN (0.1 M in 1), visible light, 24 h, rt. Yields determined by GC.
nd = not detected.
Control reactions confirm that C–H amination product 2 is not formed in the absence of either photocatalyst or visible light (Table S1). Further optimization revealed that the photocatalyst loading can be decreased to as low as 0.5 mol %, while maintaining a yield of 55%. Furthermore, the reaction can be conducted with benzene loadings as low as 1 equiv relative to 1g, albeit with a decrease in chemical yield to 23%.
We next examined the scope of arenes that participate in this C–H amination reaction.28 As summarized in Table 2, a wide range of mono-, di-, and trisubstituted arenes underwent C–H amination under these conditions. The site selectivity of these reactions is consistent with that anticipated for a radical aromatic substitution pathway.29 For instance, with arenes bearing electron-donating substituents such as alkyl and alkoxy groups, moderate to high levels of ortho/para selectivity were observed (e.g., products 4 and 5). With electron-withdrawing substituents, such as trifluoromethyl, meta-C–H functionalization products were formed with modest selectivity (3). In general, arenes bearing electron-withdrawing substituents afforded lower yields than those with electron-donor groups. This is further consistent with a pathway involving the reaction of a nitrogen-based radical with the aromatic substrates. Finally, it is noteworthy that this reaction is high yielding and selective for arene C–H amination in the presence of benzylic C–H bonds (cf., products 4, 6, and 8).
Table 2. Arene Substrate Scopea.
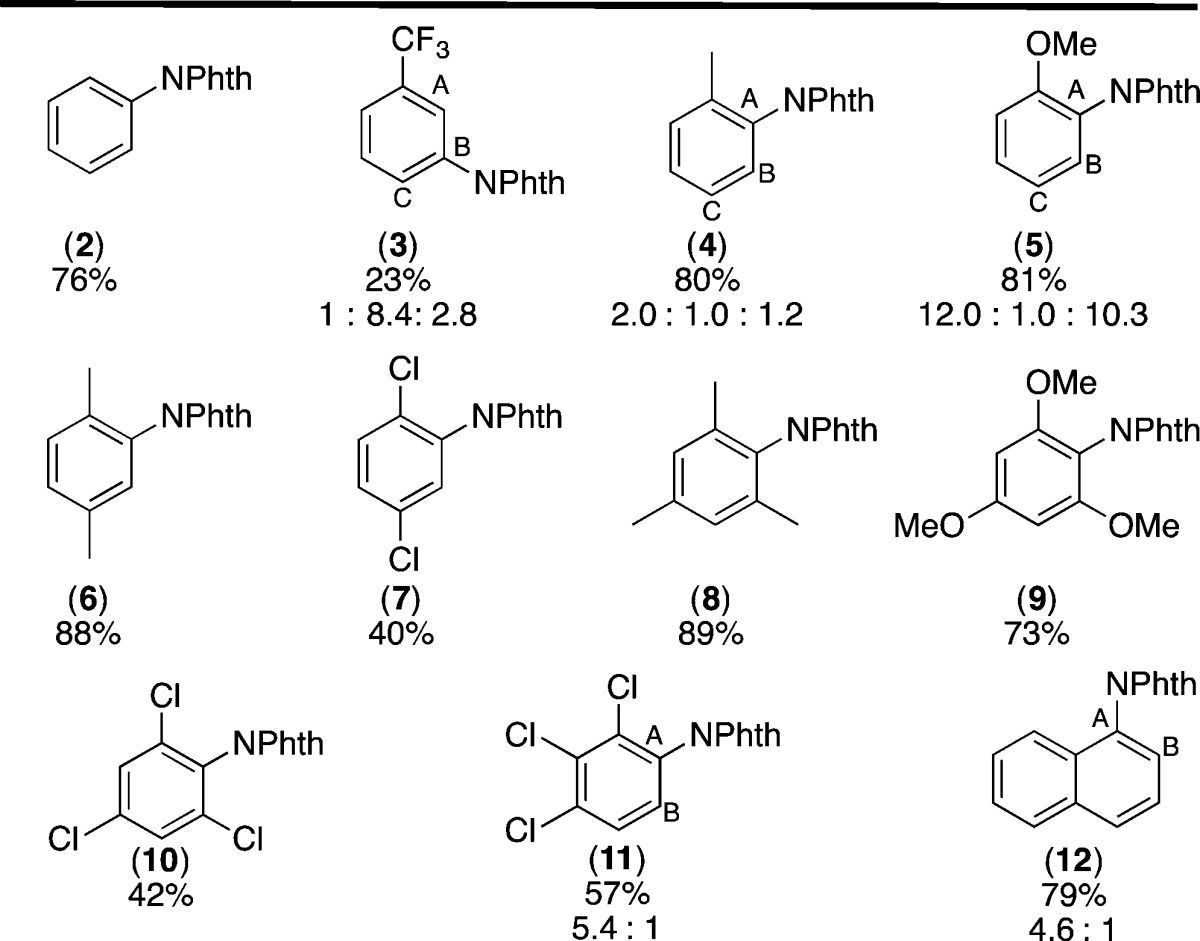
Conditions: 1g (1 equiv), Ir(ppy)3 (5 mol %), arene (10 equiv), MeCN (0.1 M in 1g), visible light, 24 h, rt. Major product shown. Yields and selectivities are of isolated products.
The C–H amination of simple arene substrates can be achieved using several other synthetic methods.14−19 In contrast, selective and high-yielding processes for the C–H amination of diverse heterocycles remain exceedingly rare.16 Due to the mild nature of the current transformation, we hypothesized that it might be compatible with heterocyles. Indeed, as summarized in Table 3, electron-rich heterocycles such as furan, thiophene, and 1-methylpyrrole all underwent C-2 selective C–H amination in moderate to good yield under our optimal conditions (products 13–15). Importantly, these substrates are not stable in the presence of the high temperatures and/or strong oxidants required for many previously reported C–H amination methods.14,15 Both electron-rich and electron-deficient pyridine derivatives are also viable substrates, providing meta-substituted products with good selectivity (17–23). Notably, there does not appear to be any catalyst inhibition with these pyridine substrates, presumably because the Ir contains strongly coordinating cyclometalated phenylpyridine ligands.
Table 3. Heteroarene Substrate Scopea.
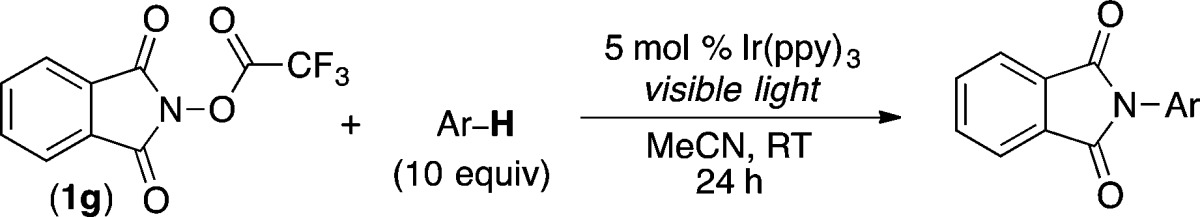
Conditions: 1g (1 equiv), Ir(ppy)3 (5 mol %), heteroarene (10 equiv), MeCN (0.1 M in 1g), visible light, 24 h, rt. Major product shown. Yields and selectivities are of isolated products.
Heteroarene (20 equiv), MeCN (0.2 M in 1g).
A proposed catalytic cycle for the current transformation is shown in Scheme 3. This cycle begins with photoexcitation of Ir(ppy)3 to generate Ir(ppy)3* (step a). Single electron transfer from Ir(ppy)3* to 1g then results in fragmentation of the N-acyloxyphthalimide to generate an N-centered phthalimidyl radical (PhthN•), trifluoroacetate anion, and Ir(ppy)3+ (step b). The PhthN• next adds to the arene, forming a neutral radical intermediate (step c) which can be oxidized by Ir(ppy)3+, regenerating the photocatalyst (step d). The trifluoroacetate anion, formed upon reductive cleavage of the N–O bond, deprotonates the cationic Wheland intermediate, providing 1 equiv of aminated product and trifluoroacetic acid (step e).30
Scheme 3. Proposed Mechanism.

Preliminary mechanistic studies show that irradiation with visible light is necessary for reaction progress. The C–H amination ceases when the reaction is removed from light and only recommences when the light is turned back on (see Scheme S1 for full details of this experiment).31 This experiment is consistent with the mechanism proposed in Scheme 3. Although a radical chain process cannot be completely ruled out, these results indicate that any chain propagation is very short-lived.18
In conclusion, we describe a mild photocatalytic method for the C–H amination of arene and heteroarene substrates. A key enabling advance in this work is the design of N-acyloxyphthalimides as precursors to nitrogen-based radical intermediates for these transformations. Our C–H amination method addresses several key limitations of prior work in the field, as it circumvents the requirement for excesses of expensive oxidants as well as elevated temperatures. The heterocyclic amine products are particularly common in biologically active molecules. Ongoing studies are focused on applying this reaction to more complex molecules as well as gaining detailed insights into the reaction mechanism.
Acknowledgments
We acknowledge NIH (GM073836) for support.
Supporting Information Available
Experimental details and complete characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Horton D. A.; Bourne G. T.; Smythe M. L. Chem. Rev. 2003, 103, 893. [DOI] [PubMed] [Google Scholar]; b Bhattacharya S.; Chaudhuri P. Curr. Med. Chem. 2008, 15, 1762. [DOI] [PubMed] [Google Scholar]; c Mcgrath N. A.; Brichacek M.; Njardarson J. T. J. Chem. Educ. 2010, 87, 1348. [Google Scholar]
- Montgomery J. H.Agrochemicals Desk Reference: Environmental Data; CRC Press: Boca Raton, FL, 1993. [Google Scholar]
- a D’Aprano G.; Leclerc M.; Zotti G.; Schiavon G. Chem. Mater. 1995, 7, 33. [Google Scholar]; b Yeh J.-M.; Liou S.-J.; Lai C.-Y.; Wu P.-C.; Tsai T.-Y. Chem. Mater. 2001, 13, 1131. [Google Scholar]
- Kremer C. B. J. Am. Chem. Soc. 1939, 61, 1321. [Google Scholar]
- a Ley S. V.; Thomas A. W. Angew. Chem., Int. Ed. 2003, 42, 5400. [DOI] [PubMed] [Google Scholar]; b Ma D.; Cai Q. Acc. Chem. Res. 2008, 41, 1450. [DOI] [PubMed] [Google Scholar]; c Evano G.; Blanchard N.; Toumi M. Chem. Rev. 2008, 108, 3054. [DOI] [PubMed] [Google Scholar]; d Hartwig J. F. Acc. Chem. Res. 2008, 41, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Surry D. S.; Buchwald S. L. Chem. Sci. 2011, 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. B.; March J.. March’s Advanced Organic Chemistry: Reactions, mechanisms, and structure, 5th ed.; Wiley: New York, 2001; p 1552–1554. [Google Scholar]
- For reviews on arene C–H amination:; a Beccalli E. M.; Broggini G.; Martinelli M.; Sottocornola S. Chem. Rev. 2007, 107, 5318. [DOI] [PubMed] [Google Scholar]; b Thansandote P.; Lautens M. Chem.—Eur. J. 2009, 15, 5874. [DOI] [PubMed] [Google Scholar]; c Sehnal P.; Taylor R. J. K.; Fairlamb I. J. S. Chem. Rev. 2010, 110, 824. [DOI] [PubMed] [Google Scholar]; d Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cho S. H.; Kim J. Y.; Kwak J.; Chang S. Chem. Soc. Rev. 2011, 40, 5068. [DOI] [PubMed] [Google Scholar]; f Engle K. M.; Mei T.-S.; Wang X.; Yu J.-Q. Angew. Chem., Int. Ed. 2011, 50, 1478. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Wendlandt A. E.; Suess A. M.; Stahl S. S. Angew. Chem. Int. Ed. 2011, 50, 11062. [DOI] [PubMed] [Google Scholar]; h Song G.; Wang F.; Li X. Chem. Soc. Rev. 2012, 41, 3651. [DOI] [PubMed] [Google Scholar]; i Dequirez G.; Pons V.; Dauban P. Angew. Chem. Int. Ed. 2012, 51, 7384. [DOI] [PubMed] [Google Scholar]; j Beletskaya I. P.; Cheprakov A. V. Organometallics 2012, 31, 7753. [Google Scholar]
- For Pd-catalyzed examples:; a Thu H.-Y.; Yu W.-Y.; Che C.-M. J. Am. Chem. Soc. 2006, 128, 9048. [DOI] [PubMed] [Google Scholar]; b Li B.-J.; Tian S.-L.; Fang Z.; Shi Z.-J. Angew. Chem., Int. Ed. 2008, 47, 1115. [DOI] [PubMed] [Google Scholar]; c Ng K.-H.; Chan A. S. C.; Yu W.-Y. J. Am. Chem. Soc. 2010, 132, 12862. [DOI] [PubMed] [Google Scholar]; d Xiao B.; Gong T.-J.; Xu J.; Liu Z.-J.; Liu L. J. Am. Chem. Soc. 2011, 133, 1466. [DOI] [PubMed] [Google Scholar]; e Sun K.; Li Y.; Xiong T.; Zhang J.; Zhang Q. J. Am. Chem. Soc. 2011, 133, 1694. [DOI] [PubMed] [Google Scholar]; f Yoo E. J.; Ma S.; Mei T.-S.; Chan K. S. L.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 7652. [DOI] [PubMed] [Google Scholar]
- For Rh-catalyzed examples:; a Ng K.-H.; Zhou Z.; Yu W.-Y. Org. Lett. 2012, 14, 272. [DOI] [PubMed] [Google Scholar]; b Grohmann C.; Wang H.; Glorius F. Org. Lett. 2012, 14, 656. [DOI] [PubMed] [Google Scholar]; c Kim J. Y.; Park S. H.; Ryu J.; Cho S. H.; Kim S. H.; Chang S. J. Am. Chem. Soc. 2012, 134, 9110. [DOI] [PubMed] [Google Scholar]
- For Cu-catalyzed examples:; a Uemura T.; Imoto S.; Chatani N. Chem. Lett. 2006, 35, 842. [Google Scholar]; b Chen X.; Hao X.-S.; Goodhue C. E.; Yu J.-Q. J. Am. Chem. Soc. 2006, 128, 6790. [DOI] [PubMed] [Google Scholar]; c John A.; Nicholas K. M. J. Org. Chem. 2011, 76, 4158. [DOI] [PubMed] [Google Scholar]
- For an Ir catalyzed example:; Ryu J.; Kwak J.; Shin K.; Lee D.; Chang S. J. Am. Chem. Soc. 2013, 135, 12861. [DOI] [PubMed] [Google Scholar]
- For Pd-catalyzed intramolecular C–H amination:; a Tsang W. C. P.; Zheng N.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 14560. [DOI] [PubMed] [Google Scholar]; b Inamoto K.; Saito T.; Katsuno M.; Sakamoto T.; Hiroya K. Org. Lett. 2007, 9, 2931. [DOI] [PubMed] [Google Scholar]; c Muñiz K. J. Am. Chem. Soc. 2007, 129, 14542. [DOI] [PubMed] [Google Scholar]; d Tsang W. C. P.; Munday R. H.; Brasche G.; Zheng N.; Buchwald S. L. J. Org. Chem. 2008, 73, 7603. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wasa M.; Yu J.-Q. J. Am. Chem. Soc. 2008, 130, 14058. [DOI] [PubMed] [Google Scholar]; f Jordan-Hore J. A.; Johansson C. C. C.; Gulias M.; Beck E. M.; Gaunt M. J. J. Am. Chem. Soc. 2008, 130, 16184. [DOI] [PubMed] [Google Scholar]; g Miura T.; Murakami M. Chem. Lett. 2009, 38, 328. [Google Scholar]; h Mei T.-S.; Wang X.; Yu J.-Q. J. Am. Chem. Soc. 2009, 131, 10806. [DOI] [PubMed] [Google Scholar]; i Tan Y.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 3676. [DOI] [PubMed] [Google Scholar]
- For Cu-catalyzed intramolecular C–H amination:; a Brasche G.; Buchwald S. L. Angew. Chem., Int. Ed. 2008, 47, 1932. [DOI] [PubMed] [Google Scholar]; b Wang H.; Wang Y.; Peng C.; Zhang J.; Zhu Q. J. Am. Chem. Soc. 2010, 132, 13217. [DOI] [PubMed] [Google Scholar]; c Cho S. H.; Yoon J.; Chang S. J. Am. Chem. Soc. 2011, 133, 5996. [DOI] [PubMed] [Google Scholar]
- a Antonchick A. P.; Samanta R.; Kulikov K.; Lategahn J. Angew. Chem., Int. Ed. 2011, 50, 8605. [DOI] [PubMed] [Google Scholar]; b Kim H. J.; Kim J.; Cho S. H.; Chang S. J. Am. Chem. Soc. 2011, 133, 16382. [DOI] [PubMed] [Google Scholar]; c Kantak A. A.; Potavathri S.; Barham R. A.; Romano K. M.; DeBoef B. J. Am. Chem. Soc. 2011, 133, 19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha R.; Mukherjee P.; Tan Y.; Litman Z. C.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 8480. [DOI] [PubMed] [Google Scholar]
- Boursalian G. B.; Ngai M.-Y.; Hojczyk K. N.; Ritter T. J. Am. Chem. Soc. 2013, 135, 13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2011, 13, 2860. [DOI] [PubMed] [Google Scholar]
- a Lüning U.; Skell P. S. Tetrahedron 1985, 41, 4289. [Google Scholar]; b Day J. C.; Govindaraj N.; McBain D. S.; Skell P. S.; Tanko J. M. J. Org. Chem. 1986, 51, 4959. [Google Scholar]
- a Cadogan J. I G.; Rowley A. G. J. Chem. Soc. Perkin 1 1975, 1069. [Google Scholar]; b Abramovitch R. A.; Beckert J. M.; Chinnassamy P.; Xiaohua H.; Pennington W.; Sanjivamurthy A. R. V. Heterocycles 1989, 28, 623. [Google Scholar]
- For analogous UV photolytic reactions of N-tosylphthalimide, see ref (19a).
- a Zeitler K. Angew. Chem., Int. Ed. 2009, 48, 9785. [DOI] [PubMed] [Google Scholar]; b Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. [DOI] [PubMed] [Google Scholar]; c Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]; d Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previous work in our group in the area of visible light photocatalysis:; a Kalyani D.; McMurtrey K. B.; Neufeldt S. R.; Sanford M. S. J. Am. Chem. Soc. 2011, 133, 18566. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ye Y.; Sanford M. S. J. Am. Chem. Soc. 2012, 134, 9034. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Neufeldt S. R.; Sanford M. S. Adv. Syn. Catal. 2012, 354, 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synthetic methods that cleanly produce nitrogen-based radicals are relatively rare. For a review on this area:Zard S. R. Chem. Soc. Rev. 2008, 37, 1603. [DOI] [PubMed] [Google Scholar]
- Saljoughian M.; Morimoto H.; Williams P. G.; Than C.; Seligman S. J. J. Org. Chem. 1996, 61, 9625. [Google Scholar]
- a Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. J. Am. Chem. Soc. 1991, 113, 9401. [Google Scholar]; b Okada K.; Okubo K.; Morita N.; Oda M. Tetrahedron Lett. 1992, 33, 7377. [Google Scholar]; c Schnermann M. J.; Overman L. E. Angew. Chem., Int. Ed. 2012, 51, 9576. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kachkovskyi G.; Faderl C.; Reiser O. Adv. Synth. Catal. 2013, 355, 2240. [Google Scholar]
- Ppy = 2-phenylpyridine
- The use of N-bromophthalimide under otherwise identical conditions afforded <10% yield of 2 along with significant quantities of bromobenzene. N-tosylphthalimide afforded 38% yield of 2 under these visible light photocatalyzed conditions.
- The 24 h reaction time was chosen to maintain general reaction conditions for all substrates. A time study demonstrated that the reaction of benzene as the substrate is complete after 7 h. See Figure S1 for more details.
- a Minisci F. Synthesis 1973, 1973, 1. [Google Scholar]; b Martínez-Barrasa V.; García de Viedma A.; Burgos C.; Alvarez-Builla J. Org. Lett. 2000, 2, 3933. [DOI] [PubMed] [Google Scholar]; c Nagib D. A.; MacMillan D. W. C. Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a related mechanism:Cheng Y.; Gu X.; Li P. Org. Lett. 2013, 15, 2664. [DOI] [PubMed] [Google Scholar]
- Nguyen J. D.; Tucker J. W.; Konieczynska M. D.; Stephenson C. R. J. J. Am. Chem. Soc. 2011, 133, 4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.