

Nitric oxide (NO) was discovered about three decades ago as an important factor in vasodilation 1, 2. Since then, NO has emerged as an important factor in signal transduction pathways that impact cell and organ functions such as survival, apoptosis, angiogenesis and endothelial permeability. In this review we will focus on issues and knowledge concerning endothelial/microvascular permeability and the emerging role of S-nitrosation (also referred to as S-nitrosylation) in the onset of the hyperpermeability response to pro-inflammatory agents.
Endothelial and Microvascular Permeability
The main function of the circulatory system is the exchange of substances between blood and tissues to support cell life. For this reason, baseline microvascular permeability is regulated by a constellation of factors. A reduction in baseline microvascular permeability, which has been demonstrated in isolated venules3 and in cultured endothelial cells4 is rarely observed in vivo5. Consequently, most of our knowledge of the regulation of microvascular permeability is derived from perturbations that increase permeability (hyperpermeability), particularly in the context of inflammation.
Microvascular permeability is regulated mainly by two major processes: a) contraction and b) modification of adherens junction (AJ) proteins. Electron microscopy observations of deformed endothelial cell (EC) nuclei 6 served as the basis for the concept that cellular contraction causes an increase in permeability, and led over the years to the discovery of the role of Rho, RhoA and associated Rho kinases in hyperpermeability mediated by contractile events7, 8. Earlier light microscopy observations provided support to the concept of the existence of a “cement substance” between adjacent endothelial cells9. The “cement substance” hypothesis was supported by electron microscopy studies of transport of macromolecules in which there were no perceptible changes in nuclear shape10, and led to the development of the concept that the major mechanism to enhance permeability is modification of the proteins that make up the endothelial cell junctions (i.e., AJ proteins). VE-cadherin, beta-catenin (2 -catenin) and p120-catenin are currently recognized as the main AJ proteins involved in the development of hyperpermeability to macromolecules11-13 (Figure 1). While S-nitrosation of contractile proteins is potentially a significant regulatory event, we will concentrate this review on NO-driven mechanisms leading to modifications of AJ proteins.
Figure 1. Illustration of Endothelial Intercellular Junctions.
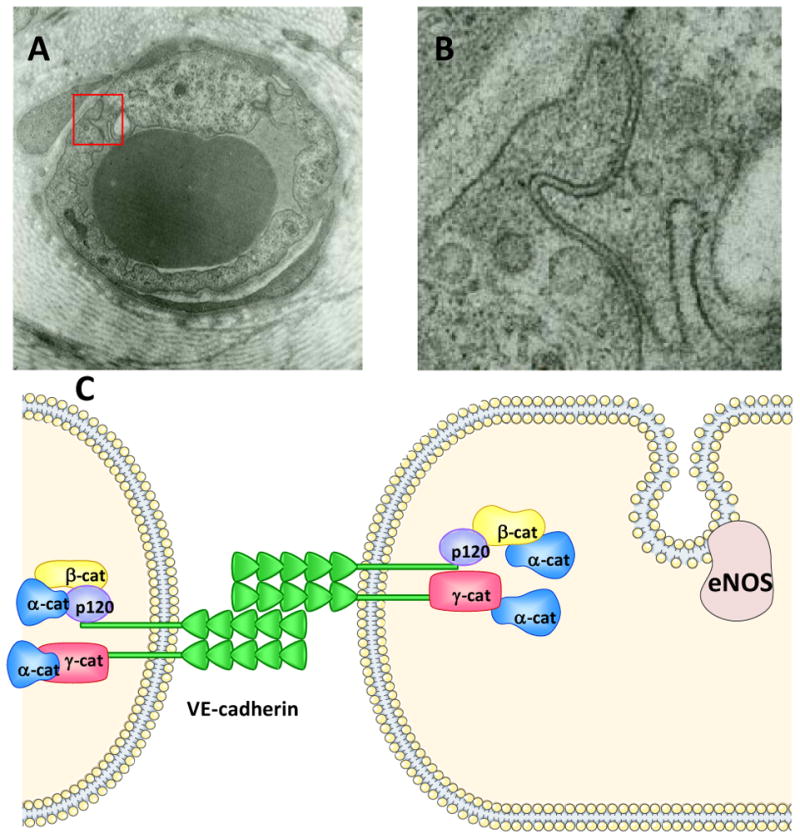
PANEL A: Electron microscopy image of postcapillary venule of the hamster cheek pouch. The image was obtained after the vasculature was stimulated topically with 10-7 M PAF. The red square identifies the insert magnified in Panel B (Durán, unpublished image). PANEL B: Insert from Panel A: Intercellular junction. A nearly constant gap separates the adjacent endothelial cells, reflecting PAF-induced hyperpermeability. PANEL C: Cartoon representing some of the known endothelial intercellular adherens junction proteins. The juxtaposition of the extracellular domains of VE-cadherin represents baseline or unstimulated state. eNOS is shown at the caveolae. Other junctional proteins are omitted due to the focus of this review.
Nitric oxide as a determinant factor in microvascular permeability
Inflammatory processes are characterized by an increase in microvascular permeability (hyperpermeability) to macromolecules. Under inflammatory conditions, microvascular permeability to macromolecules is controlled mainly at the postcapillary venules. Endothelial nitric oxide synthase (eNOS), which was recognized originally as an important regulator of vascular tone1, 2, emerged much later as a key regulator of microvascular hyperpermeability14-19. Early reports classified the main function of eNOS as an anti-inflammatory enzyme based on experiments utilizing pharmacologic agents and on the observations that NO served to decrease leukocyte adhesion to endothelium. After a period of controversial reports, evidence based on eNOS knockout mice and on eNOS-depleted EC established that microvascular hyperpermeability in response to an inflammatory challenge is regulated mainly by endothelial cells through eNOS-derived NO20-22. Our work indicates that, while eNOS is non-essential for the control of baseline permeability, eNOS-derived NO is fundamental for mounting a solid hyperpermeability response under inflammatory conditions, to the extent that NO derived from other NOS isoforms is unable to generate changes in microvascular permeability21. This pro-hyperpermeability regulation is carried out in an orchestrated synchrony with cell contraction and junctional re-organization induced by polymorphonuclear leukocytes7, 8.
Endothelial Cell Regulatory Mechanisms
The activation state of eNOS and its subcellular localization are regulated through several mechanisms. eNOS undergoes complex posttranslational modifications, including protein-protein interactions and phosphorylation or de-phosphorylation on at least five residues: serines (ser) 116, 617, 635 and 1179 (1177 in humans) and threonine (thr) 497 (495 in humans). eNOS also undergoes phosphorylation of tyrosine residues23, 24. The pathways resulting in eNOS phosphorylation have been studied in detail, and it is recognized that differential orchestrated phosphorylation/de-phosphorylation of eNOS plays an important role in the regulation of its enzymatic activity.
How the complex regulatory interactions that control the cellular functions of eNOS relate to the function of eNOS-derived NO at the tissue or organ levels is incompletely understood. Several agents (acetylcholine [ACh]; shear stress, estrogen, sphingosine1-phosphate, platelet-activating factor [PAF], vascular endothelial growth factor [VEGF], etc.) that cause different effects at the tissue/organ level phosphorylate eNOS at the same consensus sites. Because the phosphorylation of eNOS at the same site (ser1177, for instance) serves to signal for different tissue/organ functions, it appears that phosphorylation alone is not sufficient to determine the specificity of the tissue/organ function derived from NO signaling. The eNOS-derived NO signaling code for tissue function remains to be deciphered. We have advanced the novel concept that eNOS translocation from membrane to preferential intracellular compartments is linked to the resulting microvascular functions of vasodilation and microvascular/endothelial permeability22, 25-27.
Functional Significance of Differential eNOS Translocation
Some agonists cause vasodilation whereas others cause an increase in permeability in the microcirculation or both in a dose-dependent manner28, 29. However, the key differential signaling mechanisms for discriminating between vasodilation and hyperpermeability have eluded a clear answer since most agonists of endothelial cells cause significant phosphorylation of eNOS and NO release - regardless of the microvascular effect. The observation that eNOS translocates from plasma membrane to cytosol as a consequence of stimulation with bradykinin30 prompted us to investigate whether or not translocation of eNOS has functional consequences in vitro and in vivo.
We applied a strategy based on the in vivo microvascular properties of ACh (vasodilation, no effect on permeability) and PAF (vasoconstriction and hyperpermeability in hamster and humans) to assess the hypothesis that differential translocation serves as a regulatory mechanism of eNOS to determine specific microvascular responses. For direct cellular experiments, we utilized a cell line identified as ECV-304, which are derived genetically from human urinary bladder carcinoma T24 cell line, but exhibit both endothelial and epithelial markers31. An advantage of ECV-304 is that these cells lack endogenous eNOS and, thus, can be transfected with exogenous labeled and targeted eNOS constructs. Experiments conducted in ECV-eNOS-GFP cells [ECV-304 cells permanently transfected with eNOS-green fluorescent protein], human umbilical vein EC (HUVEC), human dermal microvascular EC (HMVEC) and bovine coronary postcapillary venular EC demonstrated that ACh translocated eNOS preferentially to the Golgi and that PAF translocated eNOS preferentially to the cytosol (Table 1). Similar results for differential eNOS translocation were obtained in vivo using the hamster cheek pouch as a model32. Based on these in vitro and in vivo data, we proposed the concept that PAF-induced eNOS translocation preferentially to cytosol (non-Golgi cytosolic region) reflects a differential signaling mechanism related to changes leading to the onset of hyerpermeability, whereas ACh-induced eNOS translocation to the Golgi is related to vasodilation22, 25-27. In further support of the hypothesis, we demonstrated that internalization of eNOS is required to deliver NO to subcellular locations to increase endothelial monolayer permeability to macromolecules. We showed that anchoring eNOS-containing caveolae to the plasma membrane of coronary postcapillary venular endothelial cells inhibited VEGF-induced hyperpermeability to macromolecules26.
Table 1. Subcellular distribution of eNOS.
| LOCATION | CONTROL | ACh | PAF |
|---|---|---|---|
| Membrane | 0.50 | 0.35 | 0.34 |
| Golgi | 0.38 | 0.55 | 0.43 |
| Cytosol | 0.12 | 0.10 | 0.23 |
| _____ | _____ | _____ | |
| 1.00 | 1.00 | 1.00 |
Computer-assisted image analysis of eNOS subcellular location in ECV-304 cells transfected with eNOS-GFP construct. The relative abundance of eNOS-GFP at each subcellular location is expressed as a fraction of the total detected eNOS. (Adapted from reference 27).
How does NO signal the onset of hyperpermeability?
eNOS-NO-sGC-PKG, the conventional pathway
Classically, NO interacts with soluble guanylyl cyclase (sGC) - its first described signaling receptor - to induce the production of guanosine 32:52-cyclic monophosphate (cGMP), which leads to protein kinase G (PKG) activity and stimulation of MAP kinases. This classical paradigm has been applied to explain eNOS-derived NO promoted increase in microvascular permeability (Figure 2), but some steps in the sequence of events remain elusive. Experimental evidence in support of the classical sequence pathway includes increases in permeability to water and macromolecules after stimulation of venules or endothelial cells with 8Br-cGMP, an analog of cGMP4, 33 and inhibition of permeability obtained by blocking ERK-1/24, 34. Interestingly, studies of VEGF-induced hyperpermeability indicated that VEGF-stimulated phosphorylation of ERK-1/2 and VEGF-induced activation of eNOS could represent two different and separate biochemical pathways with areas of cross-talk between them34. These findings, in association with the relatively low abundance of sGC in endothelial cells and the observations that increases in permeability to macromolecules as well as to water can also occur in the presence of inhibitors of sGC35, 36, have suggested that parallel or additional pathways exist to stimulate hyperpermeability, which can work in concert to influence the cytoskeleton, focal adhesion proteins and junctional proteins.
Figure 2. eNOS-derived NO signaling pathways.
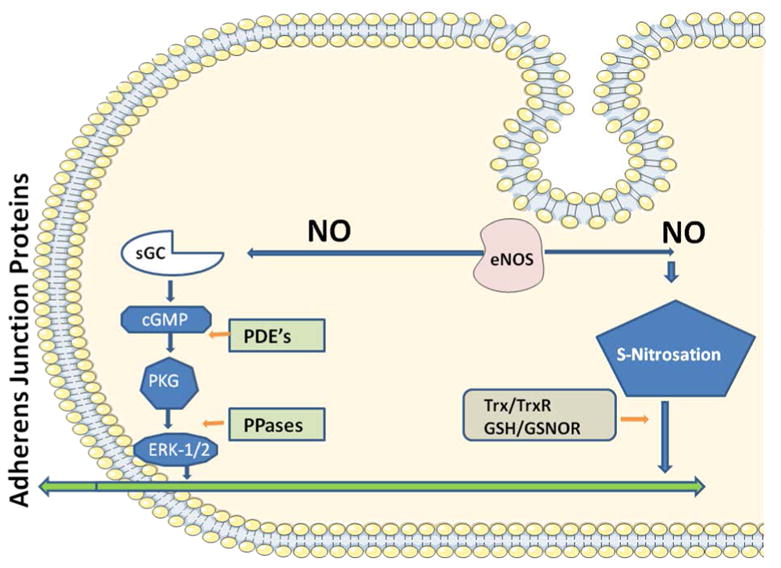
The classical cGMP-dependent pathway is illustrated on the left hand side (sGC: soluble guanylyl cyclase; cGMP: cyclic guanylyl monophosphate; PKG: protein kinase G; ERK 1/2: extracellular regulated kinase 1-2). The regulatory phosphodiesterases (PDE's) and phosphatases (PPases) are also illustrated. The cGMP-indpendent S-nitrosation pathway is shown on the right side. The denitrosating agents thioredoxin (Trx), thioredoxin reductase (TrxR); glutathione (GSH) and S-Nitrosoglutathione Reductase (GSNOR) are depicted as control for the process. Both pathways are regulated through the exquisite mechanisms that control eNOS. Both pathways appear to converge at some point(s) that modulates the adherens junctional protein complex.
S-nitrosation, the emerging alternative pathway in hyperpermeability
S-nitrosation, the covalent modification of a protein cysteine thiol by an NO group to generate an S-nitrosothiol, has emerged as the alternative, parallel or additional independent pathway for stimulating hyperpermeability to macromolecules. S-nitrosation appears to operate in a manner similar to phosphorylation in that it can activate or inactivate biological processes, reversibly, depending on the target proteins.
The sequence of biochemical events and potential cause-effect versus parallel synergism relationships between phosphorylation and S-nitrosation in the control of microvascular permeability remain important open fields for research. In regards to cell biology, the mechanisms of phosphorylation and S-nitrosation appear to be interrelated. The intricate connections have been reviewed in depth 23. While protein S-nitrosation can be demonstrated in purified enzymes and in vitro, the functional in vivo significance and consequences of S-nitrosation are still subject of investigation.
AJ proteins are primary targets for posttranslational modifications leading to endothelial hyperpermeability. In this context, special attention has been given to S-nitrosation of 2 -catenin and p120-catenin because their association with VE-cadherin is fundamental for maintenance of barrier integrity. Both AJ proteins are S-nitrosated by eNOS-derived NO in endothelial cells36, 37. VEGF causes S-nitrosation of 2 -catenin at cys619 to increase endothelial permeability. The demonstration was achieved by inhibition of S-nitrosation at this site, an intervention that led to maintenance of the association between 2 -catenin and VE-cadherin and reduction of the impact of VEGF on permeability in bovine aortic endothelial cells37. Inhibiting S-nitrosation of 2 -catenin in microvascular endothelium does not modify the phosphorylation of 2 -catenin on tyr654, a phosphorylation site required for the dissociation of 2 -catenin from VE-cadherin and promotion of hyperpermeability 37. This evidence can be interpreted as demonstration of independence of the processes, with the provocative note that S-nitrosation may play a more important role in the synergy between these two mechanisms since mutation of cys619 inhibited VEGF-induced permeability in the presence of increased phosphorylation of tyr654.
Experimental depletion of p120-catenin has emphasized the significance of this AJ protein in the maintenance of endothelial barrier integrity. Indeed, elegant experiments, measuring transendothelial electrical resistance as an index of permeability to small molecules, demonstrated that depletion of p120-catenin led to reduction in VE-cadherin expression and increased electrical conductance and that restoration of p120-catenin re-established the normal conditions, independently of VE-cadherin12. While VE-cadherin is regarded as the key AJ protein in endothelium, based on the emerging relevance of the interactions between p120-catenin and VE-cadherin through their cytoplasmic tails, we preferentially focused our attention on the S-nitrosation of p120-catenin. Using mass spectrometry and purified p120-catenin, we demonstrated that NO causes S-nitrosation of p120-catenin mainly at cys579, with partial contributions from cys429, cys450, cys618 and cys69236. Cys579 is located in the molecular stretch that interacts with E-cadherin38, 39. Based on the high homology between E-cadherin and VE-cadherin, one possible explanation for the disruption of adherens junctions is that S-nitrosation of cys579 blocks the interactions of p120 with VE-cadherin with a resulting modification of the adherens junction complex and enhanced hyperpermeability. In vitro and in vivo, PAF induces S-nitrosation of p120-catenin and 2 -catenin via activation of eNOS-derived NO36. VEGF and PAF-induced S-nitrosation of AJ proteins represent a novel mechanism to regulate hyperpermeability.
Based on our demonstration that location of eNOS in the cytosol is fundamental for the onset of agonist-induced hyperpermeability, we tested whether the subcellular location of eNOS is of significance in regards to its potential for S-nitrosation. We took advantage of the fact that ECV-304 lack endogenous eNOS and can be transfected with eNOS-targeting constructs. Using fluorescently labeled constructs (developed by Dr. David Fulton; Georgia Health Sciences University), we transfected ECV-304 with GFPeNOSG2A (ECV-GFPeNOSG2A, cells where eNOS is targeted to the cytosol) and with GFPeNOSCAAX (ECV-GFPeNOCAAX cells where eNOS is targeted to the plasma membrane). Importantly, these eNOS transfected cells are fully able to produce NO in response to challenge with inflammatory agonists25. In agreement with the requirement for eNOS translocation to the cytosol to stimulate hyperpermeability, location of eNOS in the cytosol (via GFPeNOSG2A) is fundamental to produce robust S-nitrosation of AJ proteins in response to PAF. In contrast, targeting eNOS to the plasma membrane of ECV-304 cells (via ECV-GFPeNOSCAAX) prevents PAF-induced S-nitrosation of AJ proteins. The internalization of AJ proteins into the cytosol for S-nitrosation is in agreement with the recognized concept that association of the protein with the NO source favors S-nitrosation of target proteins, the source being eNOS in this case. We used a similar reasoning of required proximity between source and target to explain the translocation of eNOS from plasma membrane (caveolae) to cytosol to implement the onset of hyperpermeability27. If so, the agonist-stimulated and signaling coordinated translocation of eNOS and internalization of AJ proteins to the cytosol would couple their S-nitrosation with the onset of hyperpermeability. These concepts are consistent with the association between S-nitrosation and regulation of microvascular/endothelial permeability.
PAF-induced S-nitrosation is associated with internalization of p120-catenin and 2 -catenin in coronary postcapillary venular endothelial cells at times compatible with early onset of hyperpermeability. In addition, PAF-induced S-nitrosation dissociates the interactions between AJ proteins as early as after 1, 3 and 5 minutes of exposure36. These results were interpreted to indicate that S-nitrosation occurred at the plasma membrane, based on the disappearance of p120-catenin and 2 -catenin from the endothelial cell plasma membrane as early as 1 minute of exposure to PAF, which coincided with the increase in S-nitrosation – measured by biotin switch (see Fig. 1 in ref. # 35). However, in the absence of continuous in vivo real-time data, these results can also be interpreted as S-nitrosation occurring in the cytosol following rapid internalization of AJ proteins. In this sequence, phosphorylation of AJ proteins would be the primary process responsible for disassembly of the junctional complex (a view that is widely accepted, but not unequivocally demonstrated) and for initiating their internalization. Quickly after internalization, AJ proteins would be S-nitrosated through the enhanced activity of eNOS, which translocates to the cytosol under agonist-stimulated conditions. In this context, S-nitrosation would serve to capture and retain AJ proteins in the cytosol, thus effectively amplifying the onset of increased permeability by preventing or delaying the reassembly of the adherens junction complexes. Should S-nitrosation of AJ proteins last longer than the period of hyperpermeability – as phosphorylation of AJ protein does, one could argue that S-nitrosated proteins are being scheduled for recycling at a later time, while restoration of the barrier integrity is implemented through traffic of newly synthesized proteins.
In this scenario, phosphorylation of AJ proteins at the plasma membrane and their S-nitrosation in the cytosol must be exquisitely coupled to determine the onset of hyperpermeability. Such required coupling would provide a basis for understanding the blockade of VEGF-induced hyperpermeability by preventing S-nitrosation of 2 -catenin through mutation of cys619 while phosphorylation of 2 -catenin on tyr654 proceeded normally37. It would similarly explain the blockade of PAF-induced hyperpermeability obtained with N-acetyl cysteine36. While phosphorylation was not measured in our experiments utilizing N-acetyl cysteine to block S-nitrosation, it is plausible that agonist-stimulated phosphorylation of AJ proteins occurred in an unopposed manner. These results suggest that phosphorylation of AJ proteins is necessary but not sufficient for the onset of robust hyperpermeability. In addition, it is possible that after PAF-induced internalization, VE-cadherin becomes subject to S-nitrosation once it reaches the appropriate cytosolic location. Since VE-cadherin becomes phosphorylated prior to its internalization, its immediately subsequent S-nitrosation would strengthen the concept that phosphorylation and S-nitrosation are finely tuned signaling mechanisms in the regulation of microvascular/endothelial permeability. These developing concepts are illustrated in Fig. 3. The driving concept is that phosphorylation and S-nitrosation are complementary, cooperative, synergistic (probably sequential) required mechanisms in the regulation of adherens junctions for the onset of hyperpermeability. Our cartoon is aimed at emphasizing the emerging role of S-nitrosation of AJ proteins and does not necessarily exclude the participation of NO-sGC interactions in hyperpermeability. As indicated previously34, parallel biochemical pathways may exist leading to hyperpermeability after agonist-stimulated eNOS activity.
Figure 3. Illustration of Hypothetical Hyperpermeability Events.
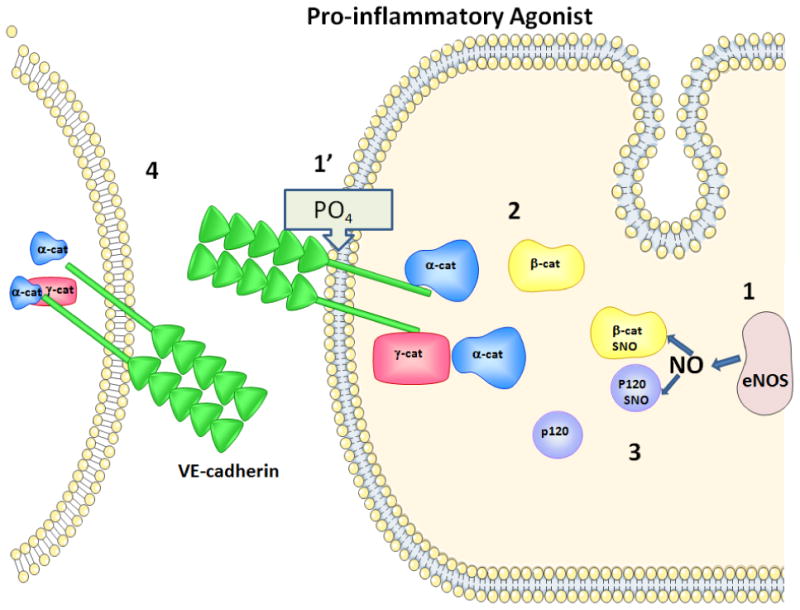
The cartoon represents developing concepts about how S-nitrosation of junctional proteins regulates the onset of hyperpermeability. The arrival of a pro-inflammatory agonist induces 1 and 1′, i.e., translocation of eNOS from the plasma membrane (via internalization of caveolae; refs. # 22, 25, 26) and phosphorylation of junctional proteins. Phosphorylated junctional proteins internalize to the cytosol (step 2). Cytosolic eNOS-derived NO causes S-nitrosation of junctional proteins (2 -catenin and p120-catenin in our studies; ref. # 35; Step 3). The combination of phosphorylation and S-nitrosation of junctional proteins is proposed as an important mechanism in the onset of pro-inflammatory agonist induced hyperpermeability, shown by a conformational change in VE-cadherin (Step 4) that would allow the passage of macromolecules.
The question that arises is how is the on-off switch of S-nitrosation regulated to control hyperpermeability? Phosphorylation is the universally accepted currency to convey on-off signals in biology. Due to intense research for decades, we know that the target specificity of the phosphorylation/de-phosphorylation in a multitude of proteins is guaranteed by the activity of well-collimated and coordinated kinases and phosphatases. Similarly, S-nitrosation is usually described as a specific reaction in most reviews of the subject. However, the “on” phase is normally ascribed to the redox state of the cell without specifying molecules or specific processes responsible for initiating S-nitrosation. It is recognized that the process requires a sensing mechanism for the redox signal and preferential oxidation of the target proteins, and that thiol-containing proteins exhibit different redox potential under normal (baseline) conditions40. The thioredoxin-thioredoxin reductase system and S-nitrosoglutathione reductase have been identified as the main enzymes responsible for implementing the “off” (de-nitrosation) signal41. The coupling between phosphorylation and S-nitrosation invoked earlier for regulation of the onset of permeability is, in principle, supported by the fact that oxidation-reduction reactions control the activity of kinases and phosphatases. In fact, the activation of eNOS - the source of NO - appears to be regulated by redox-controlled signaling42. These observations imply that factors that control cellular redox (such as mitochondria) are significantly involved through as yet unidentified mechanisms in the signaling cascade initiated by agents that induce microvascular hyperpermeability through activation/translocation of eNOS and internalization of AJ proteins.
The concept of redox driven “on/off” signaling applies well in inflammation and vascular disease, which are associated with recognized stress conditions of cells – endothelial cells in particular. S-nitrosation is now recognized to play a role under conditions of oxidative stress. Indeed, S-nitrosation of sGC limits the development of vasodilation in nitroglycerine tolerance as well as in hypertension43, 44. Under inflammatory/stress conditions there are several factors influencing the redox state (reactive oxygen and nitrogen species) that become activated; thus, it would be important to identify which factors exhibit selectivity or specificity for initiating S-nitrosation dependent hyperpermeability. Preliminary (unpublished) observations in our laboratory indicate that stimulation of endothelial cells with PAF induces S-nitrosation of many key proteins associated with the onset of hyperpermeability with the same time-course in the scale of minutes, i.e., the observed time-course of S-nitrosation does not establish a clearly defined sequence of signal activation. On the other hand, our preliminary observations support an important role for the thioredoxin-thioredoxin reductase system in the denitrosation phase. It is reasonable to postulate that the “on” phase would depend largely on the concentration of the initiating agent (as associated with the redox state of the cell), and the cell compartment where the events are taking place. These observations bring back the suggestion that the key regulator of S-nitrosation of endothelial AJ proteins is eNOS, an enzyme whose activity is exquisitely regulated by a number of factors, and whose location (via agonist-induced translocation) in the cytosol is fundamental for the onset of hyperpermeability responses.
References
- 1.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 2.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 3.Huang Q, Yuan Y. Interaction of pkc and nos in signal transduction of microvascular hyperpermeability. Am J Physiol. 1997;273:H2442–2451. doi: 10.1152/ajpheart.1997.273.5.H2442. [DOI] [PubMed] [Google Scholar]
- 4.Varma S, Breslin JW, Lal BK, Pappas PJ, Hobson RW, 2nd, Duran WN. P42/44mapk regulates baseline permeability and cgmp-induced hyperpermeability in endothelial cells. Microvasc Res. 2002;63:172–178. doi: 10.1006/mvre.2001.2381. [DOI] [PubMed] [Google Scholar]
- 5.Kramer GC, Harms BA, Bodai BI, Demling RH, Renkin EM. Mechanisms for redistribution of plasma protein following acute protein depletion. Am J Physiol. 1982;243:H803–809. doi: 10.1152/ajpheart.1982.243.5.H803. [DOI] [PubMed] [Google Scholar]
- 6.Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breslin JW, Sun H, Xu W, Rodarte C, Moy AB, Wu MH, Yuan SY. Involvement of rock-mediated endothelial tension development in neutrophil-stimulated microvascular leakage. Am J Physiol Heart Circ Physiol. 2006;290:H741–750. doi: 10.1152/ajpheart.00238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breslin JW, Yuan SY. Involvement of rhoa and rho kinase in neutrophil-stimulated endothelial hyperpermeability. Am J Physiol Heart Circ Physiol. 2004;286:H1057–1062. doi: 10.1152/ajpheart.00841.2003. [DOI] [PubMed] [Google Scholar]
- 9.Chambers R, Zweifach BW. Intercellular cement and capillary permeability. Physiol Rev. 1947;27:436–463. doi: 10.1152/physrev.1947.27.3.436. [DOI] [PubMed] [Google Scholar]
- 10.Karnovsky MJ. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J Cell Biol. 1967;35:213–236. doi: 10.1083/jcb.35.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. 2013;126(Pt 12):2545–2549. doi: 10.1242/jcs.124529. [DOI] [PubMed] [Google Scholar]
- 12.Herron CR, Lowery AM, Hollister PR, Reynolds AB, Vincent PA. P120 regulates endothelial permeability independently of its nh2 terminus and rho binding. Am J Physiol Heart Circ Physiol. 2011;300:H36–48. doi: 10.1152/ajpheart.00812.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: Differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (vecadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayhan WG. Nitric oxide accounts for histamine-induced increases in macromolecular extravasation. Am J Physiol. 1994;266:H2369–2373. doi: 10.1152/ajpheart.1994.266.6.H2369. [DOI] [PubMed] [Google Scholar]
- 15.Ramirez MM, Kim DD, Duran WN. Protein kinase c modulates microvascular permeability through nitric oxide synthase. Am J Physiol. 1996;271:H1702–1705. doi: 10.1152/ajpheart.1996.271.4.H1702. [DOI] [PubMed] [Google Scholar]
- 16.Ramirez MM, Quardt SM, Kim D, Oshiro H, Minnicozzi M, Duran WN. Platelet activating factor modulates microvascular permeability through nitric oxide synthesis. Microvasc Res. 1995;50:223–234. doi: 10.1006/mvre.1995.1055. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Chilian WM, Granger HJ, Zawieja DC. Permeability to albumin in isolated coronary venules. Am J Physiol. 1993;265:H543–552. doi: 10.1152/ajpheart.1993.265.2.H543. [DOI] [PubMed] [Google Scholar]
- 18.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am J Physiol. 1992;263:H641–646. doi: 10.1152/ajpheart.1992.263.2.H641. [DOI] [PubMed] [Google Scholar]
- 19.Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM. Histamine increases venular permeability via a phospholipase c-no synthase-guanylate cyclase cascade. Am J Physiol. 1993;264:H1734–1739. doi: 10.1152/ajpheart.1993.264.5.H1734. [DOI] [PubMed] [Google Scholar]
- 20.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatakeyama T, Pappas PJ, Hobson RW, 2nd, Boric MP, Sessa WC, Duran WN. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol. 2006;574:275–281. doi: 10.1113/jphysiol.2006.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez FA, Kim DD, Duran RG, Meininger CJ, Duran WN. Internalization of enos via caveolae regulates paf-induced inflammatory hyperpermeability to macromolecules. Am J Physiol Heart Circ Physiol. 2008;295:H1642–1648. doi: 10.1152/ajpheart.00629.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 24.Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC. Agonist-stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of enos phosphorylation at tyr83. Circ Res. 2008;102:497–504. doi: 10.1161/CIRCRESAHA.107.162933. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez FA, Rana R, Gonzalez FG, Iwahashi T, Duran RG, Fulton DJ, Beuve AV, Kim DD, Duran WN. Functional significance of cytosolic endothelial nitric-oxide synthase (enos): Regulation of hyperpermeability. J Biol Chem. 2011;286:30409–30414. doi: 10.1074/jbc.M111.234294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK, Gordon DM, Meininger CJ, Duran WN. Internalization of enos and no delivery to subcellular targets determine agonist-induced hyperpermeability. Proc Natl Acad Sci U S A. 2009;106:6849–6853. doi: 10.1073/pnas.0812694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez FA, Savalia NB, Duran RG, Lal BK, Boric MP, Duran WN. Functional significance of differential enos translocation. Am J Physiol Heart Circ Physiol. 2006;291:H1058–1064. doi: 10.1152/ajpheart.00370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gawlowski DM, Duran WN. Dose-related effects of adenosine and bradykinin on microvascular permselectivity to macromolecules in the hamster cheek pouch. Circ Res. 1986;58:348–355. doi: 10.1161/01.res.58.3.348. [DOI] [PubMed] [Google Scholar]
- 29.Gawlowski DM, Ritter AB, Duran WN. Reproducibility of microvascular permeability responses to successive topical applications of bradykinin in the hamster cheek pouch. Microvasc Res. 1982;24:354–363. doi: 10.1016/0026-2862(82)90022-x. [DOI] [PubMed] [Google Scholar]
- 30.Michel T, Li GK, Busconi L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1993;90:6252–6256. doi: 10.1073/pnas.90.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suda K, Rothen-Rutishauser B, Gunthert M, Wunderli-Allenspach H. Phenotypic characterization of human umbilical vein endothelial (ecv304) and urinary carcinoma (t24) cells: Endothelial versus epithelial features. In Vitro Cell Dev Biol Anim. 2001;37:505–514. doi: 10.1290/1071-2690(2001)037<0505:PCOHUV>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 32.Figueroa XF, Gonzalez DR, Martinez AD, Duran WN, Boric MP. Ach-induced endothelial no synthase translocation, no release and vasodilatation in the hamster microcirculation in vivo. J Physiol. 2002;544:883–896. doi: 10.1113/jphysiol.2002.021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He P, Zeng M, Curry FE. Cgmp modulates basal and activated microvessel permeability independently of [ca2+]i. Am J Physiol. 1998;274:H1865–1874. doi: 10.1152/ajpheart.1998.274.6.H1865. [DOI] [PubMed] [Google Scholar]
- 34.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, 2nd, Duran WN. Vegf increases endothelial permeability by separate signaling pathways involving erk-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 35.Lakshminarayanan S, Antonetti DA, Gardner TW, Tarbell JM. Effect of vegf on retinal microvascular endothelial hydraulic conductivity: The role of no. Invest Ophthalmol Vis Sci. 2000;41:4256–4261. [PubMed] [Google Scholar]
- 36.Marin N, Zamorano P, Carrasco R, Mujica P, Gonzalez FG, Quezada C, Meininger CJ, Boric MP, Duran WN, Sanchez FA. S-nitrosation of beta-catenin and p120 catenin: A novel regulatory mechanism in endothelial hyperpermeability. Circ Res. 2012;111:553–563. doi: 10.1161/CIRCRESAHA.112.274548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton JP. S-nitrosylation of beta-catenin by enos-derived no promotes vegf-induced endothelial cell permeability. Mol Cell. 2010;39:468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 38.Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van Roy F, Reynolds AB. A novel role for p120 catenin in e-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, Komiya S, Shimizu M, Fukunaga Y, Nagafuchi A. Involvement of p120 carboxy-terminal domain in cadherin trafficking. Cell Struct Funct. 2007;32:127–137. doi: 10.1247/csf.07023. [DOI] [PubMed] [Google Scholar]
- 40.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka T, Nakamura H, Yodoi J, Bloom ET. Redox regulation of the signaling pathways leading to enos phosphorylation. Free Radic Biol Med. 2005;38:1231–1242. doi: 10.1016/j.freeradbiomed.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Crassous PA, Couloubaly S, Huang C, Zhou Z, Baskaran P, Kim DD, Papapetropoulos A, Fioramonti X, Duran WN, Beuve A. Soluble guanylyl cyclase is a target of angiotensin ii-induced nitrosative stress in a hypertensive rat model. Am J Physiol Heart Circ Physiol. 2012;303:H597–604. doi: 10.1152/ajpheart.00138.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayed N, Kim DD, Fioramonti X, Iwahashi T, Duran WN, Beuve A. Nitroglycerin-induced s-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]