

Abstract
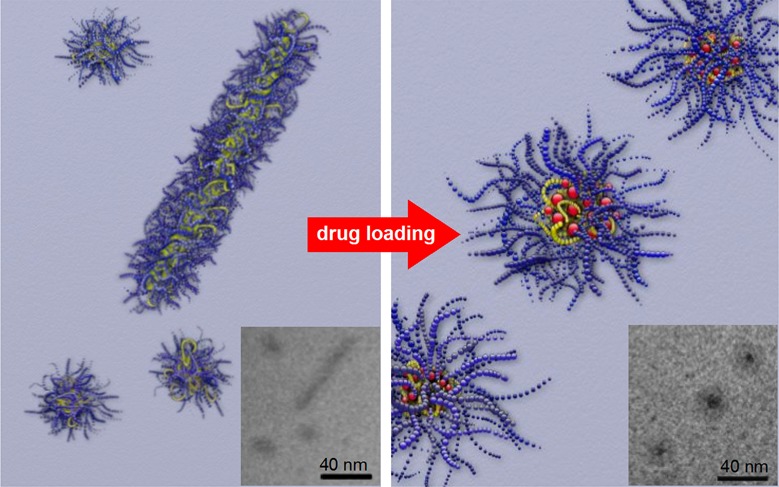
Defined aggregates of polymers such as polymeric micelles are of great importance in the development of pharmaceutical formulations. The amount of drug that can be formulated by a drug delivery system is an important issue, and most drug delivery systems suffer from their relatively low drug-loading capacity. However, as the loading capacities increase, i.e., promoted by good drug–polymer interactions, the drug may affect the morphology and stability of the micellar system. We investigated this effect in a prominent system with very high capacity for hydrophobic drugs and found extraordinary stability as well as a profound morphology change upon incorporation of paclitaxel into micelles of amphiphilic ABA poly(2-oxazoline) triblock copolymers. The hydrophilic blocks A comprised poly(2-methyl-2-oxazoline), while the middle blocks B were either just barely hydrophobic poly(2-n-butyl-2-oxazoline) or highly hydrophobic poly(2-n-nonyl-2-oxazoline). The aggregation behavior of both polymers and their formulations with varying paclitaxel contents were investigated by means of dynamic light scattering, atomic force microscopy, (cryogenic) transmission electron microscopy, and small-angle neutron scattering. While without drug, wormlike micelles were present, after incorporation of small amounts of drugs only spherical morphologies remained. Furthermore, the much more hydrophobic poly(2-n-nonyl-2-oxazoline)-containing triblock copolymer exhibited only half the capacity for paclitaxel than the poly(2-n-butyl-2-oxazoline)-containing copolymer along with a lower stability. In the latter, contents of paclitaxel of 8 wt % or higher resulted in a raspberry-like micellar core.
Keywords: amphiphilic poly(2-oxazoline)s, paclitaxel, drug delivery, rod-to-sphere transition
Drug formulation is one of the most crucial tasks in pharmaceutical technology, as many potent drugs are not water-soluble in applicable quantities. To deliver drugs, several formulation vehicles such as nano- and microparticles,1 liposomes,1,2 and polymeric micelles3−7 have been investigated. To facilitate treatment, a high capacity of these vehicles for the active pharmaceutical ingredient is desirable, as it allows a smaller volume of the formulation and a smaller amount of excipient to reach an effective dose. Paclitaxel (PTX), a potent cytostatic drug for a wide range of tumors, is one of these drugs with very low water solubility of approximately 1 μg/mL.2−5 However, a formulation (Taxol) first approved by the U.S. Food and Drug Administration (FDA) contains less than 1 wt % of PTX and uses high concentrations of the cosolvents ethanol and Cremophor EL needed to solubilize adequate amounts of PTX. This vehicle may induce a life-threatening hypersensitivity. Nevertheless, Taxol is still commonly used to treat breast, ovarian, and non-small-cell lung (NSCL) cancers.8,9 In 2005, the FDA approved an albumin-bound PTX (Abraxane), which contains 10 wt % of the active drug. It is approved for the treatment of breast and NSCL cancers that have metastasized, recurred, or cannot be treated with radiation therapy or surgery.10 In previous publications, we presented amphiphilic poly(2-oxazoline)s (POx, Figure 1) as a drug delivery platform with low toxicity, low complement activation, and very high loading capacities for a variety of hydrophobic drugs.11−15 With several drugs but especially with PTX, docetaxel (DTX), and 17-allylamino-17-demethoxygeldanamycin (17-AAG) long-term stable micellar formulations were obtained, with no significant changes of the micellar size or size distribution over a time period of at least 14 days.16 Moreover, the POx–drug micellar formulations can be freeze-dried and rehydrolyzed without a change in the drug-loading capacity, micellar size, particle dispersity, or in vitro drug activity.11
Figure 1.
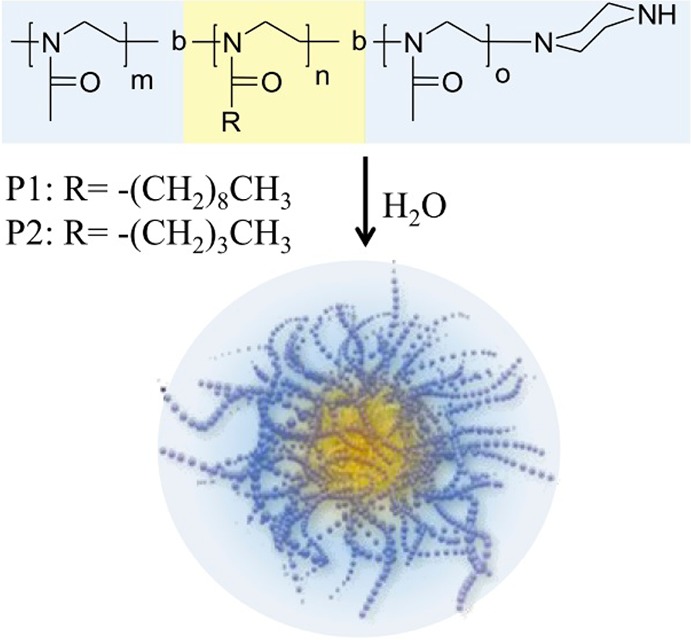
Chemical structure of amphiphilic poly(2-oxazoline)s and their schematic self-assembly to micelles.
These POx micelles not only allow unprecedented high incorporation of nearly 50 wt % PTX but also achieve high loading capacities for multiple other drugs such as 17-AAG, etoposide, and bortezomib and their combinations of up to three agents simultaneously.16 During these investigations, it became apparent that, although drugs such as PTX are hydrophobic, a pronounced hydrophobicity of the hydrophobic block is not necessarily beneficial for drug solubilization. Experiments with the fluorescent probe pyrene revealed a surprisingly polar environment within the micellar core of amphiphilic block copolymers comprising a poly(2-n-butyl-2-oxazoline) (PBuOx) block.11,17 BuOx is the first hydrophobic POx in the homologue series of poly(2-alkyl-2-oxazoline)s, and the short n-butyl side chains are probably not sufficient to shield the repeating polar amide motif in each monomer unit of the polymer backbone. In contrast, poly(2-n-nonyl-2-oxazoline) (PNOx) comprising micellar cores exhibit a nonpolar environment expected in micelles.11,18−20 We hypothesize that the highly polar BuOx core offers a highly suitable environment for the hydrophobic PTX with its many polar groups. Here, we report that the increase in the hydrophobicity of the middle block B, from barely hydrophobic BuOx to highly hydrophobic NOx, decreases the solubilization capacity for PTX by more than half. To understand this phenomenon, we investigated the morphologies of both polymer aggregates and their formulations with PTX by dynamic light scattering (DLS), atomic force microscopy (AFM), (cryo-) transmission electron microscopy ((cryo-)TEM), and small-angle neutron scattering (SANS). Moreover, the results reveal that the mutual interactions between drug and carrier strongly influence morphology, loading capacity, and stability of the drug-loaded micelles.
Results
Polymer Characterization and Drug Solubilization Capacity
The nature of the hydrophobic core in polymer micelles may influence the drug loading. For example, while amphiphilic block copolymers containing barely hydrophobic BuOx blocks promoted the solubilization of PTX extraordinarily well, Luxenhofer etal.11 also observed that an amphiphilic diblock copolymer with a strongly hydrophobic NOx block (P(MeOx32-b-NOx9)) only solubilized a fraction (7 wt %). To investigate the differences between the two types of polymers in more detail, we compare here triblock copolymers with similar hydrophilic/lipophilic balances, but different structures of the hydrophobic block (BuOx vs NOx).
Both ABA triblock copolymers were synthesized by means of living cationic ring-opening polymerization, allowing control over polymer composition and narrow polymer dispersity Đ (Mw/Mn) (Table 1). The triblock copolymer P(MeOx-b-NOx-b-MeOx) was prepared in two batches (P1′, P1″). For the sake of readability, we will not distinguish between the two batches and will refer to both polymers as P1, since we did not observe any significant differences in their physicochemical behavior and drug formulation.
Table 1. Characterization Data of ABA Triblock Copolymers Used in This Study.
| polymer compositiona | Mnb [kg/mol] | Mnc [kg/mol] | Đ | |
|---|---|---|---|---|
| P1′ | P[MeOx35-b-NOx14-b-MeOx35] | 8.8 | 8.7 | 1.28 |
| P1″ | P[MeOx34-b-NOx12-b-MeOx37] | 8.5 | 9.2 | 1.14 |
| P2 | P[MeOx33-b-BuOx26-b-MeOx45] | 10.0 | 11.4 | 1.14 |
MeOx: 2-methyl-2-oxazoline, BuOx: 2-butyl-2-oxazoline, NOx: 2-nonyl-2-oxazoline.
As determined by end group analysis from 1H NMR spectroscopy.
As determined by gel permeation chromatography.
The new batch of P(MeOx-b-BuOx-b-MeOx) (P2) displayed the same extraordinary loading capacity of PTX (49 wt %) as its predecessors, while the triblock copolymer P1 had a slightly better loading capacity (24 wt %) as compared to the diblock copolymer containing NOx reported earlier (Figure 2). We attribute this to the different architecture (AB vs ABA). First, we investigated the drug solubilization with respect to the drug feed. Interestingly, when increasing the PTX feed/POx ratio in the drug–polymer film during preparation, the efficiency of P1 to solubilize PTX was initially similar to that of P2, but dropped sharply above a ratio of 1:5 (w/w). Even more, the solubilized PTX content in the final aqueous solutions was less than in solutions prepared with lower feeding concentrations of PTX. The same phenomenon was observed for P2; however, the oversaturation of the thin film occurred only with PTX/P2 feed ratios above 1.
Figure 2.
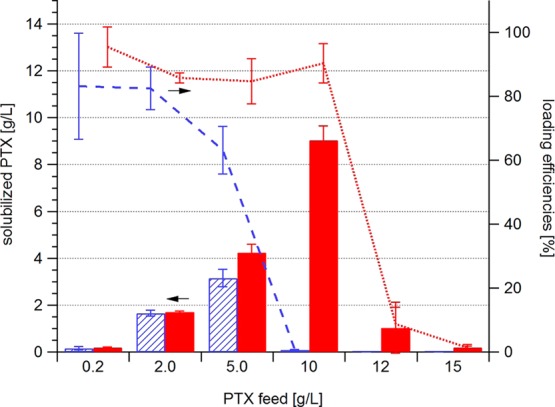
Solubilized PTX (bars; P1 striped; P2 solid, left axis) and loading efficiencies (P1 dashed line; P2 dotted line, right axis) of polymers P1 and P2 (10 g/L) at different PTX feeds. Data are presented as means ± SEM (n = 3–5).
Aggregation Behavior Studied by DLS
Earlier studies on the aggregation behavior of a P(MeOx30-b-NOx7-b-MeOx26) triblock copolymer using fluorescence correlation spectroscopy revealed micelles with a hydrodynamic radius of around 5.6 nm in deionized water.21 Here, we found similar hydrodynamic radii of the micelles in buffer solution, for both polymers at 10 g/L (Rh,mic(P1) = 8.3 ± 0.1 nm; Rh,mic(P2) = 15.3 ± 0.7 nm). However, both P1 and P2 also formed larger aggregates in solution as well (Rh,agg(P1) = 207 ± 28 nm; Rh,agg(P2) = 79 ± 25 nm) (Figure 3).
Figure 3.

Rh of micelles (open square) and aggregates (circle) of P1 (left) and P2 (right) without and with PTX. Rh values were determined by multiangle light scattering (30–150°). The scattering contribution of micelles (solid square) is derived from the measurement angle 90°. The polymer concentration was kept constant at 10 g/L. The dotted lines are added as guidance for the eye.
With the introduction of small quantities of PTX, the micelles became smaller and more defined, as evidenced by the reduced width of the peaks in the distribution of hydrodynamic radii (Figure 4). Already the presence of 3 wt % PTX (0.3 g/L) was sufficient to suppress formation of aggregates in an aqueous solution of P1. However, at a PTX content of 14.5 wt % (c(PTX) = 1.7 g/L) the micelles more than doubled in size (Rh,mic(P1+PTX) = 26.8 ± 0.2 nm) in comparison to the unloaded micelles. The same trend was observed for P2; at a maximum loading capacity of 49 wt % the largest Rh value was observed with Rh,mic(P2+PTX) = 23.8 ± 0.3 nm. Since P2 can solubilize much more PTX, the tendencies of initial compaction and subsequent growth of the micelles with increasing PTX concentration were better observable for this polymer (Figure 4).
Figure 4.

Unweighted distribution of the hydrodynamic radii Rh of P1 (left) and P2 (right) with varying concentrations of PTX and a polymer concentration of 10 g/L determined by DLS at 90°.
Again, dissolution of all larger aggregates was observed upon the addition of PTX, however in the case of P2 only at PTX contents ≥ 9 wt % (≥1.01 g/L). Bonné etal.22,23 described that large metastable aggregates for short P(MeOx-b-NOx) block copolymers at room temperature (RT) vanished after annealing the polymer solution above 40 °C. To verify that in our system the disappearance of the larger aggregates was not caused by the PTX solubilization procedure, which is carried out at 55 °C for 45 min, we carried out DLS measurements in aqueous solutions of the polymers alone that had been heated to 60 °C for over 1 h. Indeed, the scattering contribution of the micelles did increase, but the large aggregates did not dissolve completely (data not shown). It appears that in the described system the incorporation of PTX influences the aggregation of the polymers more than thermal treatment. Obviously the formation of small and well-defined micelles can be optimized by variation of the drug concentration. This effect is dependent on the nature of the hydrophobic block of the amphiphilic block copolymer.
Long-Term Stability of the PTX Formulations
In previous studies, we reported on the good stability of POx–drug formulations including the here described P2. Especially with taxanes (PTX and DTX) and 17-AAG, stable formulations were obtained with no noticeable change of the micellar size and particle dispersity for at least 14 days.16 Here, we present results of a direct comparison of the P1/PTX and P2/PTX formulation at their maximum possible drug content of 1.7 g/L PTX for P1 and 9.9 g/L for P2, respectively. The formulations were examined by eye inspection for precipitation of drug as well as by DLS to determine the development of aggregate sizes and dispersity with time. The results are summarized in Figure 5.
Figure 5.

Long-term stability study for P1/PTX and P2/PTX formulations at maximum loading conditions as determined by DLS at indicated lengths of storage time. Development of the unweighted size distribution of Rh of P1/PTX (10/1.7 g/L, above) and P2/PTX (10/9.9 g/L, below) and their corresponding intensity autocorrelation functions (right) over time. For better visibility only selected size distributions are shown with a y-offset of 0.1.
For P1/PTX excellent stability was observed, but the first changes became noticeable after 4 weeks of storage under ambient conditions. DLS indicated the formation of larger aggregates, and after 3 months, the formulation became so heterodisperse that a reasonable fit of the autocorrelation function was impossible. Moreover, some drug precipitated, and centrifugation and analysis of the clear supernatant solution resulted in a remaining PTX content of 1.4 g/L. For the P2/PTX formulation, no changes of the formulation appearance, micellar size, or particle dispersity were detectable even after 7 months of storage under ambient conditions. As shown in Figure 5, the autocorrelation function of the P2/PTX formulation did not change over a time course of more than half a year. A routine check of the drug content (centrifugation and analysis of the clear solution) after 3 months resulted in 9.8 g/L PTX, which is within the experimental error.
Aggregate Characterization by AFM and (Cryo-)TEM
While DLS gave information about the size of the micelles and aggregates, AFM was utilized to visualize their shape. It is important to note that, despite various attempts, we were unable to visualize micelles formed by bare P1 and P2. This is attributed to their disintegration upon contact with the surface or removal during washing. Only after the introduction of PTX, we were able to visualize the aggregates upon deposition on the surface (Figure 6).
Figure 6.

AFM height scans and corresponding height profiles prepared from solutions of P1/PTX (10/0.2 g/L) (a); P2/PTX (10/0.2 g/L) (b, d); and P2/PTX (10/5 g/L) (c) diluted immediately 100–1000 times prior to deposition on a freshly cleaved mica surface.
The resulting AFM scans support the previously discussed DLS data. With sufficiently high PTX content, both P1 and P2 displayed uniform spheres (Figure 6a and c) of approximately 5 nm in height and 40–50 nm in diameter. The deviation in size as compared to DLS data is attributed to the distortion of the micelles upon deposition and subsequent drying as well as to the choice of the AFM tip. Notably, P2 containing 3 wt % PTX displayed wormlike micelles (Figure 6b and d).
To verify if the wormlike micelles are also formed in the absence of PTX and to confirm their dissolution upon drug incorporation, the polymers were investigated by TEM as well. The TEM image of negatively stained P1 shows homogeneous spherical micelles with only a few wormlike structures (Figure 7a), which disappear completely upon introduction of PTX (image not shown).
Figure 7.

TEM images of uranyl acetate stained P1 (a) and P2 (b).
The image quality (Figure 7a) allowed analyzing the shape of the structures via image analysis software. The resulting regions of interest (ROIs) were further sorted by their circularity. Regions with a circularity above 4 (strong ellipsoidal form) were not included into the calculation of the micelle size, since those objects were attributed to the aggregation of micelles, which the employed image analysis algorithm could not distinguish due to insufficient threshold values in those regions. When converting the pixels into nanometers, an average radius of 4.6 ± 1.2 nm for P1 was calculated. Not surprisingly, this value is smaller than the radius determined via DLS (Rh(P1) = 8.3 ± 0.1 nm). This is attributed to the loss of the hydration shell during drying. The TEM images of bare P2 (Figure 7b) and of P2 with PTX (image not shown) were not sufficient in quality for analysis via image analysis software. Thus, the ROIs were manually defined. The size of P2 micelles at 6.7 ± 1.1 nm is slightly larger than that of P1 micelles, which corroborates the DLS results. The wormlike micelles are also discernible for bare P2 and their disappearance upon introduction of PTX. The data are summarized in Table 2.
Table 2. Dimensions (nm) of Spherical and Wormlike Micelles of P1 and P2 and Their Drug Formulations Obtained by Image Analysis of Negatively Stained (TEM) and Vitrified (cryo-TEM) Samples.
| P1 | P1/PTX 17 wt % | P2 |
P2/PTX 30 wt % |
|||
|---|---|---|---|---|---|---|
| TEM | TEM | TEM | cryo-TEM | TEM | cryo-TEM | |
| Rcore | 6.1 ± 1.2 | |||||
| Rmic | 4.6 ± 1.2 | 4.2 ± 0.6 | 6.7 ± 1.1 | 13.5 ± 2.2 | 5.2 ± 0.8 | 13.2 ± 2.8 |
| Lworms | 34–152 | 30–70 | 34–70 | |||
To overcome the effects of sample preparation that might induce artifacts due to the staining and drying procedure and to obtain images of improved quality, bare solutions of P2 and P2 with PTX (10 and 5 g/L) were investigated by cryo-TEM. Again, coexistence of wormlike structures and spherical micelles was observed when P2 was prepared alone. Upon incorporation of PTX, exclusively spherical micelles were observed (Figure 8).
Figure 8.

Cryo-TEM images of P2 (10 g/L) (a) and P2/PTX (10/5 g/L) (b). Insets show the indicated region at higher magnification.
At higher magnification the core–shell structure of the drug-loaded P2 micelles is clearly discernible (Figure 8b). In comparison to the images obtained for bare P2, the incorporation of PTX apparently increases the electron density defining the core perimeter. Again, the ROIs were manually defined. Subsequently, the size analysis of both the core and the micelles was performed (Table 2).
Small-Angle Neutron Scattering
To better understand the process of incorporation of such large quantities of PTX into the BuOx core (as opposed to the limited uptake in NOx), we briefly present results obtained by small-angle neutron scattering on a concentration series of PTX with a constant amount of P2 (10 g/L in D2O). The micelles from bare P2 and up to PTX concentrations of 5 g/L (32 wt %) were found to be spheres with radii of 4–6 nm, which excellently correlate with the size of the micellar core found in cryo-TEM measurement (Figure 9). In our present setup of the SANS experiment, the micellar shell was not resolved because of its strong hydration and its resulting low scattering contrast with respect to the solvent. Also, the contribution of wormlike micelles was not deconvoluted.
Figure 9.
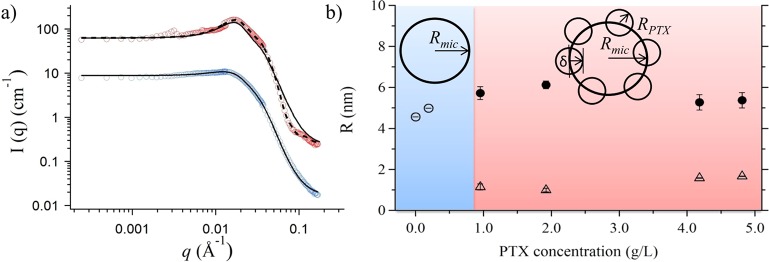
(a) Scattering curves of bare P2 (10 g/L, blue) and P2 with PTX (10 and 4.8 g/L, red) and their fitting curves assuming a homogeneous sphere (solid line) or a raspberry form (dashed line). Curves are shifted in intensity for better visibility. (b) All radii from SANS measurements of P2 solutions in D2O (c = 10 g/L at 37 °C) with varying PTX concentrations. Sketches indicate if homogeneous spheres or raspberry-like micelles were applied for the fitting process. Open circles: radii of spherical micelles, solid circles: radii of raspberry-like micelles, Rmic, open triangles: radii of the embedded PTX particles, RPTX.
The scattering profile from solutions with more than 8 wt % PTX (0.9 g/L) could not be adequately modeled by homogeneous spheres, but rather by a raspberry-like sphere as depicted in Figure 9. We interpret this as reflecting the presence of PTX-rich domains in the polymer micelles. The radius of the particles RPTX embedded into the micellar core increased along with the PTX concentration from 1.2 nm to 1.7 nm. The details of the quite complex fitting procedure of the SANS data are discussed elsewhere, as it requires a detailed presentation and is, as such, beyond the scope of this paper.24,25
Discussion
Polymeric micelles play an important role in nanomedicine as highly versatile drug delivery systems. We used such micelles based on poly(2-oxazoline) to solubilize unprecedented high loading contents of the anticancer drug paclitaxel (45 wt %). Since then, various other high-capacity micellar drug delivery systems for taxanes (29–36 wt %) have been reported in the literature.26−28 However, with the increasing success of realizing high-capacity drug formulations, it becomes important to analyze the effects of the incorporated drug on the morphology and stability of the system. Changes in the aggregation behavior of amphiphilic polymers upon drug incorporation have been previously reported for pluronic and other systems.29−32 Depending on the nature of the drug, the micelles increased29 or decreased30 in size. More interestingly, drugs also induced sphere-to-rod transitions, where the incorporated drug shifted the balance from spherical toward wormlike micelles.31,32 In the past decade, wormlike micelles have gained increased interest as a drug delivery system due to their enhanced circulation time in vivo and increased loading capacities.33−36 The presently studied triblock copolymers P[MeOx-b-BuOx-b-MeOx] and P[MeOx-b-NOx-b-MeOx] form predominantly spherical micelles with only a small fraction of wormlike micelles. However, the incorporation of PTX did not facilitate the formation of these wormlike micelles, but rather caused their disintegration. To the best of our knowledge, drug-induced rod-to-sphere transitions have so far not been reported in the literature. Tan etal. demonstrated on the basis of multiblock polyurethane micelles that defects in the micellar shell, leading to partial exposure of the hydrophobic core, initiated sphere-to-rod transition.37 In the case of P[MeOx-b-BuOx-b-MeOx], the inclusion of PTX seems to compact the micelles for PTX contents up to 32 wt %. Applying the approach of Tan etal.37 to our system, the induced compaction of the micellar core by PTX reduces the probability of hydrophobic chains being exposed on the micelle surface, thus preventing the formation of wormlike structures.
Nevertheless, at maximum drug loading both systems remained stable for a long time. While for P1 the first changes became noticeable after 4 weeks, the P2/PTX formulation displayed no measurable changes for more than half a year. This can be explained by the very effective nonpolar as well as polar drug–polymer interactions. Further increase in drug concentration above the critical loading capacity lead ultimately to a collapse in loading efficiency. While the drop occurs for the P[MeOx-b-NOx-b-MeOx] copolymer already at PTX contents of 23 wt %, P[MeOx-b-BuOx-b-MeOx] can be reproducibly loaded with up to 49 wt % PTX. In contrast to a NOx core, BuOx exhibits a considerably lower crystalline volume fraction, and the amorphous phase is well above its Tg of BuOx during the PTX solubilization process.38 The increased mobility of the chains within the core might make them more suitable to accommodate higher quantities of PTX.
The SANS experiments revealed intricate structural changes within the micelle of P[MeOx-b-BuOx-b-MeOx] upon the addition of PTX. While the scattering model suggests homogeneous spheres in the absence of PTX, small domains, likely comprising PTX, become apparent after the addition of PTX. These domains are embedded into the micellar core at an average embedment depth δ of approximately −0.3 nm, which means that they are partially submerged into the core rather than clinging as separate particles to the surface. Together with the relative surface coverage of the domains of 0.76−0.92, we can envision that the PTX-loaded P2 micelles have a core whose surface is covered by PTX-rich domains, which are partially embedded into the core. However, the domain size lies between 1 and 2 molecular diameters of PTX, and thus, even at high loading of PTX, the drug is not incorporated in the bulk state but molecularly dispersed and associated with polymer chains via nonpolar as well as polar interactions. This picture of PTX incorporation in the core is further corroborated by thermal analysis using differential scanning calorimetry (DSC, data not shown). DSC gave only the glass transition for the hydrophilic MeOx blocks of P1 or P2 ((P1/PTX19wt%) = 73 °C; Tg(P2/PTX49wt%) = 76 °C)). The hydrophobic part of the micelles (NOx or BuOx block and PTX) does not give rise to a glass or melting transition between 0 °C and decomposition.
Conclusion
The present study establishes a strong influence of the anticancer drug PTX on the morphology of poly(2-oxazoline)-based micelles. In aqueous solution, the investigated amphiphilic triblock copolymers partially form wormlike micelles. Wormlike micelles, or “filomicelles”, are currently discussed as promising drug carriers with significantly improved blood circulation times, faster endocytosis, and improved anticancer activity.39 However, drug loading of the presented triblock copolymers leads to a morphology switch from partly filomicelles to exclusively spherical micelles. They were found to be long-term stable for over half a year, well-defined, and even at high loading conditions very small. In a recent study, Kataoka etal.40 presented a conclusive study on the size effect of micellar drug delivery systems. It was shown that anticancer drug formulations of small micellar sizes are a prerequisite for the treatment of poorly permeable solid tumors. At increasing PTX concentration, localization of drug nanoparticles at the core–shell interface results in a micellar core with a raspberry-like structure. These results indicate the importance of the thorough characterization of drug carriers after drug loading, especially when pushing the limit to high-capacity systems.35,36 As the loading capacity and stability of the drug-loaded systems represent a critical aspect in micellar drug delivery, the present study provides important insights for the development of other drug delivery systems to optimize their drug delivery capabilities and stabilities.
Materials and Methods
All chemicals were purchased from Acros Organics (Geel, Belgium) and Sigma-Aldrich (Munich, Germany) and were used as received unless otherwise stated. Deuterated solvents for NMR analysis were obtained from Deutero GmbH (Kastellaun, Germany). PTX was purchased at LC Laboratories (Woburn, MA, USA). Phosphate-buffered saline (PBS) was prepared by dissolving one tablet (Fluka, 79382) in 200 mL of H2Odd to obtain a 137 mM NaCl, 2.7 mM KCl, and 10 mM phosphate buffer solution (pH 7.4 at 25 °C). NOx was received as a gift from Henkel KGaA (Düsseldorf, Germany). BuOx was prepared following the procedure by Huber etal.41 All substances used for polymerization, methyl trifluoromethylsulfonate (MeOTf), MeOx, BuOx, NOx, chlorobenzene (ClBz), and acetonitrile (ACN), were refluxed over CaH2 and distilled under nitrogen. NMR spectra were recorded on a Bruker DRX 500 P (1H: 500.13 MHz) at RT. The spectra were calibrated to the signals of residual protonated solvent signals (ACN: 1.94 ppm, MeOD: 3.31 ppm). Gel permeation chromatography (GPC) was performed on a Polymer Laboratories GPC-120 (column setup: 1× PSS GRAM analytical 1000 and 1× PSS GRAM analytical 100 obtained from Polymer Standards Services, Mainz, Germany) with N,N-dimethyl acetamide (DMAc) (5 mmol/L LiBr, 1 wt % H2O, 70 °C, 1 mL/min) as eluent and poly(methyl methacrylate) standards. Microwave-supported polymerization was performed using a CEM Discover microwave.
Polymer Synthesis
Living Cationic Ring-Opening Polymerization
Under dry and inert conditions, 1 equiv of MeOTf and 35 equiv of MeOx were dissolved in dry ACN/ClBz (50/50, v/v) at RT. The reaction mixture was irradiated in the microwave for 60 min at 150 W and 100 °C (P1’: 10 min, 150 W, 130 °C). After cooling to RT 20 equiv/13 equiv BuOx/NOx were added, and the mixture was irradiated for another 60 min (P1’: 10 min). The procedure was repeated with 35 equiv of MeOx. Termination was carried out with 3 equiv of 1-BOC-piperazine in ClBz for 1 h, 50 W and 40 °C. An excess of potassium carbonate was added, and the mixture was stirred for several hours. After filtration, the mixture was concentrated, added into a mixture of chloroform and methanol (75/25, v/v), and precipitated in cold diethyl ether. After centrifugation and removal of diethyl ether the precipitation was repeated one more time. The residue was lyophilized, and a colorless powder was obtained.
Deprotection
The obtained polymer was dissolved either in a mixture of trifluoroacetic acid (TFA), triisobutylsilane (TIBS), and water (95/2.5/2.5, v/v/v) or in chloroform, methanol, and phosphoric acid (25/50/25, v/v/v) and stirred for 1 h and overnight at RT, respectively. The volatiles were removed under fine vacuum. The residue was dissolved in deionized water (if necessary neutralized with sodium hydroxide and salts removed after lyophilization in ACN), transferred into a dialysis bag (MWCO 3500 g/mol), and dialyzed against deionized water. The solution was recovered from the bag and lyophilized, and a colorless powder was obtained.
Methyl-P[MeOx35-b-NOx14-b-MeOx35]-piperazine (P1′):
21.4 mg (0.13 mmol, 1 equiv) of MeOTf, 0.40 g (4.71 mmol, 36 equiv) of MeOx, 0.34 g (1.70 mmol, 13 equiv) of NOx, 0.39 g (4.62 mmol, 36 equiv) of MeOx, and 75.0 mg (4.03 mmol, 3 equiv) of 1-BOC-piperazine. A colorless powder was obtained (0.79 g, 72%). GPC (DMAc): Mn = 11.8 kg/mol (Đ = 1.13). A 0.50 g amount of the polymer was deprotected with 3 mL of TFA/TIBS/H2O. A 0.46 g (92%) yield of a colorless powder was obtained. GPC (DMAc): Mn = 8.7 kg/mol (Đ = 1.28).
1H NMR (MeOD, 300 K): δ [ppm] 3.53 (m, 334H, N-CH2CH2); 3.10/3.06/2.95 (m, 3H, N-CH3Ini); 2.75–2.33(m, 33H, CO-CH2nonyl, CH2Pip); 2.15–2.09 (m, 210H, CO-CH3); 1.59 (br, 28H, CH2-CH2-CH2−); 1.31 (br, 168H, −CH2−); 0.91 (br, 41H, CH3nonyl).
Methyl-P[MeOx35-b-NOx11-b-MeOx36]-piperazine (P1″):
0.19 g (1.17 mmol, 1 equiv) of MeOTf, 3.49 g (0.04 mol, 35 equiv) of MeOx, 2.69 g (0.01 mol, 12 equiv) of NOx, 3.37 g (0.04 mmol, 34 equiv) of MeOx, and 0.70 g (4.03 mmol, 3 equiv) of 1-BOC-piperazine. A colorless powder was obtained (7.85 g, 79%). GPC (DMAc): Mn = 9.5 kg/mol (Đ = 1.17). A 7.70 g amount of the polymer was deprotected with 20 mL of CHCl3/MeOH/H3PO4. A 4.51 g (59%) yield of a colorless powder was obtained. GPC (DMAc): Mn = 9.2 kg/mol (Đ = 1.14).
1H NMR (ACN, 300 K): δ [ppm] 3.38 (br, 334H, N-CH2CH2); 2.99/2.96/2.84 (m, 3H, N-CH3Ini); 2.47–2.22(m, 31H, CO-CH2nonyl, CH2Pip); 2.04–1.98 (m, 213H, CO-CH3); 1.51 (br, 24H, CH2-CH2-CH2−); 1.41 (br, 5H, CH3BOC); 1.27 (br, 141H, −CH2−); 0.88 (br, 34H, CH3nonyl).
Methyl-P[MeOx33-b-BuOx26-b-MeOx45]-piperazine (P2):
0.30 g (1.82 mmol, 1 equiv) of MeOTf, 5.44 g (63.9 mmol, 35 equiv) of MeOx, 4.57 g (35.9 mmol, 20 equiv) of BuOx, 5.34 g (62.7 mmol, 34 equiv) of MeOx, 1.03 g (5.6 mmol, 3 equiv) of 1-BOC-piperazine. A colorless powder (13.65 g, 82%) was obtained. GPC (DMAc): Mn = 10.6 kg/mol (Đ = 1.16). A 9.92 g amount of the obtained polymer was deprotected with 60 mL with TFA/TIBS/H2O. A 7.19 g (72%) yield of a colorless powder was obtained. GPC (DMAc): Mn = 11.4 kg/mol (Đ = 1.14).
1H NMR (ACN, 300 K): δ [ppm] 3.43 (br, 418H, N-CH2CH2); 2.98/2.84 (m, 3H, N-CH3Ini); 2.33–2.22 (m,86 H, CO-CH2butyl, CH2Pip); 2.04–1.08 (m, 241H, CO-CH3); 1.51 (br, 51H, CH2-CH2-CH2−); 1.31 (br, 52 H, CH2-CH3); 0.89 (br, 77 H, CH3butyl).
Paclitaxel Solubilization
Ethanolic stock solutions were mixed in desired ratios. Ethanol was evaporated with the help of a steady hot air flow (100 °C, ∼20 min). The generated thin films were further dried for >10 min in vacuo to ensure complete removal of the solvent. After the addition of PBS, the solutions were heated to 55 °C and shaken at 1000–1200 rpm for 30–45 min with the help of a thermoshaker. Nonsolubilized PTX (if any) was removed via centrifugation (10 000 min–1, 10 min). The quantification was performed on a Varian 920-LC HPLC with a stepwise gradient using a Kinetex 2.6 μm PFP, 100 × 4.60 mm column by Phenomenex at RT and UV detection at 254 nm. Within the first 15 min, the ratio of H2O/MeOH (40/60, v/v) was decreased to 5:95, followed by 10 min purging of the column with 95% MeOH and subsequent reversion of the ratio to the start settings. The loading capacity (LC) is defined as the mass of solubilized PTX divided by the total mass (LC = mPTX/mtotal), while the loading efficiency (LE) is the percentage of solubilized PTX from the original PTX feed (LE = (mPTX/mPTX,0) × 100%). The POx concentration was kept constant at 10 g/L for all solubilization experiments. Solubilization experiments were repeated 3 to 5 times, and results are presented as means ± standard error of the mean (SEM).
Dynamic Light Scattering
The DLS experiments were performed on an ALV/DLS/SLS-5000 compact goniometer system with an ALV/SO-SIPD/DUAL photomultiplier detection unit and a JDS Uniphase He–Ne-laser with a wavelength λ = 632.8 nm used at 22 mW. The toluene bath was temperature controlled by a Julabo thermostat and kept constant at 25 °C. Angle-dependent measurements were performed between 30° and 150° in 5° steps with a measurement time of 120 s.
Polymer solutions were prepared in PBS. The polymer concentration was kept constant at 10 g/L, while the PTX concentration ranged from 0 to 9.9 g/L. All solutions were filtered (0.45 μm PVDF), and PTX content was determined prior to measurement via HPLC.
The DLS data were evaluated according to Schärtl.42 The field autocorrelation functions were either modeled with a stretched exponential form,
| 2 |
for stretched exponents β ≥ 0.9 assuming a monomodal distributions or fitted with a double-exponential function (polydisperse sample).
| 1 |
Here bmic and bagg are the relative scattering contributions from different species (micelles and aggregates), respectively, while Γ is the decay rate of the respective correlation functions.
The obtained decay rates are related to the diffusion coefficient D via
| 3 |
where q is the scattering vector with q = 4π sin(θ/2)/λ and θ the scattering angle. To test the validity of a monomodal or bimodal assumption, we checked whether Γ ∝ q2 and whether the extrapolation of the data to q = 0 gave Γ = 0.
From the resulting diffusion coefficient, we calculated the hydrodynamic radii according to the Stokes–Einstein relation
| 4 |
where kB is Boltzmann’s constant, T the temperature, and η the viscosity of the solvent. Uncertainties of Rh were derived from the confidence interval of 99% of the regression lines. The distribution of Rh at θ = 90° was obtained by a regularized inverse Laplace transformation algorithm—which is incorporated in the ALV-5000 program—of the correlation function.
Atomic Force Microscopy
The AFM scans were obtained with an NT-MDT NTEGRA Spectra system. All scans were performed in tapping mode with standard AFM tips (NSG03/NT-MDT) under ambient conditions. The sample solutions were diluted (100–1000 times) immediately prior to deposition on the freshly cleaved mica surface and incubated for <1 min. Depending on the dilution, the surface was additionally washed with a drop of DI water or dried under an argon atmosphere right away. The stock solutions contained 10 g/L POx and varying concentrations of PTX (0.2–5.0 g/L). The data were processed with the Gwyddion 2.3 freeware.
Transmission Electron Microscopy
Vitrified specimens for cryo-TEM were prepared by a blotting procedure, performed in a chamber with controlled temperature and humidity using a LEICA grid plunger. A drop of the sample solution (1 g/L) was placed onto an EM grid coated with a holey carbon film (C-flat, Protochips Inc., Raleigh, NC, USA). Excess solution was then removed with a filter paper, leaving a thin film of the solution spanning the holes of the carbon film on the EM grid. Vitrification of the thin film was achieved by rapid plunging of the grid into liquid ethane held just above its freezing point. The vitrified specimen were kept below 108 K during both transfers to the microscope and investigation. Specimens were examined with a LIBRA 120 PLUS instrument (Carl Zeiss Microscopy GmbH, Oberkochen, Germany), operating at 120 kV. The microscope is equipped with a Gatan 626 cryotransfer system. Images were taken with a BM-2k-120 dual-speed on axis SSCCD camera (TRS, Moorenweis, Germany).
The negatively stained samples were prepared by spreading 5 μL of the dispersion onto a Cu grid coated with a Formvar-film (PLANO, Wetzlar, Germany). After 1 min, excess liquid was blotted off with filter paper and 5 μL of a 1% aqueous uranyl acetate solution was placed onto the grid and drained off after 1 min. The dried specimens were examined with an EM 900 transmission electron microscope (Carl Zeiss Microscopy GmbH). Micrographs were taken with an SSCCD SM-1k-120 camera (TRS, Moorenweis, Germany).
The images were analyzed with Igor Pro 6.31 using the ImageAnalyzeParticles operation or manual detection of regions of interest. The circularity of the ROIs was defined by the ratio of the square of the perimeter to 4π A, where A is the object area. For a perfect circle the value 1 is approached, while elongated objects result in values larger than 1.
Small-Angle Neutron Scattering
SANS experiments were carried out at KWS-1, Forschungsneutronenquelle Heinz Maier-Leibnitz, FRM II, in Garching. A wavelength of λ = 6.9 Å (Δ λ/λ = 10%) and sample–detector distances (SDD) of 1.72, 7.72, and 19.72 m were used, which resulted in a q-range of 0.002–0.221 Å–1. The detector was a 6Li glass scintillation detector with an active area of 60 × 60 cm. Exposure times were 5 min at SDD = 1.72 m, 15 min at SDD = 7.72, and 30 min at SDD = 19.72 m. The samples were mounted in Hellma quartz cuvettes with a light path of 2 mm in an oven. This oven was made from aluminum with a plastic cover and was temperature controlled by a Julabo thermostat. The experiments were performed at a physiological temperature of 37 °C. The scattering of boron carbide was used for correcting the intensities for dark current and background. The scattering of D2O and the empty cell were subtracted from the sample scattering, taking the transmissions into account. The resulting intensities were azimuthally averaged. Good agreement was found in the overlap region of the curves measured of all SDDs. All data reduction was performed with the software QtiKWS provided by JCNS.
Samples were prepared in D2O with concentrations as given in Table 3. The fitting procedures were written in IGOR Pro with the NIST NCNR SANS analysis package.43
Table 3. Polymer and PTX Concentrations in Samples for SANS Measurements.
| c(P2) (g/L) | 9.9 | 9.0 | 10.0 | 9.9 | 8.6 | 10.0 |
| c(PTX) (g/L) | 0 | 0.2 | 0.9 | 1.9 | 4.2 | 4.8 |
Acknowledgments
This work was supported by a Cancer Nanotechnology Platform Partnership grant (U01 CA116591) of the National Cancer Institute Alliance for Nanotechnology in Cancer.
Author Present Address
△ Jülich Centre for Neutron Science at FRM II, Forschungszentrum Jülich GmbH, Lichtenbergstraße 1, 85748 Garching, Germany.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wu J.; Liu Q.; Lee R. J. A Folate Receptor-Targeted Liposomal Formulation for Paclitaxel. Int. J. Pharm. 2006, 316, 148–153. [DOI] [PubMed] [Google Scholar]
- Yang T.; Cui F.-D.; Choi M.-K.; Cho J.-W.; Chung S.-J.; Shim C.-K.; Kim D.-D. Enhanced Solubility and Stability of Pegylated Liposomal Paclitaxel: In Vitro and in Vivo Evaluation. Int. J. Pharm. 2007, 338, 317–326. [DOI] [PubMed] [Google Scholar]
- Savić R.; Eisenberg A.; Maysinger D. Block Copolymer Micelles as Delivery Vehicles of Hydrophobic Drugs: Micelle–Cell Interactions. J. Drug Targeting 2006, 14, 343–355. [DOI] [PubMed] [Google Scholar]
- Kabanov A. V.; Vinogradov S. V. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angew. Chem., Int. Ed. 2009, 48, 5418–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P. Targeted Polymeric Micelles for Delivery of Poorly Soluble Drugs. Cell. Mol. Life Sci. 2004, 61, 2549–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K.; Harada A.; Nagasaki Y. Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance. Adv. Drug Delivery Rev. 2001, 47, 113–131. [DOI] [PubMed] [Google Scholar]
- Matsumoto S.; Christie R. J.; Nishiyama N.; Miyata K.; Ishii A.; Oba M.; Koyama H.; Yamasaki Y.; Kataoka K. Environment-Responsive Block Copolymer Micelles with a Disulfide Cross-Linked Core for Enhanced siRNA Delivery. Biomacromolecules 2009, 10, 119–127. [DOI] [PubMed] [Google Scholar]
- Weiss R. B.; Donehower R. C.; Wiernik P. H.; Ohnuma T.; Gralla R. J.; Trump D. L.; Baker J. R.; van Echo D. A.; von Hoff D. D.; Leyland-Jones B. Hypersensitivity Reactions from Taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Gelderblom H.; Verweij J.; Nooter K.; Sparreboom A. Cremophor EL: The Drawbacks and Advantages of Vehicle Selection for Drug Formulation. Eur. J. Cancer 2001, 37, 1590–1598. [DOI] [PubMed] [Google Scholar]
- Pazdur; Richard. http://www.cancer.gov/cancertopics/druginfo/fda-nanoparticle-paclitaxel (accessed July 29, 2013).
- Luxenhofer R.; Schulz A.; Roques C.; Li S.; Bronich T. K.; Batrakova E. V.; Jordan R.; Kabanov A. V. Doubly Amphiphilic Poly(2-Oxazoline)s as High-Capacity Delivery Systems for Hydrophobic Drugs. Biomaterials 2010, 31, 4972–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenhofer R.; Sahay G.; Schulz A.; Alakhova D.; Bronich T. K.; Jordan R.; Kabanov A. V. Structure-property Relationship in Cytotoxicity and Cell Uptake of Poly(2-Oxazoline) Amphiphiles. J. Controlled Release 2011, 153, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenhofer R.; Han Y.; Schulz A.; Tong J.; He Z.; Kabanov A. V.; Jordan R. Poly(2-oxazoline)s as Polymer Therapeutics. Macromol. Rapid Commun. 2012, 33, 1613–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas T. X.; Bentley M. D.; Harris J. M.; Fang Z.; Yoon K.; Dizman B.; Weimer R.; Mero A.; Pasut G.; Veronese F. M. Polyoxazoline: Chemistry, Properties, and Applications in Drug Delivery. Bioconjugate Chem. 2011, 22, 976–986. [DOI] [PubMed] [Google Scholar]
- Tong J.; Zimmerman M. C.; Li S.; Yi X.; Luxenhofer R.; Jordan R.; Kabanov A. V. Neuronal Uptake and Intracellular Superoxide Scavenging of a Fullerene (C60)-Poly(2-Oxazoline)s Nanoformulation. Biomaterials 2011, 32, 3654–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; He Z.; Schulz A.; Bronich T. K.; Jordan R.; Luxenhofer R.; Kabanov A. V. Synergistic Combinations of Multiple Chemotherapeutic Agents in High Capacity Poly(2-oxazoline) Micelles. Mol. Pharmaceutics 2012, 9, 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi K.; Takasu A.; Okada M.; Imae T. Synthesis and Assembly of Novel Chitin Derivatives Having Amphiphilic Polyoxazoline Block Copolymer as a Side Chain. Macromol. Chem. Phys. 1999, 200, 1112–1120. [Google Scholar]
- Kalyanasundaram K.; Thomas J. K. Environmental Effects on Vibronic Band Intensities in Pyrene Monomer Fluorescence and Their Application in Studies of Micellar Systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar]
- Ananthapadmanabhan K. P.; Goddard E. D.; Turro N. J.; Kuo P. L. Fluorescence Probes for Critical Micelle Concentration. Langmuir 1985, 1, 352–355. [DOI] [PubMed] [Google Scholar]
- Si Z. H.; Wang G. C.; Li H. X.; Yuan J. L.; He B. L. Studies on Micellization of a Polystyrene-b-poly(acrylic acid) Copolymer in Aqueous Media by Pyrene Fluorescence. Chin. Chem. Lett. 2003, 14, 39–42. [Google Scholar]
- Bonné T. B.; Lüdtke K.; Jordan R.; Papadakis C. M. Effect of Polymer Architecture of Amphiphilic Poly(2-oxazoline) Copolymers on the Aggregation and Aggregate Structure. Macromol. Chem. Phys. 2007, 208, 1402–1408. [Google Scholar]
- Bonné T. B.; Lüdtke K.; Jordan R.; Štěpánek P.; Papadakis C. M. Aggregation Behavior Of Amphiphilic Poly(2-Alkyl-2-Oxazoline) Diblock Copolymers in Aqueous Solution Studied by Fluorescence Correlation Spectroscopy. Colloid Polym. Sci. 2004, 282, 833–843. [Google Scholar]
- Bonné T. B.; Lüdtke K.; Jordan R.; Štěpánek P.; Papadakis C. M. Aggregation Behavior Of Amphiphilic Poly(2-Alkyl-2-Oxazoline) Diblock Copolymers in Aqueous Solution Studied by Fluorescence Correlation Spectroscopy. Colloid Polym. Sci. 2004, 282, 1425. [Google Scholar]
- Jaksch S.Phasenverhalten von Poly(2-Oxazolinen) in wässriger Lösung. PhD Thesis, Technische Universität München, 2013. [Google Scholar]
- Jaksch S.; Schulz A.; Brüningk S. C.; Di Z.; Luxenhofer R.; Jordan R.; Papadakis C. M. In preparation.
- Shi Y.; van Steenbergen M. J.; Teunissen E. A.; Novo L.; Gradmann S.; Baldus M.; van Nostrum C. F.; Hennink W. E. π–π Stacking Increases the Stability and Loading Capacity of Thermosensitive Polymeric Micelles for Chemotherapeutic Drugs. Biomacromolecules 2013, 14, 1826–1837. [DOI] [PubMed] [Google Scholar]
- Zhang L.; He Y.; Ma G.; Song C.; Sun H. Paclitaxel-loaded Polymeric Micelles Based on Poly(ε-caprolactone)-Poly(ethylene glycol)-Poly(ε-caprolactone) Triblock Copolymers: In Vitro and in Vivo Evaluation. Nanomedicine 2012, 8, 925–934. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xu X.; Shen Y.; Qian C.; Lu F.; Guo S. Preparation and Evaluation of Copolymeric Micelles with High Paclitaxel Contents and Sustained Drug Release. Colloids Surf., A 2013, 429, 12–18. [Google Scholar]
- Sharma P. K.; Reilly M. J.; Jones D. N.; Robinson P. M.; Bhatia S. R. The Effect of Pharmaceuticals on the Nanoscale Structure of PEO–PPO–PEO Micelles. Colloids Surf., B 2008, 61, 53–60. [DOI] [PubMed] [Google Scholar]
- Valero M.; Dreiss C. A. Growth, Shrinking, and Breaking of Pluronic Micelles in the Presence of Drugs and/or β-Cyclodextrin, a Study by Small-Angle Neutron Scattering and Fluorescence Spectroscopy. Langmuir 2010, 26, 10561–10571. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; D’Emanuele A.; Attwood D. Solubility Enhancement Of Paclitaxel Using a Linear-Dendritic Block Copolymer. Int. J. Pharm. 2013, 452, 173–179. [DOI] [PubMed] [Google Scholar]
- Khimani M.; Ganguly R.; Aswal V. K.; Nath S.; Bahadur P. Solubilization of Parabens in Aqueous Pluronic Solutions: Investigating the Micellar Growth and Interaction as a Function of Paraben Composition. J. Phys. Chem. B 2012, 116, 14943–14950. [DOI] [PubMed] [Google Scholar]
- Chaibundit C.; Ricardo N. M. P. S.; Crothers M.; Booth C. Micellization of Diblock(oxyethylene/oxybutylene) Copolymer E 11 B 8 in Aqueous Solution. Micelle Size and Shape. Drug Solubilization. Langmuir 2002, 18, 4277–4283. [Google Scholar]
- Christian D. A.; Cai S.; Garbuzenko O. B.; Harada T.; Zajac A. L.; Minko T.; Discher D. E. Flexible Filaments for in Vivo Imaging and Delivery: Persistent Circulation of Filomicelles Opens the Dosage Window for Sustained Tumor Shrinkage. Mol. Pharmaceutics 2009, 6, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S.; Vijayan K.; Cheng D.; Lima E. M.; Discher D. E. Micelles of Different Morphologies—Advantages of Worm-like Filomicelles of PEO-PCL in Paclitaxel Delivery. Pharm. Res. 2007, 24, 2099–2109. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Cheetham A. G.; Lin Y.-A.; Cui H. Self-Assembled Tat Nanofibers as Effective Drug Carrier and Transporter. ACS Nano 2013, 7, 5965–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H.; Wang Z.; Li J.; Pan Z.; Ding M.; Fu Q. An Approach for the Sphere-to-Rod Transition of Multiblock Copolymer Micelles. ACS Macro Lett. 2013, 2, 146–151. [DOI] [PubMed] [Google Scholar]
- Rettler E. F.-J.; Kranenburg J. M.; Lambermont-Thijs H. M.; Hoogenboom R.; Schubert U. S. Thermal, Mechanical, and Surface Properties of Poly(2-N-alkyl-2-oxazoline)s. Macromol. Chem. Phys. 2010, 211, 2443–2448. [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Shape Effects of Filaments versus Spherical Particles in Flow and Drug Delivery. Nat. Nanotechnol. 2007, 2, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral H.; Matsumoto Y.; Mizuno K.; Chen Q.; Murakami M.; Kimura M.; Terada Y.; Kano M. R.; Miyazono K.; Uesaka M.; Nishiyama N.; Kataoka K. Accumulation of Sub-100 nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size. Nat. Nanotechnol. 2011, 6, 815–823. [DOI] [PubMed] [Google Scholar]
- Huber S.; Jordan R. Modulation of the Lower Critical Solution Temperature of 2-Alkyl-2-oxazoline Copolymers. Colloid Polym. Sci. 2008, 286, 395–402. [Google Scholar]
- Schärtl W.Light Scattering from Polymer Solutions and Nanoparticle Dispersions; Springer: Berlin, 2007. [Google Scholar]
- Kline S. R. Reduction and Analysis of SANS and USANS Data Using IGOR Pro. J. Appl. Crystallogr. 2006, 39, 895–900. [Google Scholar]