

Abstract
The mechanistic aspects of hydration of guanine radical cations, G•+ in double- and single-stranded oligonucleotides were investigated by direct time-resolved spectroscopic monitoring methods. The G•+ radical one-electron oxidation products were generated by SO4•– radical anions derived from the photolysis of S2O82– anions by 308 nm laser pulses. In neutral aqueous solutions (pH 7.0), after the complete decay of SO4•– radicals (∼5 μs after the actinic laser flash) the transient absorbance of neutral guanine radicals, G(-H)• with maximum at 312 nm, is dominant. The kinetics of decay of G(-H)• radicals depend strongly on the DNA secondary structure. In double-stranded DNA, the G(-H)• decay is biphasic with one component decaying with a lifetime of ∼2.2 ms and the other with a lifetime of ∼0.18 s. By contrast, in single-stranded DNA the G(-H)• radicals decay monophasically with a ∼ 0.28 s lifetime. The ms decay component in double-stranded DNA is correlated with the enhancement of 8-oxo-7,8-dihydroguanine (8-oxoG) yields which are ∼7 greater than in single-stranded DNA. In double-stranded DNA, it is proposed that the G(-H)• radicals retain radical cation character by sharing the N1-proton with the N3-site of C in the [G•+:C] base pair. This [G(-H)•:H+C ⇆ G•+:C] equilibrium allows for the hydration of G•+ followed by formation of 8-oxoG. By contrast, in single-stranded DNA, deprotonation of G•+ and the irreversible escape of the proton into the aqueous phase competes more effectively with the hydration mechanism, thus diminishing the yield of 8-oxoG, as observed experimentally.
Introduction
Chronic inflammation is one of the major risk factors for initiation of many human cancers.1 Persistent oxidative stress developed at sites of chronic inflammation is characterized by overproduction of free radicals, electrophiles, and other reactive species, which target cellular DNA and as a consequence may lead to mutations and cancer. The primary target of one-electron oxidation in DNA is guanine, the most easily oxidizable nucleic acid base in DNA.2 The best known oxidation product of guanine is 8-oxo-7,8-dihydroguanine (8-oxoG), which is ubiquitous in cellular DNA and is used widely as a biomarker of oxidative stress.3 The 8-oxoG lesion is genotoxic, and failure to remove 8-oxoG before replication induces G:C → T:A transversion mutation.4,5 Formation of 8-oxoG can be initiated by oxidation of guanine bases with hydroxyl,6 oxyl,7,8 and peroxyl9,10 radicals, one-electron oxidants,11−13 and singlet oxygen.14,15 The oxidation of guanine bases by hydroxyl radicals (•OH) and by one-electron oxidants involves a common intermediate, the 8-hydroxy-7,8-dihydroguanyl radical (8-HO-G•), which is reducing in nature and is rapidly oxidized by molecular oxygen (4 × 109 M–1 s–1) or by other weak oxidants, to yield the 8-oxoG lesion (Figure 1).16 It is worth noting that the competing process of 8-HO-G• decay involves purine ring-opening, followed by one-electron reduction and formation of 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG). Thus, the formation of Fapy, relative to 8-oxoG may be significant in cellular DNA.3,15 However, the yield of FapyG is much lower than that of 8-oxoG in isolated DNA upon one-electron oxidation in aerated aqueous solutions,17 and therefore, this particular guanine decomposition product is not considered in the present study.
Figure 1.

Mechanisms of 8-oxoG formation initiated by one-electron oxidation or by •OH addition to guanine at C8 in DNA (under oxidative conditions FapyG is a minor product17 and its formation is not shown).
The 8-HO-G• radicals can be generated by either the addition of •OH to C8 of guanine16 or by hydration11 of the guanine radical cation (G•+) derived from the one-electron oxidation of guanine (Figure 1).12,15
In acid solutions, the G•+ radicals generated by one-electron oxidation of guanine bases in single-stranded oligonucleotides are hydrated with the rate constant kaq ∼ 3 × 102 s–1 at pH 2.5 determined by time-resolved spectroscopic monitoring of the decay of G•+ radicals.18 However, in neutral solutions, the yields of 8-oxoG in single-stranded DNA and in free 2′-deoxyguanosine (dGuo) are negligible11,16,18 because the hydration of G•+ cannot compete effectively with the faster deprotonation mechanism in neutral solutions (Figure 1). Deprotonation of dGuo•+ gives rise to the neutral nucleoside radical dGuo(-H)• or the G(-H)• base in single-stranded DNA. Indeed, at the nucleoside level, dGuo•+ is a Brønsted acid (pKa = 3.9), and thus the dGuo(-H)• radical is the dominant one-electron oxidation product in neutral pH solutions.19 Pulse radiolysis studies20 have shown that deprotonation of dGuo•+ to dGuo(-H)• occurs rapidly with the rate constant kdp = 1.8 × 107 s–1 which is 5 orders of magnitude greater than kaq.18 The major final oxidation products of dGuo have been identified as 2,2,4-triamino-5-(2H)-oxazolone and its 2,5-diamino-4H-imidazol-4-one precursor21 that arise from the addition of superoxide anion radical to the C8 atom of dGuo(-H)•.22 In double-stranded oligonucleotides the rates of G•+ deprotonation remain very high (kdp ≥ 3 × 106 s–1),20,23 and negligible yields of 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxodGuo) per G•+ radical cation (<0.01%) are expected because kdp ≫ kaq.18 Nevertheless, in spite of this difference in the kaq and kdp values, the production of 8-oxodGuo lesions initiated by the one-electron oxidation of guanine remains very efficient in double-stranded DNA.11 This apparent paradox is re-enforced by the observation that a hydration mechanism is indeed involved in the formation of 8-oxodGuo in double-stranded DNA since the experiments conducted in H218O demonstrated that 18O is incorporated into the 8-oxodGuo formed.10,11 Thus, the high yields of 8-oxodGuo observed in double-stranded DNA present a significant challenge for understanding the mechanism of 8-oxodGuo formation in double-helical DNA under neutral physiological conditions.
The objectives of this work were to directly monitor the kinetics of decay of G•+ and G(-H)• radicals in single- and double-stranded 2′-deoxyribooligonucleotides using transient absorption spectroscopy and to measure, in parallel, the formation of 8-oxodGuo using HPLC methods of analysis. This novel approach allowed us to, for the first time, directly correlate the kinetics of guanine radical decay with 8-oxodGuo formation induced by the one-electron oxidation of guanine in a single- and double-stranded oligonucleotide model system. The sequences of single oligonucleotide strands employed are shown below, and the double-stranded oligonucleotides (duplex 1) were formed by annealing strands 1a and 1b:
| 1a |
| 1b |
The same duplex sequence was previously used in studies of sequence-dependent guanine oxidation in riboflavin-mediated photosensitized oxidation24 and nitrosoperoxycarbonate oxidation25 experiments. Our studies with these model sequences reveal striking differences in the decay kinetics of G(-H)• radicals and yields of 8-oxodGuo lesions in the double- and single-stranded DNA forms. These results suggest that in double-stranded DNA, the G(-H)• radicals retain some cationic character because G-C Watson–Crick base-pairing diminishes the probability of escape of the proton that governs the decay of the G•+ radical cation. The hydration of these cation radicals competes with the release of the protons into the bulk solution and leads to the formation of 8-oxodGuo. On the other hand, the reactivity of the G(-H)• radical with water is much lower as evidenced by the long lifetime of these neutral radicals that decay on the time scale of seconds in single-stranded DNA.
Experimental Methods
Materials
All chemicals (analytical grade) were used as received. The oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). All oligonucleotides were purified and desalted using reversed-phase HPLC. The integrity of the oligonucleotides was confirmed by MALDI-TOF/MS analysis. Duplex 1 was prepared by annealing 1:1 mixtures of sequences 1a and 1b by heating the sample to 90 °C in a water bath followed by slow cooling to room temperature over a 4–5 h time period. Duplex 1 (20 μM) in 20 mM phosphate buffer solution (pH 7.0) containing 50 mM Na2SO4 exhibits a single, well-defined cooperative melting behavior with a melting point of 71.5 ± 0.5 °C. Calf thymus DNA was from Sigma Chemical; the concentrations of nucleobases in solutions of calf thymus DNA were determined using a molar extinction coefficient ε260 = 6.6 × 103 M–1 cm–1.26
Laser Flash Photolysis
The kinetics of oxidative reactions initiated by SO4•– radicals were monitored directly using a fully computerized kinetic spectrometer system (∼7 ns response time) described elsewhere.18 The rate constants, determined by least-square fits of the appropriate kinetic equations to the experimentally measured transient absorption profiles, have been described earlier.18 The values reported are averages of five independent measurements.
Generation of Guanine Radicals in DNA
The SO4•– radicals have been widely used to produce guanine radicals in DNA.19,20,23 The cascade of events was initiated by nanosecond 308 nm XeCl excimer laser pulses (Table 1). This mode of excitation induces the selective photodissociation of peroxodisulfate anions S2O82– and the formation of SO4•– radicals (reaction 1). The latter rapidly oxidize guanines in duplex 1 or the single strands 1a and 1b with formation of G(-H)• radicals (reactions 3–5). The kinetic parameters obtained in the transient absorption experiments are summarized in Table 1.
Table 1. Generation of G(-H)• Radicals in DNA: Reaction Scheme and Rate Constantsa.
| N | reaction | kn, M–1 s–1b | |
|---|---|---|---|
| 1 | φ308 = 0.55c | ||
| 2 | (1.6 ± 0.2) × 109 | ||
| 3 | (10.9 ± 0.9) × 109d | ||
| 4 | (8.6 ± 0.9) × 109d | ||
| 5 | (10.4 ± 0.9) × 109d |
The kinetic parameters were measured in air-equilibrated 10 mM phosphate buffer solutions, pH 7.0 containing 50 mM Na2SO4 at 24 ± 1 °C.
The rate constants were obtained from the best least-squares fits of the appropriate kinetic equations to the transient absorption profiles describing the decay of SO4•– radicals at 445 nm (ε445 = 1.6 × 103 M–1cm–1)27 and the formation of G(-H)• radicals at 312 nm (ε312 = 7.2 × 103 M–1cm–1).18,28
Quantum yield of SO4•– radicals at 308 nm.29
The G(-H)• radicals form with the yields of 0.55 ± 0.10 calculated from the ratio of the transient absorbances of G(-H)• and SO4•– radicals at 312 and 445 nm, respectively.
Quantitation of 8-oxodGuo Lesions in DNA
The 80 μL air-equilibrated samples (100 μM single- or double-stranded oligonucleotides, 20 mM Na2S2O8, 50 mM Na2SO4 in 20 mM phosphate buffer solution) were placed in a 0.2 × 1.0 cm quartz cell and photolyzed through a rectangular aperture (0.3 × 1.0 cm) by a predetermined number of 308 nm XeCl excimer laser pulses. The irradiated oligonucleotides were placed on ice, washed 3 times with 400 μL of cold water using Amicon Ultra-0.5 centrifugal filters (3 kDa MWCO), split into several aliquots containing ∼60 μg DNA, and dried under vacuum. The DNA samples (∼60 μg) were digested with nuclease P1 (2 units) in 40 μL of 30 mM sodium acetate buffer (pH 5.2) containing 0.1 mM ZnCl2 and 3 mM deferoxamine for 3 h at 37 °C. Following digestion, 4 μL of 1 M Tris-HCl buffer solution (pH 8.2) containing 100 mM MgCl2 was added, and the samples were incubated with 2 units of alkaline phosphatase and 0.04 units of snake venom phosphodiesterase for 2 h at 37 °C. The digestion mixtures were passed through Amicon Ultra-0.5 centrifugal filters (10 kDa MWCO) to remove the enzymes. The 2′-deoxyribonucleosides were separated by reversed-phase HPLC column using a 0–24% linear gradient of acetonitrile in 20 mM ammonium acetate for 60 min at a flow rate of 1 mL/min. The amounts of 8-oxodGuo were quantified by integration of the chromatograms monitored at 300 nm using for calibration an authentic standard of 8-oxodGuo.
Results
Kinetics of Decay of Guanine Radicals in Double- and Single-Stranded DNA
In 100 μM solutions of duplex 1 the lifetime of SO4•– radicals is shortened from ∼35 μs in the absence of DNA to ∼1 μs, and the decay of the SO4•– absorption band at 445 nm is well correlated with the growth of new absorption bands in the 300–750 nm spectral range. The transient observed after the complete decay of SO4•– radicals (5 μs curve shown in red in Figure 2A) was assigned to the guanine neutral radical, G(-H)•, characterized by a narrow absorption band at 312 nm and two less intense bands near 390 and 510 nm.19
Figure 2.
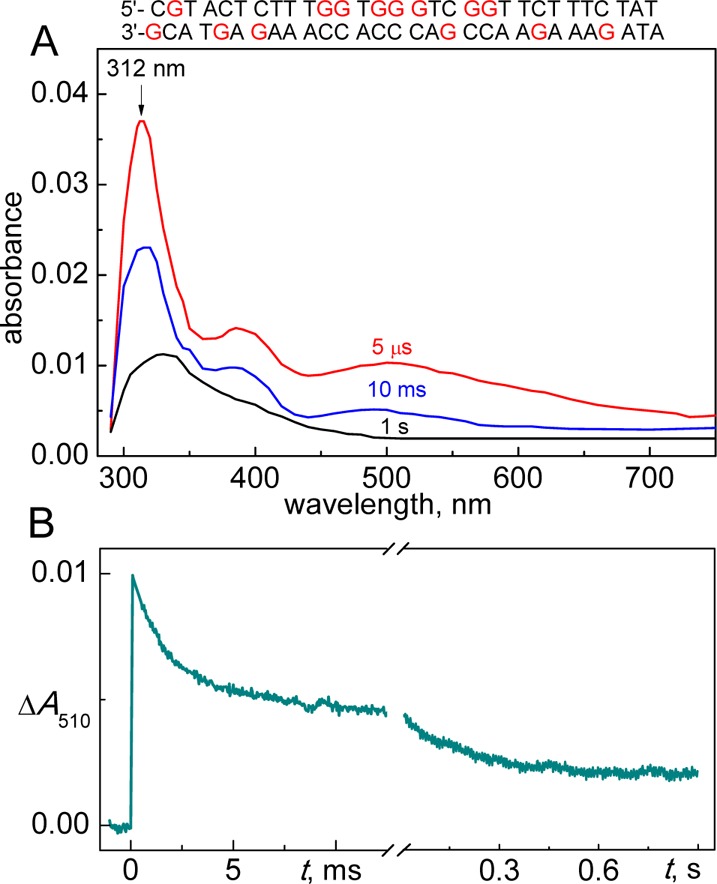
(A) Transient absorption spectra recorded after a nanosecond single 308 nm XeCl laser pulse excitation of samples containing 100 μM duplex 1, 20 mM Na2S2O8, and 50 mM Na2SO4 in air-equilibrated 20 mM phosphate buffer solutions, pH 7.0. (B) The transient kinetic traces at 510 nm are attributed to the decay of G(-H)• radicals in duplex 1.
The spectrum of the guanine radical cation, G•+ is markedly different:19 (1) the narrow absorption band of the G•+ radical is at 300 nm instead of 312 nm in the case of the G(-H)• radical; and (2) at wavelengths >600 nm, the absorbance of G•+ radicals becomes negligible in comparison to the absorbance of G(-H)• radicals (for additional details, see Figure S1). The predominance of G(-H)• absorption in the 5 μs transient absorption spectrum is in excellent agreement with the pulse radiolysis studies of G•+ deprotonation kinetics by the Kobayashi group.20,23 The latter have shown that in double-stranded DNA, deprotonation of G•+ is very fast (kdp ≥ 3 × 106 s–1) and is completed before the start of registration of the transient absorbance in our experiments (5 μs curve, Figure 2A).
In duplex 1, the kinetics of G(-H)• radical decay are heterogeneous and biphasic with lifetimes in the millisecond and second time domains (Figure 2B). In the millisecond time window, the decay of the G(-H)• absorbance occurs with the characteristic decay time of ∼2.2 ms, which is practically the same in the 310–700 nm spectral range. Furthermore, the transient absorption spectrum observed at the 10 ms time point displays all of the spectral features of the G(-H)• radicals (as shown by the 10 ms curve shown in blue in Figure 2A). In turn, in the one-second time domain, the decay of G(-H)• radicals occurs with the characteristic time of ∼0.18 s (Figure 2B). The residual absorbance is characterized by a broad asymmetric absorption band around 330 nm (1 s curve shown in black in Figure 2).
In the case of the single-strand 1a, the kinetics of G(-H)• formation are also correlated with the decay of SO4•– radicals, and kinetic parameters obtained are very close to those observed in duplex 1 (Table 1). The intermediate observed after the complete decay of SO4•– radicals is the G(-H)• radical that is characterized by the transient absorption spectrum shown in Figure 3A (5 μs curve shown in magenta).
Figure 3.
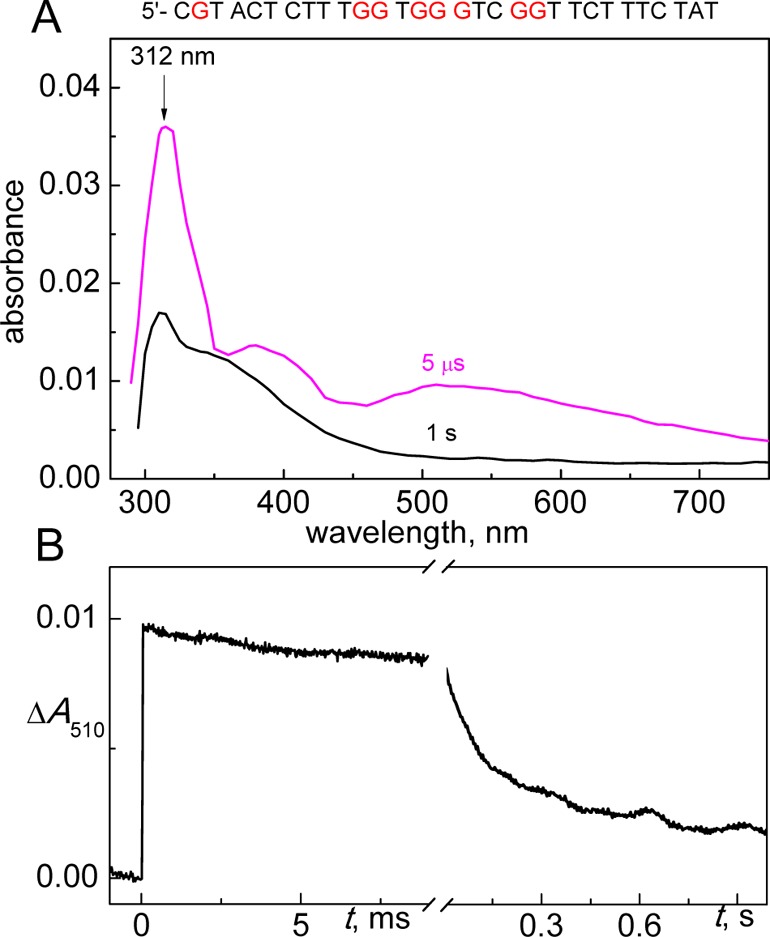
(A) Transient absorption spectra recorded after excitation with a single 308 nm XeCl nanosecond laser pulse. The samples contained 100 μM sequence 1a, 20 mM Na2S2O8, and 50 mM Na2SO4 in air-equilibrated 20 mM phosphate buffer solutions, pH 7.0. (B) The transient kinetic traces at 510 nm are attributed to the decay of G(-H)• radicals in sequence 1a.
Indeed, this 5 μs transient absorption spectrum of sequence 1a shows all of the characteristic features of the G(-H)• spectrum including a narrow absorption band at 312 nm and two less intense bands near 390 and 510 nm.19
Although, the spectral characteristics of G(-H)• radicals in sequence 1a (Figure 3A) and duplex 1 (Figure 2A) are very close, the kinetics of their decays are quite different. In sequence 1a, the decay of G(-H)• radicals is mostly monophasic, because G(-H)• radicals decay on the time scale of 1 s with the characteristic lifetime of 0.28 s (Figure 3B); the kinetic component clearly observed in duplex 1 on the ms time scale (Figure 2B) was not detected in sequence 1a (Figure 3B).
Other experiments with double-stranded calf thymus DNA and single-stranded sequence 1b clearly showed that the behavior of guanine radicals produced by one-electron oxidation is mostly determined by the DNA secondary structure rather than by the DNA sequence context (see details in Supporting Information). In both calf thymus DNA (Figure S2) and sequence 1b (Figure S3) the absorption spectrum of G(-H)• radicals dominates the 5 μs transient absorption spectra. Moreover, in calf thymus DNA, the kinetics of G(-H)• decay remain heterogeneous with pronounced millisecond and ∼1 s kinetic components (inset in Figure S2). In turn, G(-H)• radicals in sequence 1b, which is the complementary strand to sequence 1a, decay in the 1 s time window, and the millisecond kinetic component is not observed (inset in Figure S3). In the following section we show that this difference in the kinetics of G(-H)• radical decay in single- and double-stranded DNA on the ms time scale is correlated with the yield of 8-oxodGuo lesions.
Yields of 8-oxodGuo Lesions in Double- and Single-Stranded DNA
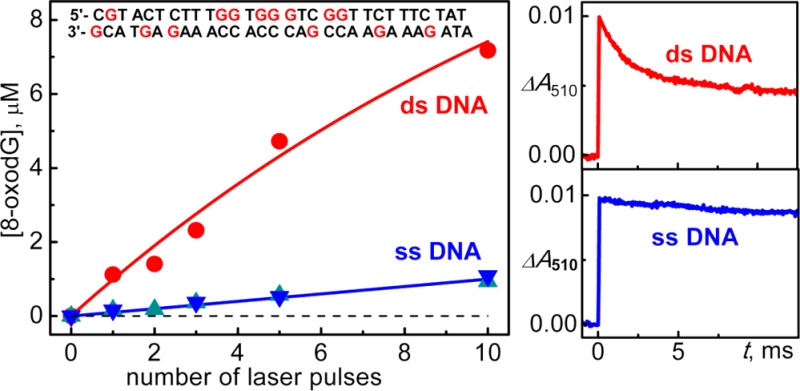
The samples containing 100 μM duplex 1, 20 mM Na2S2O8, and 50 mM Na2SO4 in air-equilibrated 20 mM phosphate buffer solution, pH 7.0 were irradiated by defined numbers of successive 308 nm XeCl excimer laser pulses. After complete enzymatic digestion to the nucleoside level, the 8-oxodGuo lesions were quantified by reversed-phase HPLC. The yields of 8-oxodGuo lesions as a function of the number of laser pulses are shown in Figure 4.
Figure 4.
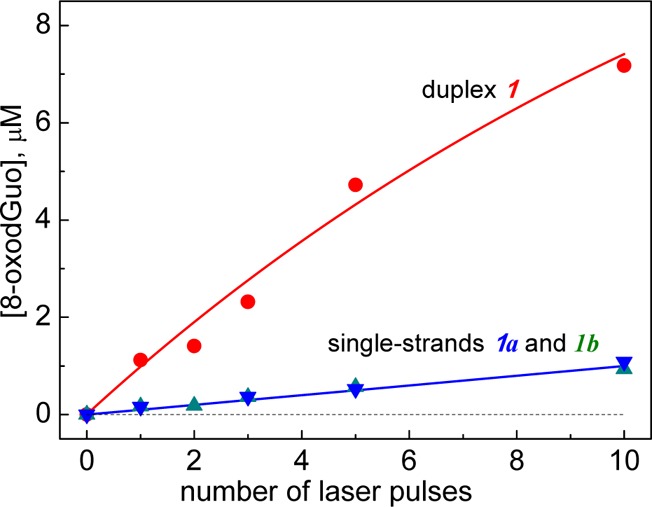
Dependence of the yields of 8-oxodGuo lesions in double- and single-stranded DNA on the number of successive 308 nm laser pulses. The 8-oxodGuo yields were estimated by integrating the areas under the bands of 8-oxodGuo in the HPLC elution profiles recorded at 300 nm.
Increasing the number of laser pulses induces a monotonic rise in the yields of the 8-oxoGuo lesions. In duplex 1, the initial 8-oxodGuo yields are ∼1 μM per laser pulse that correspond to ∼20% per G(-H)• radical generated. In calf thymus DNA, the initial 8-oxodGuo yields were at the level of ∼2 μM per laser pulse (Figure S4). In contrast, in the single-stranded sequences 1a and 1b the yields of 8-oxodGuo are smaller by a factor of ∼7 than in duplex 1. These results clearly show that the double-stranded secondary structure of DNA is a crucial factor that enhances the formation of 8-oxodGuo lesions from G(-H)• radicals.
Discussion
In this work, we explored the effects of DNA secondary structure on the formation of 8-oxodGuo lesions and the kinetics of decay of guanine radicals produced by the one-electron oxidation of guanine bases with SO4•– radicals generated by single laser pulses excitation. The SO4•– radical is a very strong one-electron oxidant that unselectively oxidizes all four natural nucleic acid bases (A, G, C, and T).30,31 Although, all four nucleobases can be oxidized by SO4•– radicals with rate constants that are close to diffusion controlled rates,30 the primary damage is localized at guanine sites,20,23 because guanine is the most easily oxidizable nucleobase among the four natural ones.2,32 Thus, guanines can be rapidly oxidized by neighboring adenine and pyrimidine radical cations that are indiscriminately generated by SO4•– radicals.30,31 Following the one-electron oxidation of any of the four DNA bases, the “hole” is ultimately localized on guanines by hole-hopping mechanisms over long distances.33−38 However, the mechanisms and efficiencies of hole localization on G-sites in double- and single-stranded DNA can be different. Single-stranded DNA is flexible, and direct contacts between guanine and neighbor adenine or pyrimidine radical cations can occur.18,30 In our 30-mer strands 1a and 1b with eight or six guanines, the distance from any randomly injected hole to the closest G-site does not exceed 3 bases, which is sufficient to provide high yields of guanine radicals per SO4•– oxidant in both oligonucleotides (Table 1). The predominant damage of guanine by SO4•– radicals has been also confirmed by gel electrophoresis methods.39−42
Extensive ESR studies in aqueous solutions43,44 and in glassy media at low temperatures45−47 have shown that the unpaired electron in the G•+ radical cation is mostly delocalized with spin densities distributed among the N2, N3, and C8–H atoms, but not on N1. In free nucleosides and single-stranded DNA, deprotonation of G•+ occurs via escape of the proton from N1-G•+ to the surrounding water to form the G(N1–H)• radical, in which there is also no spin density on N1.43−47 By contrast, in double-stranded DNA, G:C Watson–Crick base pairing suggests that the proton is at least partially shielded from bulk water and that the proton is shared between the Watson–Crick paired G and C as proposed by Steenken48,49 (Figure 5).
Figure 5.

Mechanisms of G•+ deprotonation and hydration in double-stranded DNA.
The equilibrium constant of proton transfer (Kpt = 0.25) estimated from the pKa values of 3.9 (dGuo•+)19 and 4.3 (dCyd)50 suggests that the guanine radical in DNA, on average, has 30% ionic character [G•+:C pair] and ∼70% neutral guanine radical character [G(-H)•:H+C)].48,49 The rise of the transient absorption band of G(-H)• radicals at 600–700 nm in double-stranded DNA reported by the Kobayashi group20,23 provides a measure of the characteristic time scale for establishing the proton-transfer equilibrium (Figure 5). In free nucleoside and single-stranded DNA, the escape of protons directly into the bulk solution occurs with rates that are faster than in the case of double-stranded DNA.20,23 Thus, it is not surprising that the neutral guanine radical with an absorption maximum at 312 nm dominates the transient absorption spectrum of both single and double-stranded DNA (Figures 2A, 3A, S2, and S3).
In all experiments (Figures 2, 3, and S2), the concentrations of G(-H)• radicals generated by a single laser pulse (∼5 μM/pulse) were ∼20-fold lower than the concentrations of DNA molecules (∼100 μM). These so-called “single-hit” conditions exclude the occurrence of sequential one-electron oxidation events at a particular guanine site that would give rise to the formation of secondary radicals such as 8-oxoG•+/8-oxoG(-H)• radicals that were detected in gamma-irradiated glassy media at low temperatures.51 We further hypothesize that the ms kinetic component, which appears only in double-stranded DNA (Figure 2B and inset in Figure S2), can be assigned to the hydration of the [G(-H)•:H+C ⇆ G•+:C] pair because the guanine radical retains cationic character in this species (Figure 5). The C8-site of guanine is exposed to the aqueous environment because of its location in the major groove of B-DNA.52 Thus, it is not necessary for the guanine base to flip out of the interior of the double-helix in order to facilitate the nucleophilic addition of water to its C8-site.53 The 8-HO-G• radicals formed are reducible and are readily oxidized by weak oxidants to yield the observed 8-oxodGuo lesions.16 Hydration of the [G(-H)•:H+C ⇆ G•+:C] pair competes with proton escape to the surrounding water and formation of the [G(-H)•:C] pair (Figure 5), which has the same spectrum (10 ms curve shown in blue in Figure 2A) and decays in the 1 s time scale (Figure 2B and inset in Figure S2). Thus, competitive hydration and proton escape can account for the biphasic kinetics of G(-H)• decay in double-stranded DNA; in single-stranded DNA, deprotonation of G•+ dominates and kinetics of G(-H)• decay becomes monophasic (Figures 3B and S3 inset).
Proton escape from [G(-H)•:H+C] to the bulk water that suppresses hydration and reduces 8-oxodGuo yields can occur by the following mechanisms: (1) opening of [G(-H)•:H+C] pair; and (2) tautomeric transformation of [G(-H)•:H+C] to the [G(N2-H)•:C] pair (Figure 5).53,54 The detailed kinetics of base pair opening of GC base pairs in oligonucleotide duplexes were derived from 1H NMR measurements of imino proton exchange rates.55−61 These studies have shown that base pair opening rates depend on DNA sequence context and can be described in terms the rate constant kop = (τop)−1, where τop is characteristic time scale in which base pair opening occurs. Base pair opening in double-stranded DNA can thus contribute to proton escape and the formation of G(-H)• radicals. Typical values of τop in double-stranded DNA are of the order of milliseconds. For example, in runs of contiguous guanines, the values of τop in GC tracts are about 5 ms or shorter,61 while in isolated GC base pairs the values of τop are typically in the range of 10–60 ms.55−60 The latter exceeds the characteristic lifetimes of G(-H)• radicals in the ms time range (Figures 2 and S2).
In contrast, proton escape via the formation of the [G(N2-H)•:H+C] intermediate does not require base pair opening because the G(N2-H)• radicals are exposed to the aqueous phase.53,54 The existence of the G(N2-H)• radicals (Figure 5) is supported using different approaches (pulse radiolysis,62 absorption and ESR spectroscopy,45−47 and extensive DFT calculations).45,47,53,54 It has been demonstrated by pulse radiolysis experiments that G(N2-H)• tautomers produced by the reduction of 8-bromo-2′-deoxyguanosine transform to G(N1–H)• radicals within ∼5 μs.62 This observation suggests that in double-stranded DNA, the equilibrium between these tautomers (Kt) is shifted to the [G(-H)•:H+C] form. Furthermore, DFT calculations suggest that in hydrated DNA, G(N1–H)• is more stable than G(N2-H)• by ∼3.3 kcal/mol;45 consequently, the driving force of proton escape from the duplex is the deprotonation of G(N2-H)• (kt) coupled with base displacement and reorganization of the hydrogen-bonding network (Figure 5).53,54
Conclusions
The radical species detected after the complete decay of SO4•– radicals (5 μs after the laser pulse) exhibit the known spectroscopic characteristics of neutral guanine radicals. In double-stranded DNA, the kinetics of radical decay are heterogeneous and involve two kinetic components that are clearly observed in the millisecond–second time scale windows. In contrast, in single-stranded DNA, the decay of guanine radicals occurs on the time range of several seconds, and the ms kinetic component observed in double-stranded DNA is missing in single-stranded DNA. This difference in the kinetics of radical decay is correlated with a significant reduction of the yields of 8-oxodGuo lesions in single-stranded DNA by a factor of ∼7 relative to double-stranded DNA. In the latter, the deprotonation of guanine radical is attributed to the loss of a proton from the [G(-H)•:H+C] pair. In this structure, the guanine radical retains cationic character and decays by two competitive pathways: (1) hydration, leading to 8-oxodGuo lesions; and (2) proton escape to the surrounding water to form the [G(-H)•:C] base pair which does not readily undergo water addition. In single-stranded DNA, the deprotonation of guanine radical cations dominates over the direct escape of the proton into the bulk water, and the yields of 8-oxodGuo become negligible in comparison with double-stranded DNA.
Acknowledgments
This work was supported by the National Institute of Environmental Health and Sciences Grant RO1ES011589. Components of this work were conducted in the Shared Instrumentation Facility at NYU that was constructed with support from a Research Facilities Improvement Grant (C06 RR-16572) from the National Center for Research Resources, National Institutes of Health. The acquisition of the MALDI-TOF mass spectrometer was supported by the National Science Foundation (CHE-0958457).
Supporting Information Available
The transient absorption spectra of dGuo•+, dGuo(-H)• free nucleoside radicals, and G(-H)• radicals in sequence 1b and calf thymus DNA are shown. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Yale University, Department of Chemical and Environmental Engineering, 9 Hillhouse Avenue, New Haven, Connecticut 06511, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lonkar P.; Dedon P. C. Int. J. Cancer 2011, 128, 1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar]
- Cadet J.; Douki T.; Ravanat J. L. Free Radic. Biol. Med. 2010, 49, 9–21. [DOI] [PubMed] [Google Scholar]
- Shibutani S.; Takeshita M.; Grollman A. P. Nature 1991, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- Bjelland S.; Seeberg E. Mutat. Res. 2003, 531, 37–80. [DOI] [PubMed] [Google Scholar]
- Kasai H.; Tanooka H.; Nishimura S. Gann 1984, 75, 1037–1039. [PubMed] [Google Scholar]
- Joffe A.; Geacintov N. E.; Shafirovich V. Chem. Res. Toxicol. 2003, 16, 1528–1538. [DOI] [PubMed] [Google Scholar]
- Crean C.; Geacintov N. E.; Shafirovich V. Angew. Chem., Int. Ed. Engl. 2005, 44, 5057–5060. [DOI] [PubMed] [Google Scholar]
- Bourdat A.-G.; Douki T.; Frelon S.; Gasparutto D.; Cadet J. J. Am. Chem. Soc. 2000, 122, 4549–4556. [Google Scholar]
- Bergeron F.; Auvre F.; Radicella J. P.; Ravanat J. L. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H.; Yamaizumi Z.; Berger M.; Cadet J. J. Am. Chem. Soc. 1992, 114, 9692–9694. [Google Scholar]
- Cadet J.; Douki T.; Ravanat J. L. Nat. Chem. Biol. 2006, 2, 348–349. [DOI] [PubMed] [Google Scholar]
- Ravanat J. L.; Saint-Pierre C.; Cadet J. J. Am. Chem. Soc. 2003, 125, 2030–2031. [DOI] [PubMed] [Google Scholar]
- Ravanat J. L.; Di Mascio P.; Martinez G. R.; Medeiros M. H.; Cadet J. J. Biol. Chem. 2000, 275, 40601–40604. [DOI] [PubMed] [Google Scholar]
- Cadet J.; Douki T.; Ravanat J. L. Acc. Chem. Res. 2008, 41, 1075–1083. [DOI] [PubMed] [Google Scholar]
- Candeias L. P.; Steenken S. Chem.—Eur. J. 2000, 6, 475–484. [DOI] [PubMed] [Google Scholar]
- Douki T.; Cadet J. Int. J. Radiat. Biol. 1999, 75, 571–581. [DOI] [PubMed] [Google Scholar]
- Rokhlenko Y.; Geacintov N. E.; Shafirovich V. J. Am. Chem. Soc. 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candeias L. P.; Steenken S. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar]
- Kobayashi K.; Tagawa S. J. Am. Chem. Soc. 2003, 125, 10213–10218. [DOI] [PubMed] [Google Scholar]
- Cadet J.; Berger M.; Buchko G. W.; Joshi P. C.; Raoul S.; Ravanat J.-L. J. Am. Chem. Soc. 1994, 116, 7403–7404. [Google Scholar]
- Misiaszek R.; Crean C.; Joffe A.; Geacintov N. E.; Shafirovich V. J. Biol. Chem. 2004, 279, 32106–32115. [DOI] [PubMed] [Google Scholar]
- Kobayashi K.; Yamagami R.; Tagawa S. J. Phys. Chem. B 2008, 112, 10752–10757. [DOI] [PubMed] [Google Scholar]
- Saito I.; Nakamura T.; Nakatani K.; Yoshioka Y.; Yamaguchi K.; Sugiyama H. J. Am. Chem. Soc. 1998, 120, 12686–12687. [Google Scholar]
- Margolin Y.; Cloutier J. F.; Shafirovich V.; Geacintov N. E.; Dedon P. C. Nat. Chem. Biol. 2006, 2, 365–366. [DOI] [PubMed] [Google Scholar]
- Tamm C.; Hodes M. E.; Chargaff E. J. Biol. Chem. 1952, 195, 49–63. [PubMed] [Google Scholar]
- McElroy W. J. J. Phys. Chem. 1990, 94, 2435–2441. [Google Scholar]
- Steenken S.; Jovanovic S. V.; Bietti M.; Bernhard K. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar]
- Ivanov K. L.; Glebov E. M.; Plyusnin V. F.; Ivanov Y. V.; Grivin V. P.; Bazhin N. M. J. Photochem. Photobiol., A 2000, 133, 99–104. [Google Scholar]
- Candeias L. P.; Steenken S. J. Am. Chem. Soc. 1993, 115, 2437–2440. [Google Scholar]
- Wolf P.; Jones G. D.; Candeias L. P.; O’Neill P. Int. J. Radiat. Biol. 1993, 64, 7–18. [DOI] [PubMed] [Google Scholar]
- Seidel C. A. M.; Schulz A.; Sauer M. H. M. J. Phys. Chem. 1996, 100, 5541–5553. [Google Scholar]
- Hall D. B.; Holmlin R. E.; Barton J. K. Nature 1996, 382, 731–735. [DOI] [PubMed] [Google Scholar]
- Nunez M. E.; Hall D. B.; Barton J. K. Chem. Biol. 1999, 6, 85–97. [DOI] [PubMed] [Google Scholar]
- Giese B. Acc. Chem. Res. 2000, 33, 631–636. [DOI] [PubMed] [Google Scholar]
- Schuster G. B. Acc. Chem. Res. 2000, 33, 253–260. [DOI] [PubMed] [Google Scholar]
- Berlin Y.; Burin A. L.; Ratner M. A. J. Am. Chem. Soc. 2001, 123, 260–268. [DOI] [PubMed] [Google Scholar]
- Lewis F. D.; Zhu H.; Daublain P.; Fiebig T.; Raytchev M.; Wang Q.; Shafirovich V. J. Am. Chem. Soc. 2006, 128, 791–800. [DOI] [PubMed] [Google Scholar]
- Kawanishi S.; Yamamoto K.; Inoue S. Biochem. Pharmacol. 1989, 38, 3491–3496. [DOI] [PubMed] [Google Scholar]
- Muller J. G.; Zheng P.; Rokita S. E.; Burrows C. J. J. Am. Chem. Soc. 1996, 118, 2320–2325. [Google Scholar]
- Muller J. G.; Hickerson R. P.; Perez R. J.; Burrows C. J. J. Am. Chem. Soc. 1997, 119, 1501–1506. [Google Scholar]
- Lee Y. A.; Liu Z.; Dedon P. C.; Geacintov N. E.; Shafirovich V. ChemBioChem. 2011, 12, 1731–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildenbrand K.; Schulte-Frohlinde D. Free Radical Res. Commun. 1990, 11, 195–206. [DOI] [PubMed] [Google Scholar]
- Bachler V.; Hildenbrand K. Radiat. Phys. Chem. 1992, 40, 59–68. [Google Scholar]
- Adhikary A.; Kumar A.; Becker D.; Sevilla M. D. J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Khanduri D.; Sevilla M. D. J. Am. Chem. Soc. 2009, 131, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Munafo S. A.; Khanduri D.; Sevilla M. D. Phys. Chem. Chem. Phys. 2010, 12, 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenken S. Free Radical Res. Commun. 1992, 16, 349–379. [DOI] [PubMed] [Google Scholar]
- Steenken S. Biol. Chem. 1997, 378, 1293–1297. [PubMed] [Google Scholar]
- In Basic Principles in Nucleic Acid Chemistry; Ts’o P. O. P., Ed.; Academic Press: New York, 1974; Vol. 1, pp 454–584. [Google Scholar]
- Shukla L. I.; Adhikary A.; Pazdro R.; Becker D.; Sevilla M. D. Nucleic Acids Res. 2004, 32, 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson R. E.; Drew H. R.; Conner B. N.; Wing R. M.; Fratini A. V.; Kopka M. L. Science 1982, 216, 475–485. [DOI] [PubMed] [Google Scholar]
- Reynisson J.; Steenken S. Phys. Chem. Chem. Phys. 2002, 4, 527–532. [Google Scholar]
- Reynisson J.; Steenken S. Phys. Chem. Chem. Phys. 2002, 4, 5346–5352. [Google Scholar]
- Kochoyan M.; Leroy J. L.; Gueron M. J. Mol. Biol. 1987, 196, 599–609. [DOI] [PubMed] [Google Scholar]
- Leroy J. L.; Kochoyan M.; Huynh-Dinh T.; Gueron M. J. Mol. Biol. 1988, 200, 223–238. [DOI] [PubMed] [Google Scholar]
- Folta-Stogniew E.; Russu I. M. Biochemistry 1994, 33, 11016–11024. [DOI] [PubMed] [Google Scholar]
- Gueron M.; Leroy J. L. Methods Enzymol. 1995, 261, 383–413. [DOI] [PubMed] [Google Scholar]
- Leijon M.; Leroy J. L. Biochimie 1997, 79, 775–779. [DOI] [PubMed] [Google Scholar]
- Leijon M.; Sehlstedt U.; Nielsen P. E.; Graslund A. J. Mol. Biol. 1997, 271, 438–455. [DOI] [PubMed] [Google Scholar]
- Dornberger U.; Leijon M.; Fritzsche H. J. Biol. Chem. 1999, 274, 6957–6962. [DOI] [PubMed] [Google Scholar]
- Chatgilialoglu C.; Caminal C.; Altieri A.; Vougioukalakis G. C.; Mulazzani Q. G.; Gimisis T.; Guerra M. J. Am. Chem. Soc. 2006, 128, 13796–13805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.