

Abstract
Symptoms originating from the central nervous system (CNS) frequently occur in patients with systemic lupus erythematosus (SLE). These symptoms are extremely diverse, including a state of dementia. The aim of this study was to examine the cerebrospinal fluid (CSF) content of soluble molecules indicating axonal degeneration and amyloidogenesis.
One hundred and fourteen patients with SLE and age-matched controls were evaluated clinically, with magnetic resonance imaging of the brain and CSF analyses. Levels of tau, amyloid precursor protein (APP), β-amyloid protein (Aβ42), and transforming growth factor beta (TGF-β) were all determined using sandwich ELISAs.
APP and Aβ42 levels were significantly decreased in SLE patients irrespective of their CNS involvement, as compared with healthy controls. Patients with neuropsychiatric SLE who underwent a second lumbar puncture following successful cyclophosphamide treatment showed further decreases of Aβ42. CSF-tau levels were significantly increased in SLE patients showing magnetic resonance imaging-verified brain pathology as compared with SLE patients without such engagement. Importantly, tau levels displayed significant correlation to Aβ42 levels in the CSF. Finally, TGF-β levels were significantly increased in patients with neuropsychiatric SLE as compared with those without.
Low intrathecal levels of Aβ42 found in SLE patients seem to be a direct consequence of a diminished production of APP, probably mediated by heavy anti-inflammatory/immuno-suppressive therapy. Furthermore, our findings suggest that CSF tau can be used as a biochemical marker for neuronal degeneration in SLE. Finally, the increased TGF-β levels observed may support a notion of an ongoing anti-inflammatory response counteracting tissue injury caused by CNS lupus.
Keywords: amyloid precursor protein, β-amyloid protein, cerebrospinal fluid, neuropsychiatric systemic lupus erythematosus, tau
Introduction
Central nervous system (CNS) involvement has been reported to occur in 14–75% of all systemic lupus erythematosus (SLE) patients [1-3]. The large differences regarding the frequency depend on the diagnostic criteria applied. CNS lupus can occur at any time during the course of SLE and its symptoms are extremely diverse. The features of this condition may include seizures, stroke, depression, psychoses and disordered mentation. Dementia, a common state in the population in general, is occasionally also reported in SLE patients [4]. Neuropsychiatric involvement in SLE (NPSLE) has been shown to predict a high frequency of flares and is considered a major cause of longstanding functional impairment as well as a cause of mortality [5]. CNS lupus in recent decades has been treated with cytotoxic drugs that improve the disease outcome [6,7]. However, acquisition of valuable treatment remedies increases the need for early recognition of CNS manifestations in lupus and continuing evaluation of local (i.e. intrathecal) responses to the medication.
Due to the multiple pathogenic mechanisms causing manifestations of CNS lupus, there is no single confirmatory diagnostic test available. Several clinical, laboratory, and radiographic test findings are reported to be abnormal in some but not all patients with CNS lupus. Magnetic resonance imaging (MRI) of the brain has been shown valuable in detecting even minor lesions caused by CNS lupus and correlated to CNS manifestations in SLE [8]. Pleocytosis and elevated protein levels are found in some but not all patients with CNS lupus [9,10]. Elevated concentrations of IgG in the cerebrospinal fluid (CSF), of the IgG–albumin ratio and of the IgG index, and the presence of oligoclonal bands have all been described with varying frequencies in patients with NPSLE [11-13]. A few studies have demonstrated elevated IL-6 levels in the CSF from patients with CNS lupus [14-18]. Some other reports have described increased levels of IL-1 [14], of IL-8 [16] and of interferon gamma [19] in CSF from patients with CNS lupus. All these biochemical indices are indirect measures of brain inflammation. In contrast to a multitude of studies analyzing local inflammatory response in NPSLE, the evaluation of neuronal damage and the formation of toxic metabolic products such as β-amyloid protein (Aβ42) have not been assessed at all in this condition.
Protein tau is a microtubuli-associated protein that promotes assembly and stability of microtubuli [20]. Elevated CSF-tau levels are found in neurodegenerative disorders such as Alzheimer disease (AD) and in acute CNS disorders such as stroke [21], and they reflect neuronal and axonal degeneration and damage. In contrast, decreased CSF-Aβ42 levels reflect either increased deposition of this molecule in senile plaques and cerebral blood vessels (e.g. commonly occurring in AD) and/or decreased synthesis of β-amyloid precursor protein (APP) [22], a large transmembrane protein. Numerous studies have found increased CSF-tau levels, decreased CSF-Aβ42 levels but variable levels of CSF-APP in AD [22-25].
Transforming growth factor beta (TGF-β), produced by both glial cells and neuronal cells within the brain [26], is a multifunctional cytokine of importance in many physiological processes, including vascular development, immune responses and fibrosis [27-29]. TGF-β in AD is present at increased levels in the CSF [30], and has also been detected in senile plaques.
The aim of the present study was to measure the levels of brain-specific proteins directly or indirectly related to dementia disorders (tau, Aβ42, APP and TGF-β) in the CSF of SLE patients with and without CNS involvement, and to assess these data prospectively with respect to immunosuppressive treatment. Our results indicate that the SLE patients studied display significantly decreased levels of Aβ42 and APP, irrespective of their CNS engagement. Prospective evaluation of a subgroup of these patients indicates that combined anti-inflammatory/immunosuppressive treatment further downregulated expression of these molecules. Despite this clear down-regulation of Aβ42, none of the SLE patients included in the study fulfilled the criteria of dementia of any type.
Materials and methods
Participants
One hundred and fourteen patients fulfilled four or more of the American Rheumatism Association 1987 updated revised criteria for the classification of SLE [31]. The 96 females and 18 males, 17–75 years old (mean age ± standard deviation, 40 ± 13 years), were all patients at the Departments of Rheumatology at Sahlgrenska University Hospital or at Karolinska University Hospital. The patients were consecutively incorporated into the study. The patients underwent a thorough clinical examination by an experienced staff rheumatologist, neurologist and neuropsychologist. Examination of the CNS signs and symptoms included lumbar puncture, neuropsychological tests and MRI of the brain. Nine of the patients underwent lumbar puncture twice, four patients on three occasions and one patient on four occasions.
The proposed definition for CNS lupus in the American Rheumatism Association's criteria for SLE [31] appears inadequate, given that only two elements (psychosis and seizures) are included. As previously described [32], we defined CNS lupus as the presence of at least two of the following seven items occurring in association with clinical evidence of disease progression: recent onset psychosis, transverse myelitis, aseptic meningitis, seizures, pathological brain MRI, severely abnormal neuropsychiatric test [33] or oligoclonal IgG bands in the CSF. The pathogenesis of antiphospholipid antibody-mediated brain damage is a thrombotic complication rather than an inflammatory complication of SLE, so we decided to exclude this condition from the definition of CNS lupus. Non-SLE causes of neurological events (e.g. cerebral infections) were also ruled out. Based on the earlier criteria, the patients were divided into three distinct groups: group I, patients with CNS lupus (n = 36); group II, patients with SLE but without any signs of CNS engagement (n = 71); and group III, patients with SLE complicated by antiphospholipid syndrome (n = 7). The exact frequency of various CNS manifestations and the patient treatment found in our population of SLE patients are presented in Tables 1 and 2. The study was approved by the ethical committee of the University of Göteborg.
Table 1.
Pharmacological treatment of patients with systemic lupus erythematosus included in the study, at the time the lumbar puncture was performed
| Treatment | NPSLE (n = 36) | No NPSLE (n = 71) | Antiphospholipid antibody syndrome (n = 7) |
| Prednisolone (≤ 10 mg) | 15 (42%) | 39 (55%) | 1 (14%) |
| Prednisolone (> 10 mg) | 3 (8%) | 13 (18%) | 0 (0%) |
| No prednisolone | 16 (58%) | 21 (30%) | 5 (29%) |
| Antimalarials | 3 (8%) | 7 (10%) | 0 (0%) |
| Azathioprine | 2 (6%) | 8 (11%) | 2 (29%) |
| Azathioprine+ cyclosporin A | 2 (6%) | 2 (3%) | 0 (0%) |
| Methotrexate | 2 (6%) | 1 (1.5%) | 0 (0%) |
| Cyclosporin A | 1 (3%) | 6 (8%) | 1 (14%) |
| Cyclophosphamide | 11 (31%) | 11 (15%) | 0 (0%) |
| Cyclophosphamide+ cyclosporin A | 3 (8%) | 1 (1.5%) | 0 (0%) |
| Cyclophosphamide+antimalarials | 1 (3%) | 1 (1.5%) | 0 (0%) |
| No cytotoxic drug | 11 (31%) | 34 (48%) | 4 (57%) |
| Antihypertensive treatment | 12 (33%) | 18 (25%) | 1 (14%) |
| Low-dose aspirin | 7 (19%) | 17 (24%) | 3 (43%) |
| Warfarin | 3 (8%) | 0 (0%) | 2 (29%) |
NPSLE, neuropsychiatric systemic lupus erythematosus.
Table 2.
Clinical central nervous system (CNS) manifestations in systemic lupus erythematosus patients included in the study
| CNS manifestations | NPSLE (n = 36) | No NPSLE (n = 71) | Antiphospholipid antibody syndrome (n = 7) |
| Acute confusional state | 2 (6%) | 1 (1.5%) | 0 (0%) |
| Anxiety disorder | 1 (3%) | 5 (7%) | 0 (0%) |
| Aseptic meningitis | 1 (3%) | 0 (0%) | 0 (0%) |
| Cerebrovascular disease | 5 (14%) | 7 (10%) | 4 (57%) |
| Cognitive dysfunction | 7 (19%) | 19 (27%) | 0 (0%) |
| Demyelinating syndrome | 3 (8%) | 1 (1.5%) | 0 (0%) |
| Headache | 8 (22%) | 28 (39%) | 3 (43%) |
| Mood disorders | 7 (19%) | 10 (14%) | 1 (14%) |
| Movement disorder | 2 (6%) | 0 (0%) | 0 (0%) |
| Myastenia gravis | 0 (0%) | 2 (3%) | 0 (0%) |
| Myelopathy | 2 (6%) | 0 (0%) | 0 (0%) |
| Polyneuropathy | 0 (%) | 2 (3%) | 0 (0%) |
| Mononeuropathy | 0 (0%) | 1 (1.5%) | 0 (0%) |
| Neuropathy | 0 (0%) | 2 (3%) | 1 (14%) |
| Psychosis | 2 (6%) | 1 (1.5%) | 0 (0%) |
| Seizure disorders | 6 (17%) | 0 (0%) | 0 (0%) |
Each patient may have had multiple clinical manifestations of the CNS involvement. NPSLE, neuropsychiatric systemic lupus erythematosus.
Control subjects
Forty-one females and 12 males, without a previous history of neurological disorder and with a normal neurological status, served as controls in the present study. CSF-tau levels and CSF-Aβ42 levels from the 53 patients, and CSF-APP levels from 35 of these 53 healthy subjects (mean age ± standard deviation, 42 ± 17 years), were measured. There were no significant differences between males and females with respect to intrathecal levels of either tau (179 ± 121 pg/ml versus 151 ± 98 pg/ml, not significant), Aβ42 (828 ± 190 pg/ml versus 842 ± 173 pg/ml, P = 0.8), or APP (7178 ± 866 pg/ml versus 6644 ± 1318 pg/ml, not significant).
CSF analyses
Levels of tau were determined using a sandwich ELISA (Innotest hTAU-Ag; Innogenetics, Gent, Belgium), constructed to measure both phosphorylated tau and non-phosphorylated tau [34,35]. The level of Aβ42 was determined using an ELISA (Innotest β-amyloid(1–42); Innogenetics) specific for Aβ42 [36,37].
The levels of total APP were determined using a novel sandwich ELISA (unpublished data) based on the monoclonal antibody 22C11 (Chemicon, Temecula, CA, USA) and the biotinylated monoclonal LN27 (Zymed, San Francisco, CA, USA). Capturing antibody 22C11, recognizing an epitope within amino acids 66–81 of the N-terminus of APP, was used to coat plastic dishes, whereas LN27, which is reactive to an epitope within the first 200 amino acids of APP, was used as a detection antibody. The concentration of APP in samples of CSF was calculated from the linear part of a standard curve. In contrast to other analyses, only 86 SLE patients were analyzed regarding APP.
A sandwich ELISA (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK) was used to measure TGF-β in the CSF of 104 SLE patients. The detection level was 4 pg/ml. All values below the detection levels were considered negative. Paired serum and CSF samples were analyzed for albumin and IgG levels using nephelometry. As an indicator of blood–brain barrier function, the quotient of CSF albumin × 103/serum albumin was analyzed (normal values: < 6.8, younger than 45 years of age; < 10.2, older than 45 years of age) [38]. The CSF/serum IgG index was used as a measure of intrathecal IgG production and calculated using the formula:
![]()
(normal value < 0.7). All CSF samples were also analyzed by isoelectric focusing to permit detection of oligoclonal IgG bands. These bands represent the brain-infiltrating oligoclonal B-cell population.
MRI analyses
Neuroimaging was performed to evaluate the extent and localization of brain lesions. The neuroimaging technique used was multiplanar MRI. The MRI examinations (Philips Gyroscan NT5, Einhoven, The Netherlands) were performed with axial proton density and T2-weighted images of the brain. The following findings were considered to be pathological: myelitis, brain atrophy leading to expansion of the ventricles, multiple high signal changes in the white matter, multiple sclerosis-like changes and infarctions. MRI abnormalities were seen in 67% of cases with CNS lupus and in 30% of SLE cases classified as cerebrally healthy.
Neuropsychological assessment
The neuropsychiatric tests were carried out by a professional neuropsychologist and judged as pathological or not. The test battery included neuropsychological assessment of the following categories: memory and learning, attention, psychomotor speed, and visiospatial ability. If at least one of the categories was pathological, the patient tested was found to have an abnormal neuropsychiatric test. Results regarding MRI and neuropsychological assessment were not accessible for the respective investigators.
Statistical analysis
Statistical comparisons were made using the nonparametric Mann–Whitney U test, or the Wilcoxon's test for paired data in the case of follow-up data. Results are presented as means ± standard error of the mean. P ≤ 0.05 was considered statistically significant. Spearman rank correlation was used for calculation of correlations. The statistical analyses were carried out using the Statview® program.
Results
One hundred and fourteen patients met the inclusion criteria for SLE diagnosis. Thirty-six patients were found to have NPSLE in accordance with the criteria presented in Materials and methods, seven patients were found to have phospholipid antibody syndrome and the remaining 71 SLE patients were considered cerebrally healthy.
Mild pleocytosis was seen in patients with CNS lupus (10 × 106 ± 7 × 106 cells/l) compared with cerebrally healthy SLE patients (2 × 106 ± 0.5 × 106 cells/l) (not significant). As previously validated [39], we found an increased number of oligoclonal bands in the CSF of the CNS lupus group (2.2 ± 0.4) as compared with SLE patients without CNS involvement (0.4 ± 0.2) (P ≤ 0.0005). The mean level of the CSF/serum albumin ratio was not increased in patients with NPSLE (5.5 ± 0.4 mg/dl) as compared with cerebrally healthy SLE subjects (5.2 ± 0.4 mg/dl) (not significant). There were no significant differences regarding levels of serum antibodies specific for native DNA, or regarding complement levels (C3 and C4) between SLE patients with or without CNS involvement.
Intrathecal Aβ42 levels were decreased in all the SLE patients compared with healthy controls (mean ± standard error of the mean, 574 ± 17 pg/ml versus 831 ± 25 pg/ml; P < 0.0001) (Fig. 1a). Patients with NPSLE had slightly but not significantly decreased Aβ42 levels as compared with patients without CNS lupus (541 ± 34 pg/ml versus 583 ± 18 pg/ml, not significant).
Figure 1.
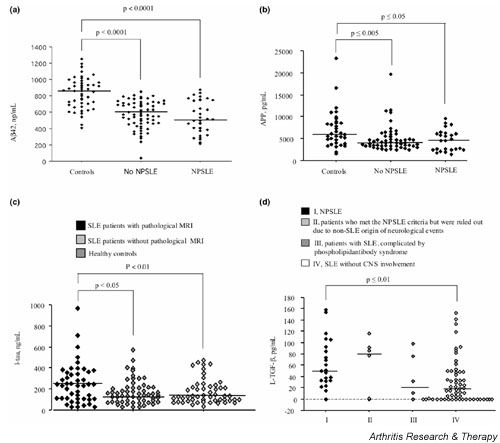
Decreased levels of soluble amyloid β-protein precursor and β-amyloid protein but increases of intrathecal axonal degradation products and TGF-β in patients with cerebral lupus. (a) Cerebrospinal fluid (CSF) content of soluble amyloid β-protein (Aβ42) in patients with systemic lupus erythematosus (SLE) with and without central nervous system (CNS) engagement, as well as in cerebrally healthy control subjects. (b) CSF content of amyloid precursor protein (APP) in patients with SLE with and without CNS engagement, as well as in cerebrally healthy control subjects. (c) CSF content of tau in patients with SLE stratified with respect to brain magnetic resonance imaging (MRI)-verifiable changes, and in cerebrally healthy control subjects. (d) CSF content of transforming growth factor beta (TGF-β) in SLE patients stratified with respect to the presence/absence and type of CNS engagement. NPSLE, neuropsychiatric systemic lupus erythematosus.
A second lumbar puncture was performed in nine patients who met the criteria of NPSLE and were successfully treated with cyclophosphamide. The CSF levels of Aβ42 were further decreased on the second occasion as compared with the first (702 ± 45 pg/ml versus 621 ± 45 pg/ml, P ≤ 0.05). In contrast, CSF levels of tau were not affected by treatment (275 ± 77 pg/ml versus 273 ± 61 pg/ml, not significant). To investigate whether the decreased Aβ42 levels in SLE patients are due to a diminished production or to its local deposition in the brain, we analyzed the intrathecal levels of APP. We found decreased CSF-APP levels (Fig. 1b) in SLE patients compared with healthy controls (4834 ± 314 pg/nl versus 7010 ± 714 pg/nl, P < 0.001), clearly indicating decreased production of this precursor molecule.
There were no statistically significant differences regarding CSF-tau levels between healthy controls and patients with NPSLE (173 ± 16 pg/ml versus 237 ± 35 pg/ml). However, upon stratification of all the SLE patients into two groups, according to the presence or absence of MRI findings, we found a statistical difference regarding CSF-tau levels in SLE patients with MRI pathology compared with those without (261 ± 26 pg/ml versus 164 ± 15 pg/ml, P < 0.01) (Fig. 1c). Next we assessed the possible relationship between decreased Aβ42 and increased tau levels in the whole patient population. Our results show clearly that these two molecules, related to neuronal toxicity and damage, display a significant relationship (r = 0.48, P < 0.0001).
We finally decided to assess CSF levels of TGF-β, another amyloidogenic protein with anti-inflammatory properties. In the CNS lupus group, intrathecal levels of TGF-β were significantly increased compared with those of SLE patients without overt CNS disease (mean ± standard error of the mean, 54.3 ± 8.4 pg/ml versus 31.6 ± 4.8 pg/ml; P ≤ 0.01), as seen in Fig. 1d.
Discussion
Patients with SLE present with a wide array of neuropsychiatric features, although evidence for dementia in these patients is scarce.
The precise pathogenic mechanisms of neuropsychiatric manifestations are still the subject of intense investigations, but autoantibody and cytokine-mediated neural dysfunction, intracranial angiopathy and coagulopathy have all been implicated. Although there is no diagnostic golden standard for CNS lupus, there is a wide selection of non-invasive tests that are of value in the assessment and monitoring of the individual patients. Autopsy studies of brains from SLE patients have revealed vasculopathy [40] as a pathogenic event either directly responsible for clinical neuropsychiatric events or, alternatively, by altering blood–brain barrier permeability and facilitating the access of pathogenic antibodies from the circulation into an immunologically privileged site that is normally protected from aberrant immune responses. The extracellular fluid of the brain is in direct contact with the CSF, and biochemical processes in the brain can be reflected therein. Analysis of degradation products from neuronal cells found in CSF would be of value to improve the clinical evaluation of NPSLE and to study the ongoing brain parenchyme status in living subjects with NPSLE.
In the current study, we found significantly decreased levels of Aβ42 in the CSF of SLE patients compared with healthy control subjects. Low levels of CSF-Aβ42 may be either due to their accumulation and deposition (e.g. as diffuse plaques, as in the case of AD) and/or to a decreased production of APP by neuronal cells. Our results showed significantly decreased levels of APP in SLE patients included in the study compared with neurologically healthy subjects, indicating decreased production rather than increased tissue deposition. What would be the cause of the decreased APP production in SLE patients? At first glance, there were no differences in the CSF-APP levels with respect to the medication (corticosteroids, immunosuppressive drugs). However, the variability of the patient material as well as differences in duration and intensity of the treatment throw doubt on this conclusion. Interestingly, a subpopulation of nine patients was followed prospectively after immunosuppressive treatment. In this group such a treatment resulted in significantly decreased Aβ42 levels. This finding indicates that treatment with alkylating agents such as cyclophosphamide might affect either the production of APP or the activity of secretases. Indeed, it has been recently demonstrated that anti-inflammatory drugs may directly modulate γ-secretase activity and thereby decrease Aβ42 production [41].
We showed that SLE patients with MRI findings compared with those without MRI findings had significantly higher CSF-tau levels. These increases reflect the neuronal damage of the brain in SLE patients. Indeed, our recent study [32] supports this conclusion since another neuronal protein, neurofilament protein, was also increased.
Importantly, a significant relationship was noted between the occurrence of tau and of Aβ42. This finding, together with the correlation between tau and MRI-verified brain damage, indicates that Aβ42 might have exerted its toxicity locally, leading to sequels. Finally, as previously found in AD [30] and in low-pressure hydrocephalus-induced dementia (unpublished observation), levels of TGF-β in the CSF of NPSLE patients were clearly increased. Such an increase may be very well related to the disease process itself since, at least in the case of patients with low-pressure hydrocephalus, a shunt operation will simultaneously lead to decreased levels of TGF-β and increased mentation (unpublished observation).
Altogether, our results show that there is increased production of TGF-β and an ongoing destruction of brain parenchyma (manifested as increased tau levels) in the brains of SLE patients. We suggest that levels of toxic Aβ42 and its precursor APP reflect efficient cytotoxic therapy, as shown in a limited subgroup analysis. Notably, while our findings shed some light on the pathogenesis of cerebral lupus, none of the findings reported in the present study may be used as a diagnostic parameter, since many other brain diseases show similar patterns.
Conclusion
Our findings indicate that intrathecal levels of tau, a marker of neuronal degeneration, are clearly increased in SLE patients with MRI-verifiable brain lesions, both as compared with CSF findings of healthy controls as well as of lupus patients without MRI-verified brain pathology. This finding, together with increased intrathecal levels of TGF-β, indicates an ongoing destructive parenchymatous process in the SLE brain.
In addition, the present study provides evidence of decreased intrathecal levels of Aβ42 in SLE patients irrespective of CNS engagement. We interpret this finding not as a sign of early dementia, but rather as a sign of decreased APP synthesis, a precursor of Aβ42, as verified in the present study. We suggest that this decrease may be an outcome of an intense immunosuppressive treatment to which the majority of SLE patients included in the study are being subjected.
Competing interests
None declared.
Abbreviations
AD = Alzheimer disease; Aβ42 = β-amyloid protein; APP = amyloid precursor protein; CNS = central nervous system; CSF = cerebrospinal fluid; ELISA = enzyme-linked immunosorbent assay; IL = interleukin; MRI = magnetic resonance imaging; NPSLE = neuropsychiatric systemic lupus erythematosus; SLE = systemic lupus erythematosus; TGF-β = transforming growth factor beta.
Acknowledgments
Acknowledgements
The work was supported by the Göteborg Medical Society, the Swedish Association against Rheumatism, the King Gustaf V Foundation, the Swedish Medical Research Council, the Nanna Svartz Foundation, Börje Dahlin's Foundation, the Swedish National Inflammation Network, the Swedish National Infection and Vaccination Network, the AME Wolff Foundation, and the University of Göteborg.
References
- McCune WJ, Golbus J. Neuropsychiatric lupus. Rheum Dis Clin North Am. 1988;14:149–167. [PubMed] [Google Scholar]
- Hanly JG, Liang MH. Cognitive disorders in systemic lupus erythematosus. Epidemiologic and clinical issues. Ann NY Acad Sci. 1997;823:60–68. doi: 10.1111/j.1749-6632.1997.tb48379.x. [DOI] [PubMed] [Google Scholar]
- Feinglass EJ, Arnett FC, Dorsch CA, Zizic TM, Stevens MB. Neuropsychiatric manifestations of systemic lupus erythematosus: diagnosis, clinical spectrum, and relationship to other features of the disease. Medicine (Baltimore) 1976;55:323–339. doi: 10.1097/00005792-197607000-00004. [DOI] [PubMed] [Google Scholar]
- Croake JW, Pursley M, Hardin JG, Michalski JP. Systemic lupus erythematosus and dementia. Psychol Rep. 1998;83:1034. doi: 10.2466/pr0.1998.83.3.1034. [DOI] [PubMed] [Google Scholar]
- Jonsson H, Nived O, Sturfelt G. Outcome in systemic lupus erythematosus: a prospective study of patients from a defined population. Medicine (Baltimore) 1989;68:141–150. [PubMed] [Google Scholar]
- McCune WJ, Golbus J, Zeldes W, Bohlke P, Dunne R, Fox DA. Clinical and immunologic effects of monthly administration of intravenous cyclophosphamide in severe systemic lupus erythematosus. N Engl J Med. 1988;318:1423–1431. doi: 10.1056/NEJM198806023182203. [DOI] [PubMed] [Google Scholar]
- Boumpas DT, Yamada H, Patronas NJ, Scott D, Klippel JH, Balow JE. Pulse cyclophosphamide for severe neuropsychiatric lupus. Q J Med. 1991;81:975–984. doi: 10.1093/qjmed/81.3.975. [DOI] [PubMed] [Google Scholar]
- Oku K, Atsumi T, Furukawa S, Horita T, Sakai Y, Jodo S, Amasaki Y, Ichikawa K, Amengual O, Koike T. Cerebral imaging by magnetic resonance imaging and single photon emission computed tomography in systemic lupus erythematosus with central nervous system involvement. Rheumatology (Oxford) 2003;42:773–777. doi: 10.1093/rheumatology/keg216. [DOI] [PubMed] [Google Scholar]
- Sergent JS, Lockshin MD. Editorial: treatment of central nervous system lupus erythematosus. Ann Intern Med. 1974;80:413–414. doi: 10.7326/0003-4819-80-3-413_2. [DOI] [PubMed] [Google Scholar]
- Abel T, Gladman DD, Urowitz MB. Neuropsychiatric lupus. J Rheumatol. 1980;7:325–333. [PubMed] [Google Scholar]
- Ernerudh J, Olsson T, Lindstrom F, Skogh T. Cerebrospinal fluid immunoglobulin abnormalities in systemic lupus erythematosus. J Neurol Neurosurg Psychiatry. 1985;48:807–813. doi: 10.1136/jnnp.48.8.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HAd, Koffler D. Intrathecal IgG synthesis and blood–brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983;74:837–844. doi: 10.1016/0002-9343(83)91075-6. [DOI] [PubMed] [Google Scholar]
- Hirohata S, Hirose S, Miyamoto T. Cerebrospinal fluid IgM, IgA, and IgG indexes in systemic lupus erythematosus. Their use as estimates of central nervous system disease activity. Arch Intern Med. 1985;145:1843–1846. doi: 10.1001/archinte.145.10.1843. [DOI] [PubMed] [Google Scholar]
- Alcocer-Varela J, Aleman-Hoey D, Alarcon-Segovia D. Interleukin-1 and interleukin-6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T-cell activation markers. Lupus. 1992;1:111–117. doi: 10.1177/096120339200100209. [DOI] [PubMed] [Google Scholar]
- Hirohata S, Tanimoto K, Ito K. Elevation of cerebrospinal fluid interleukin-6 activity in patients with vasculitides and central nervous system involvement. Clin Immunol Immunopathol. 1993;66:225–229. doi: 10.1006/clin.1993.1029. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9:498–503. doi: 10.1177/096120330000900704. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Tsai ST, Chen KH, Thajeb P, Lin WM, Yu HS, Yu CL. Cerebrospinal fluid interleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand J Rheumatol. 1994;23:57–63. doi: 10.3109/03009749409103028. [DOI] [PubMed] [Google Scholar]
- Yeh TS, Wang CR, Jeng GW, Lee GL, Chen MY, Wang GR, Lin KT, Chuang CY, Chen CY. The study of anticardiolipin antibodies and interleukin-6 in cerebrospinal fluid and blood of Chinese patients with systemic lupus erythematosus and central nervous system involvement. Autoimmunity. 1994;18:169–175. doi: 10.3109/08916939409007993. [DOI] [PubMed] [Google Scholar]
- Svenungsson E, Andersson M, Brundin L, van Vollenhoven R, Khademi M, Tarkowski A, Greitz D, Dahlstrom M, Lundberg I, Klareskog L, Olsson T. Increased levels of proinflammatory cytokines and nitric oxide metabolites in neuropsychiatric lupus erythematosus. Ann Rheum Dis. 2001;60:372–379. doi: 10.1136/ard.60.4.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75:835–864. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- Hesse C, Rosengren L, Vanmechelen E, Vanderstichele H, Jensen C, Davidsson P, Blennow K. Cerebrospinal fluid markers for Alzheimer's disease evaluated after acute ischemic stroke. J Alzheimers Dis. 2000;2:199–206. doi: 10.3233/jad-2000-23-402. [DOI] [PubMed] [Google Scholar]
- Galasko D, Chang L, Motter R, Clark CM, Kaye J, Knopman D, Thomas R, Kholodenko D, Schenk D, Lieberburg I, Miller B, Green R, Basherad R, Kertiles L, Boss MA, Seubert P. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol. 1998;55:937–945. doi: 10.1001/archneur.55.7.937. [DOI] [PubMed] [Google Scholar]
- Sjogren M, Davidsson P, Gottfries J, Vanderstichele H, Edman A, Vanmechelen E, Wallin A, Blennow K. The cerebrospinal fluid levels of tau, growth-associated protein-43 and soluble amyloid precursor protein correlate in Alzheimer's disease, reflecting a common pathophysiological process. Dement Geriatr Cogn Disord. 2001;12:257–264. doi: 10.1159/000051268. [DOI] [PubMed] [Google Scholar]
- Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26:231–245. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Vanmechelen E, Van de Voorde A, Davidsson P, Hesse C, Tarvonen S, Raiha I, Sourander L, Winblad B, Blennow K. Cerebrospinal fluid tau protein as a biochemical marker for Alzheimer's disease: a community based follow up study. J Neurol Neurosurg Psychiatry. 1998;64:298–305. doi: 10.1136/jnnp.64.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt BM, McPherson JM. TGF-beta in the central nervous system: potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor Rev. 1997;8:267–292. doi: 10.1016/S1359-6101(97)00018-X. [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA. TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int. 1997;51:1388–1396. doi: 10.1038/ki.1997.190. [DOI] [PubMed] [Google Scholar]
- Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/S1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Perrella MA, Jain MK, Lee ME. Role of TGF-beta in vascular development and vascular reactivity. Miner Electrolyte Metab. 1998;24:136–143. doi: 10.1159/000057361. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002;23:237–243. doi: 10.1016/S0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter] [see comments] Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Nylén K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48:2881–2887. doi: 10.1002/art.11279. [DOI] [PubMed] [Google Scholar]
- Breitbach SA, Alexander RW, Daltroy LH, Liang MH, Boll TJ, Karlson EW, Partiridge AJ, Roberts WN, Stern SH, Wacholtz MC, Straaton KV. Determinants of cognitive performance in systemic lupus erythematosus. J Clin Exp Neuropsychol. 1998;20:157–166. doi: 10.1076/jcen.20.2.157.1166. [DOI] [PubMed] [Google Scholar]
- Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26:231–245. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- Vandermeeren M, Mercken M, Vanmechelen E, Six J, van de Voorde A, Martin JJ, Cras P. Detection of tau proteins in normal and Alzheimer's disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. 1993;61:1828–1834. doi: 10.1111/j.1471-4159.1993.tb09823.x. [DOI] [PubMed] [Google Scholar]
- Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- Vanderstichele H, Van Kerschaver E, Hesse C, Davidsson P, Buyse MA, Andreasen N, Minthon L, Wallin A, Blennow K, Vanmechelen E. Standardization of measurement of beta-amyloid(1–42) in cerebrospinal fluid and plasma. Amyloid. 2000;7:245–258. doi: 10.3109/13506120009146438. [DOI] [PubMed] [Google Scholar]
- Blennow K, Fredman P, Wallin A, Gottfries CG, Karlsson I, Langstrom G, Skoog I, Svennerholm L, Wikkelso C. Protein analysis in cerebrospinal fluid. II. Reference values derived from healthy individuals 18–88 years of age. Eur Neurol. 1993;33:129–133. doi: 10.1159/000116919. [DOI] [PubMed] [Google Scholar]
- Moore BW. A soluble protein characteristic of the nervous system. Biochem Biophys Res Commun. 1965;19:739–744. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Walsh NM, Sangalang V. Brain pathology in systemic lupus erythematosus. J Rheumatol. 1992;19:732–741. [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003;278:31831–31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]