

Abstract
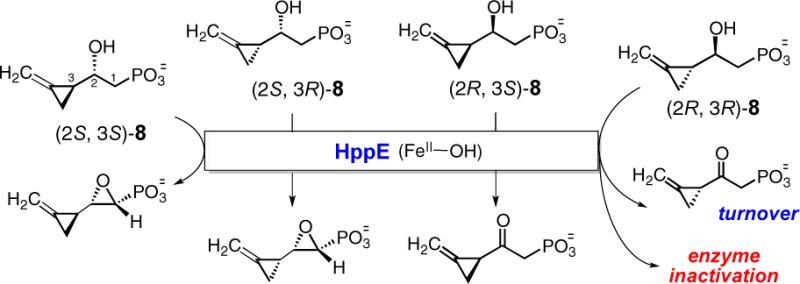
(S)-2-Hydroxypropylphosphonic acid [(S)-HPP] epoxidase (HppE) is a mononuclear iron enzyme that catalyzes the last step in the biosynthesis of the antibiotic fosfomycin. HppE also processes the (R)-enantiomer of HPP but converts it to 2-oxo-propylphosphonic acid. In this study, all four stereoisomers of 3-methylenecyclopropyl-containing substrate analogues, (2R, 3R)-8, (2R, 3S)-8, (2S, 3R)-8, and (2S, 3S)-8, were synthesized and used as radical probes to investigate the mechanism of the HppE-catalyzed reaction. Upon treatment with HppE, (2S, 3R)-8 and (2S, 3S)-8 were converted via a C1 radical intermediate to the corresponding epoxide products, as anticipated. In contrast, incubation of HppE with (2R, 3R)-8 led to enzyme inactivation, and incubation of HppE with (2R, 3S)-8 yielded the 2-keto product. The former finding is consistent with the formation of a C2 radical intermediate, where the inactivation is likely triggered by radical-induced ring cleavage of the methylenecyclopropyl group. Reaction with (2R, 3S)-8 is predicted to also proceed via a C2 radical intermediate, but no enzyme inactivation and no ring-opened product were detected. These results strongly suggest that an internal electron transfer to the iron center subsequent to C–H homolysis competes with ring-opening in the processing of the C2 radical intermediate. The different outcomes of the reactions with (2R, 3R)-8 and (2R, 3S)-8 demonstrate the need to carefully consider the chirality of substituted cyclopropyl groups as radical reporting groups in studies of enzymatic mechanisms.
Mononuclear non-heme iron enzymes are an important class of biocatalysts that are widespread in nature.1 The catalytic functions of these enzymes are remarkably diverse, but the involvement of a high valent Fe(IV)-oxo (ferryl) species as a reactive intermediate is common to all their catalytic mechanisms. This reactive iron–oxygen species abstracts a hydrogen atom, typically from an unactivated carbon of the substrate, to generate a substrate radical, which then undergoes further transformations en route to the final product.2 Despite the proposed intermediacy of substrate radicals in various mononuclear non-heme iron enzyme mechanisms, only a limited number of studies provide evidence for the radical intermediates during turnover.3
(S)-2-Hydroxypropylphosphonic acid epoxidase (HppE), a mononuclear non-heme iron enzyme, performs an epoxidation reaction on (S)-2-hydroxypropylphosphonic acid ((S)-HPP, 1) to form fosfomycin (2) in the last step of the biosynthesis of this clinically useful antibiotic.4,5 Previous studies showed that the epoxide ring of 2 is generated by an initial abstraction of 1-HR from (S)-HPP and subsequent C–O bond formation between C1 and the 2-OH group of (S)-HPP in a dehydrogenation–cyclization manner (Scheme 1A).4−7 When HppE is incubated with (R)-HPP (3), the 2-H is selectively abstracted, leading to direct oxidation of the alcohol moiety to give 2-oxopropylphosphonic acid (4) as the sole product (Scheme 1B).8 Crystallographic and spectroscopic experiments have shown that both (S)- and (R)-HPP bind to the iron center of the enzyme as bidentate ligands.9 The C2 chirality of the substrates governs the binding geometry such that regio- and stereoselective hydrogen atom abstraction (1-HR for (S)-HPP and 2-H for (R)-HPP) is facilitated by the reactive iron–oxygen species10,11 giving different radical intermediates (5 or 6).12
Scheme 1. Proposed Mechanism for the Oxidation of (S)- and (R)-HPP by HppE.
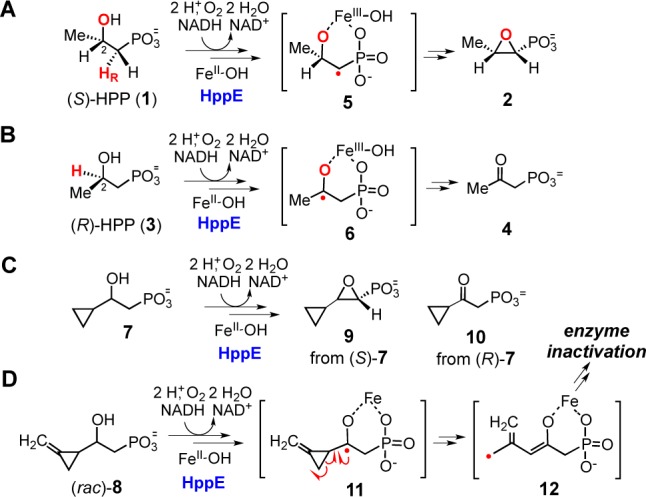
(A) Conversion of (S)-HPP (1) to fosfomycin (2). (B) Conversion of (R)-HPP (3) to 2-oxopropylphosphonic acid (4). (C) Conversion of (S)-7 to 9 and (R)-7 to 10 by HppE. (D) Proposed mechanisms for the inactivation of HppE by 8.
Recently, we reported mechanistic studies of HppE with the 2-hydroxyl-3-cyclopropyl and 2-hydroxyl-3-methylenecyclopropyl compounds (S)-7, (R)-7, and (rac)-8.13 Both (S)- and (R)-7 were efficiently converted by HppE to the epoxide and ketone products 9 and 10, respectively (Scheme 1C). In contrast, when HppE was incubated with (rac)-8, only a small portion of the substrate was converted to new products before the enzyme was inactivated. It was suggested that inactivation results from ring-opening of the C3 methylenecyclopropyl group triggered by the C2 radical (11) generated during turnover (Scheme 1D). These studies provided preliminary evidence for a radical intermediate in the HppE-catalyzed reactions.13 However, (rac)-8 used in the experiment was a mixture of four stereoisomers, which complicated the analysis of the reaction outcomes and thus hampered investigation of the inactivation mechanism. To gain further mechanistic insight and verify our previous observations, we synthesized all four stereoisomers of 8 and studied their reactions with HppE. Our results, reported herein, show that HppE reactivity toward different diasteromers of 8 is dictated not only by the chirality of the C2 hydroxyl group but also the methylenecyclopropyl substituent at C3. Specifically, it was found that incubation of the (2R, 3R)-isomer with HppE leads to enzyme inactivation, whereas reaction with the (2R, 3S)-isomer gives rise to complete turnover to a 2-keto product. These results are mechanistically informative and shed light on the likely formation of a carbocation intermediate in HppE catalysis.
The syntheses of all four diastereomers of 8 began by addressing the chirality of the C3 methylenecyclopropyl group. This was achieved by following the previously reported enantioselective synthetic method for alcohols (S)- and (R)-13.14 Oxidation of (R)-13 produced aldehyde (R)-14, and subsequent nucleophilic addition with methyldiethylphosphonate gave (2S, 3R)- and (2R, 3R)-15 as a pair of diastereomeric alcohols (Scheme 2). Direct separation or enzymatic resolution (using lipases or esterases) of these two diastereomers did not give satisfactory results. Consequently, we focused on chemical resolution by derivatizing the C2 hydroxyl group with a TBDPS group to inflict internal steric hindrance and to introduce a UV chromophore for product detection during purification. It was hoped that the attachment of such a bulky substituent would limit the free rotation of the C2–C3 bond connecting the two neighboring chiral centers and, therefore, magnify the diasteromeric difference. This strategy generated two TBDPS-ethers, (2S, 3R)- and (2R, 3R)-16, which were successfully separated by silica gel chromatography. Removal of the TBDPS group yielded pure (2S, 3R)- and (2R, 3R)-15, enabling chirality and enantiomeric excess determination of the C2 alcohol by the Mosher ester method.15 Subsequent deprotection by TMSBr gave the desired substrate analogues (2S, 3R)- and (2R, 3R)-8. Following the same procedure, (2S, 3S)- and (2R, 3S)-8 were obtained using (S)-13 as the starting material.
Scheme 2. Synthetic Scheme for the Preparation of (2S, 3R)- and (2R, 3R)-8.

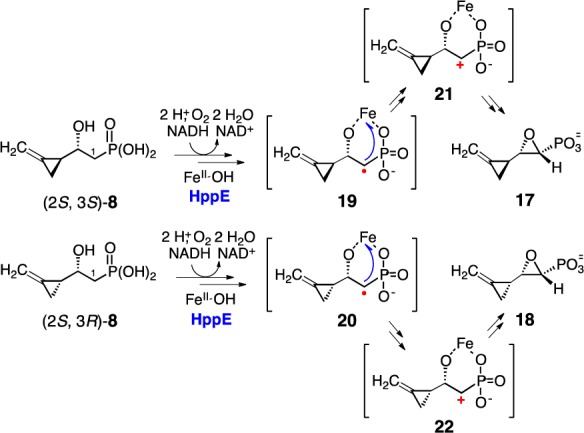
The four purified diastereomers of 8 were each incubated with HppE, and the reactions were monitored using real-time 1H NMR spectroscopy. All four compounds were substrates for HppE, but each reaction gave a different result. The 1,2-epoxide 17 was produced when (2S, 3S)-8 was employed as the substrate, whereas formation of a different 1,2-epoxide, 18, was detected in the reaction with (2S, 3R)-8 (see Figures S3, S4).16 These results are consistent with what was previously observed for (rac)-8 with HppE13 and support the regio- and stereoselective conversions of (2S, 3S)- and (2S, 3R)-8 proposed in Scheme 1A. Specifically, when these substrate analogues bearing C2-(S) hydroxyl group react with HppE, the putative C1 centered radicals (19 and 20) are produced by selective hydrogen abstraction with the high-valent Fe(IV)-oxo species. These C1 radicals are not affected by the methylenecyclopropyl group at C3 position and undergo cyclization to the corresponding epoxides 17 and 18 in a manner similar to the formation of 2 from 1 (Scheme 3). Our more recent results suggest that cyclization of the resulting radical intermediate (19 and 20) to the epoxide product likely involves a carbocation intermediate (21 and 22) via internal electron transfer to reduce the iron center.17
Scheme 3. Conversion of (2S, 3S)-8 to 17 and (2S, 3R)-8 to 18 by HppE.
Based on the mechanism proposed in Scheme 1B, incubation of the C2-(R) analogue, (2R, 3R)-8, with HppE is expected to proceed with regioselective C-2 hydrogen atom abstraction, where the resulting C2 radical will trigger ring-opening of the neighboring methylenecyclopropyl group18 leading to enzyme inactivation. Interestingly, incubation of (2R, 3R)-8 with HppE produces a small amount of new product as detected by 1H NMR spectroscopy (see Figure S1). However, product formation stopped with no further consumption of the substrate after ∼10 min. Moreover, no fosfomycin could be detected when the natural substrate, (S)-HPP (1), was incubated with HppE that had been preincubated with (2R, 3R)-8, suggesting complete inactivation of HppE by the pretreatment. The turnover product of this process was later determined to be ketone 23, which had been previously shown not to be an inactivator of HppE.13 Thus, a logical explanation for the inactivation of HppE by (2R, 3R)-8 is a mechanism initiated by a radical-triggered ring-opening of the C3-(R)-methylenecyclopropyl group (Scheme 4A). The fact that (2R, 3R)-8 is an inactivator and also a substrate for HppE reveals a competition between the radical-induced ring-opening (24 → 12) and the internal electron-transfer (24 → 25) reactions.
Scheme 4. (A) HppE Inactivation by (2R, 3R)-8 and (B) Conversion of (2R, 3S)-8 to 26 by HppE.
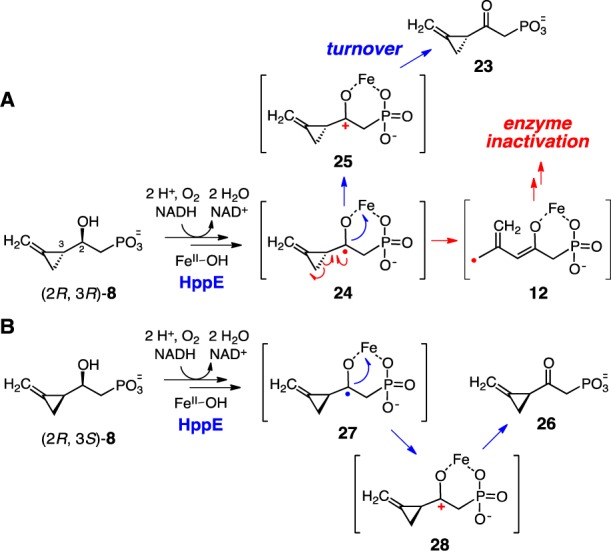
For the other C2-(R) diastereomer, (2R, 3S)-8, a similar C2 radical-triggered ring-opening reaction with subsequent enzyme inactivation was also expected. However, HppE remained active after incubation with (2R, 3S)-8, and NMR analysis of the incubation showed that (2R, 3S)-8 was completely transformed into ketone 26 without formation of ring-opened products (Scheme 4B and Figure S2). Given that the methylenecyclopropyl group is a highly sensitive radical probe,18,19 the different outcomes of these two reactions may lie in the orientation of the methylenecyclopropyl group of 8 in the enzyme active site. Specifically, the three-membered ring is more readily opened when the C3–C4 bond of the methylenecyclopropyl group and the singly occupied orbital of the radical adopt an anticoplanar conformation18 (the ring-opening rate of a methyenecyclopropyl carbinyl radical in free solution is ∼6 × 109 s–1).19 It is possible that such an anticoplanar conformation is inaccessible for (2R, 3S)-8 due to steric interference between the methylenecyclopropyl moiety and the groups at the enzyme active site. Thus, electron transfer from the radical intermediate 27 to the iron center (27 → 28) becomes the path of choice leading to a 2-keto product (26) instead of the ring-opened product(s).
To gain more insight into the mechanism of inhibition by (2R, 3R)-8, the kinetics of the reaction were determined using HPLC. When HppE was incubated with 10-fold excess of (2R, 3R)-8, time-dependent inactivation was observed, and the enzyme completely lost activity within 3 min (Figure S6). When (S)-HPP (1) was coincubated with (2R, 3R)-8 (molar ratio of 1:8 = 20:1), fosfomycin production was still discernible after 8 min, indicating a reduction of the rate of inactivation in the presence of native substrate. The observed substrate protection implies that the inhibition is active-site directed. When HppE was treated with different amounts of (2R, 3R)-8, a plot of the residual enzyme activity versus the equivalents of (2R, 3R)-8 employed resulted in a partition ratio of 0.5 (Figure S6), reflecting that ∼1.5 equiv of (2R, 3R)-8 are required to fully inactivate 1.0 equiv of HppE. Moreover, the inactivation appears to be irreversible since enzyme activity could not be regenerated after extensive dialysis.
Tritiated (rac)-[3-3H]-8 was also synthesized and incubated with HppE to determine the stoichiometry of inactivation.16 After HppE was completely inactivated by (rac)-[3-3H]-8, the reaction mixture was subjected to size exclusion chromatography followed by dialysis to remove unbound small molecules. About 1.1 equiv of tritiated compound per enzyme were found to be associated with the inactivated HppE.16 Interestingly, when this protein sample was denatured by boiling, over 80% of the radioactivity was lost from the protein fraction after size exclusion centrifugal filtration.16 In addition, no covalently modified peptide fragment was detected by mass spectrometry analysis of the inactivated HppE after denaturation and tryptic digestion (see Figure S7). Taken together, these results strongly suggest that the inactivation is not due to covalent modification of HppE, as previously proposed,13 but is likely due to tight binding of a ring-opened product to the enzyme active site.
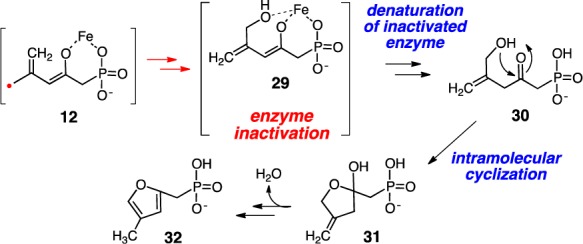
It is conceivable that the causative agent for the observed inactivation is compound 29, which arises from canonical hydroxyl group rebound to the initially formed acyclic radical intermediate (12, Scheme 5). The tridentate coordination of 29 to the iron center could render 29 to bind strongly in the active site of HppE, resulting in enzyme inactivation. However, efforts to isolate and identify this putative tight binding species after protein denaturation were unsuccessful. It was later found during our attempt to chemically synthesize 30 (the keto tautomer of 29) that this compound readily undergoes intramolecular cyclization to give 32 as the main product.16 The fact that a low abundance signal at m/z 175 [M – 1]− was detected by LC-MS analysis in the denatured enzymatic sample (but not in the control) is consistent with the presence of 32 in the protein-free solution after enzyme denaturation. Unfortunately, the low abundance of this signal prevented MS-MS analysis to further verify its structure.
Scheme 5. Proposed HppE Inactivation by the Ring-Opened Product Derived From (2R, 3R)-8.
The results presented herein are significant for three reasons. First, a highly effective inhibitor for HppE, (2R, 3R)-8, has been identified. The inactivation is mechanism-based, since assays using NMR, HPLC, MS, and radioactive methods indicate that the inactivation arises from the methylenecyclopropyl ring-opening triggered by the C2-centered radical. However, unlike most known suicide inhibitors, the inactivation by (2R, 3R)-8 does not involve covalent modification of the targeted enzyme but is likely due to tight binding of a ring-opened product in the active site.
Second, to “capture” radical intermediate(s) in a reaction with transient lifetimes, the sensitivity of the cyclopropylcarbinyl radical probe can be fine-tuned by adding substituent(s) to the cyclopropyl ring.18 Such modifications usually introduce new chiral center(s) and inflict steric demands upon binding to the chiral environment of the active site. However, the effect of the chirality of the substituted cyclopropyl ring of the probe on the outcomes of the radical triggered ring-opening reaction has rarely been addressed.20,21 In this study, enzymatic reactions with (2R, 3R)- and (2R, 3S)-8 led to different results, even though both are believed to involve a C2 radical intermediate. The distinct outcomes may simply reflect different binding geometries of (2R, 3R)- and (2R, 3S)-8 in the enzyme active site, and underscores the need to consider the chirality of substituted cyclopropyl moiety in a radical probe and its binding mode in the active site in studies of enzyme mechanisms. One also needs to be cautious to interpret the experimental data when no predicated ring-opening/rearrangement happens.
Third, the intermediacy of a substrate-derived cation in the catalytic cycle of HppE was recently proposed based on the observed 1,2-phosphono migration with an alternative substrate (R)-1-HPP.17 The observation of both enzyme inactivation and 2-keto product formation upon incubation of HppE with (2R, 3S)-8 is mechanistically significant, as it reveals that an internal electron transfer (leading to the 2-keto product 23) is a competing pathway with ring-opening (leading to enzyme inactivation) in the processing of the C2 radical intermediate. Thus, these results provide additional evidence supporting the formation of a cation intermediate in HppE catalysis and permit estimation of the rate constant for internal electron transfer to be on the order of ∼6 × 109 s–1, which is the reported rate constant for ring-opening of the methylenecyclopropylcarbinyl radical.19
Acknowledgments
We thank Steve Sorey for his help in acquiring the NMR spectra. This work is supported in part by grants from the National Institutes of Health (GM040541 to H.-W.L.) and the Welch Foundation (F-1511 to H.-W.L. and A-1176 to D.H.R.).
Supporting Information Available
Details of experimental conditions and procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bruijnincx P. C. A.; van Koten G.; Klein Gebbink R. J. M. Chem. Soc. Rev. 2008, 37, 2716. [DOI] [PubMed] [Google Scholar]
- a Fitzpatrick P. F. Annu. Rev. Biochem. 1999, 68, 355. [DOI] [PubMed] [Google Scholar]; b Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Chem. Rev. 2004, 104, 939. [DOI] [PubMed] [Google Scholar]; c Bollinger J. M. Jr.; Krebs C. Curr. Opin. Chem Biol. 2007, 11, 151. [DOI] [PubMed] [Google Scholar]; d Kovaleva E. G.; Lipscomb J. D. Nat. Chem. Biol. 2008, 4, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]; e van der Donk W. A.; Krebs C.; Bollinger J. M. Jr. Curr. Opin. Struct. Biol. 2010, 20, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples:; a Baldwin J. E.; Adlington R. M.; Domaynehayman B. P.; Knight G.; Ting H. H. J. Chem. Soc., Chem. Commun. 1987, 1661. [Google Scholar]; b Ryle M. J.; Liu A.; Muthukumaran R. B.; Ho R. Y. N.; Koehntop K. D.; McCracken J. Jr.; Que L.; Hausinger R. P. Biochemistry 2003, 42, 1854. [DOI] [PubMed] [Google Scholar]; c Howard-Jones A. R.; Elkins J. M.; Clifton I. J.; Roach P. L.; Adlington R. M.; Baldwin J. E.; Rutledge P. J. Biochemistry 2007, 46, 4755. [DOI] [PubMed] [Google Scholar]
- Hidaka T.; Goda M.; Kuzuyama T.; Takei N.; Hidaka M.; Seto H. Mol. Gen. Genet. 1995, 249, 274. [DOI] [PubMed] [Google Scholar]
- a Liu P.; Murakami K.; Seki T.; He X.; Yeung S. M.; Kuzuyama T.; Seto H.; Liu H.-w. J. Am. Chem. Soc. 2001, 123, 4619. [DOI] [PubMed] [Google Scholar]; b Liu P.; Liu A.; Yan F.; Wolfe M. D.; Lipscomb J. D.; Liu H.-w. Biochemistry 2003, 42, 11577. [DOI] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Chang W.-c.; Liu H.-w. Chem. Rev. 2012, 112, 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hammerschmidt F.; Bovermann G.; Bayer K. Liebigs Ann. Chem. 1990, 1055. [Google Scholar]; b Hammerschmidt F. J. Chem. Soc. Perkin Trans. 1 1991, 1993. [Google Scholar]
- Zhao Z.; Liu P.; Murakami K.; Kuzuyama T.; Seto H.; Liu H.-w. Angew. Chem., Int. Ed. Engl. 2002, 41, 4529. [DOI] [PubMed] [Google Scholar]
- a Higgins L. J.; Yan F.; Liu P.; Liu H.-w.; Drennan C. L. Nature 2005, 437, 838. [DOI] [PubMed] [Google Scholar]; b Yun D.; Dey M.; Higgins L. J.; Yan F.; Liu H.-w.; Drennan C. L. J. Am. Chem. Soc. 2011, 133, 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Chang W.-c.; Guo Y.; Huang H.; Peck S. C.; Pnadelia M. E.; Lin G.-m.; Liu H.-w.; Krebs C.; Bollinger J. M. Jr. Science 2013, 342, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HppE had previously been considered an oxidase that uses Fe(III)-superoxo for H• abstraction. However, a recent study revealed that the preferred cosubstrate of HppE is H2O2 instead of O2.10 Hence, HppE should now be classified as a non-heme iron peroxidase that uses Fe(IV)-oxo complex for hydrogen atom abstraction. It was also shown that slow infusion of H2O2 into the assay mixture is essential for high turnover and minimization of HppE inactivation during catalysis. Since H2O2 is generated in situ when O2 is used with the NADH/FMN reducing system as described herein (and in our early work), the slow evolution of H2O2 from O2 by the chemical reducing system remains an effective approach to assay HppE activity.
- a Woschek A.; Wuggenig F.; Peti W.; Hammerschmidt F. ChemBioChem 2002, 3, 829. [DOI] [PubMed] [Google Scholar]; b Yan F.; Moon S.-J.; Liu P.; Zhao Z.; Lipscomb J. D.; Liu A.; Liu H.-w. Biochemistry 2007, 46, 12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Chang W.-c.; Pai P.-j.; Romo A.; Mansoorabadi S.; Russell D.; Liu H.-w. J. Am. Chem. Soc. 2012, 134, 16171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Okuma K.; Tanaka Y.; Yoshihara K.; Ezaki A.; Koda G.; Ohta H.; Hara K.; Kashimura S. J. Org. Chem. 1993, 58, 5915. [Google Scholar]; b Le Corre M.; Hercouet A.; Bessieres B. J. Org. Chem. 1994, 59, 5483. [Google Scholar]; c Chen X.; Zemlicka J. J. Org. Chem. 2002, 67, 286. [DOI] [PubMed] [Google Scholar]
- a Ohtani I.; Kusumi T.; Ishitsuka M. O.; Kakisawa H. Tetrahedron Lett. 1989, 3147. [Google Scholar]; b Hoye T. R.; Jeffrey C. S.; Shao F. Nat. Protoc. 2007, 2, 2451. [DOI] [PubMed] [Google Scholar]
- See SI for experimental details.
- a Chang W.-c.; Dey M.; Liu P.; Manssorabadi S. O.; Moon S.-J.; Ahao Z. K.; Drennan C. L.; Liu H.-w. Nature 2013, 496, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chang W.-c.; Mansoorabadi S. O.; Liu H.-w. J. Am. Chem. Soc. 2013, 135, 11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Griller D.; Ingold K. U. Acc. Chem. Res. 1980, 13, 317. [Google Scholar]; b Nonhebel D. C. Chem. Soc. Rev. 1993, 347. [Google Scholar]; c Newcomb M. Tetrahedron 1993, 49, 1151. [Google Scholar]; d Silverman R. B. Acc. Chem. Res. 1995, 28, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Newcomb M.; Toy P. H. Acc. Chem. Res. 2000, 33, 449. [DOI] [PubMed] [Google Scholar]; f Ortiz de Montellano P. R. Chem. Rev. 2010, 110, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner J. H.; Johnson C. C.; Lai M. T.; Lin H.-w.; Martinesker A. A.; Newcomb M.; Oh E. Bioorg. Med. Chem. Lett. 1994, 4, 2693. [Google Scholar]
- Spence E. L.; Langley G. J.; Bugg T. D. H. J. Am. Chem. Soc. 1996, 118, 8336. [Google Scholar]
- For examples of covalent enzyme modification by a ring-opened butenyl radical:; a Silverman R. B.; Zhou J. P.; Eaton P. E. J. Am. Chem. Soc. 1993, 115, 8841. [Google Scholar]; b Lai M. T.; Liu L. D.; Liu H.-w. J. Am. Chem. Soc. 1991, 113, 7388. [Google Scholar]; c Ziering D. L.; Pascal R. A. J. Am. Chem. Soc. 1990, 112, 834. [Google Scholar]; d Bishwajit Paul B.; Das D.; Ellington B.; Marsh E. N. G. J. Am. Chem. Soc. 2013, 135, 5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.