

Abstract
A lipid/calcium/phosphate (LCP) nanoparticle (NP) formulation (particle diameter ~25 nm) with superior siRNA delivery efficiency was developed and reported previously. Here, we describe the successful formulation of 111In into LCP for SPECT/CT imaging. Imaging and biodistribution studies showed that, polyethylene glycol grafted 111In-LCP preferentially accumulated in the lymph nodes at ~70% ID/g in both C57BL/6 and nude mice when the improved surface coating method was used. Both the liver and spleen accumulated only ~25% ID/g. Larger LCP (diameter ~67 nm) was less lymphotropic. These results indicate that 25 nm LCP was able to penetrate into tissues, enter the lymphatic system, and accumulate in the lymph nodes via lymphatic drainage due to 1) small size, 2) a well-PEGylated lipid surface, and 3) a slightly negative surface charge. The capability of intravenously injected 111In-LCP to visualize an enlarged, tumor-loaded sentinel lymph node was demonstrated using a 4T1 breast cancer lymph node metastasis model. Systemic gene delivery to the lymph nodes after IV injection was demonstrated by the expression of red fluorescent protein cDNA. The potential of using LCP for lymphatic drug delivery is discussed.
1. Introduction
The lymphatic system is a central component of the immune system and serves as the secondary circulation system responsible for the drainage of fluid, proteins, and waste products from tissues into the blood. Lymph nodes also play an important role in infectious disease, inflammation, and cancer [1]. Primary tumors commonly invade draining lymph nodes, which then serve as a reservoir for further metastatic spread of cancer cells [2–4].
The delivery of genes and drugs to both local, draining lymph nodes and the lymphatic system as a whole is a challenging task. Certain lipophilic compounds such as long-chain fatty acids, cholesterol esters, triglycerides, and lipid-soluble vitamins can be transported through the lymphatic channels [1, 5]. However, most chemotherapy agents do not gain access to the lymphatic system and lymph node metastases after conventional intravenous (IV) infusion [1, 5]. Consequently, the development of clinical chemotherapy for lymph node metastasis has remained elusive.
Many different types of nanoparticle (NP), including liposomes, silica NPs, and other polymer-based drug delivery systems, have exhibited improved efficiency in regionally delivering drugs to the lymphatic system [6–10]. For example, intraperitoneally (IP) injected liposomes containing doxorubicin result in an 8- to 14-fold (4 h post injection) and a 3- to 6-fold (24 h post injection) increase in doxorubicin concentration in the draining lymph nodes in rats compared to free doxorubicin [6]. However, no significant difference was observed after IV administration. Thus, methods for effective delivery of IV administered NP to the lymphatic system are still needed for the detection of lymph node metastasis.
Non-invasive imaging techniques using nuclear medicine, such as single photon emission computer tomography (SPECT) and positron emission tomography (PET) have been developed [11, 12]. Among the radionuclides used in clinical practice, 111In is the second most widely used radionuclide in clinical practice, surpassed only by 99mTc. 111In displays major decay at photon energy levels of 171.3 and 245.4 keV, within the ideal range of the detector device. The half-life of 111In (2.83 days) is also advantageous because prolonged exposure to the radionuclide may cause undesired toxicity and should be prevented. Many studies have demonstrated in vivo imaging of tumors with various types of NPs using SPECT/CT or PET/CT technique [12–14].
Lipid/calcium/phosphate (LCP) NPs were first developed for siRNA delivery [15–17] and can also successfully deliver plasmid DNA to hepatocytes [18]. Based on the mechanism of formation of the CaP core, we hypothesized that any drug or radionuclide that can form co-precipitates with CaP has the potential to be formulated into LCP. Since indium (In) can form precipitates with phosphate very efficiently (solubility product constant, Ksp, of InPO4 = 2.3×10−22) in a manner similar to that of calcium (Ksp of Ca3(PO4)2 = 1.0×10−25), we hypothesize that 111In will be a good candidate to add to LCP formulations to provide in vivo imaging capability.
Systemic accumulation of NPs in the lymph nodes after IV administration has rarely been reported. A 25 nm, dextran-coated, ultrasmall super-paramagnetic iron oxide (USPIO, Feridex®) NP has shown systemic lymphotropism after IV administration and has been evaluated for MRI of clinical lymph node metastasis [19–22]. However, larger iron oxide NPs coated with the same dextran coating were found to preferentially accumulate in the liver and spleen [19]. Another self-luminescing, near-infrared semiconductor polymer NP (30~40 nm) capable of accumulating in the lymph node at a certain level has been described recently for lymph node mapping and tumor imaging [23]. However, quantitative data showing the level of accumulation is missing and liver showed the highest accumulation according to near-infrared fluorescent imaging.
Other NPs of similar size (25~30 nm) or smaller may exhibit prolonged circulation time in the blood. No preferential accumulation in lymph nodes has been reported, however. For example, one recent publication reported that PET imaging demonstrated a long-circulating, 15 nm, micellar NP had minimal accumulation in the liver and spleen, with ~6% ID/g tumor accumulation. However, this micellar NP did not show lymphotropism [24]. Another recent publication reported 30 nm Au nanocages for use in tumor PET imaging also exhibit minimal accumulation in the liver and spleen but no lymphotropism [25]. Thus, NP lymphotropism may be correlated not only with NP size but also other surface properties.
In this paper, we demonstrate that LCP were able to achieve both systemic delivery of genes to the lymphatic system and imaging of lymph node metastasis by a mechanism similar to the aforementioned Feridex®. The systemic lymphotropism of LCP and Feridex® was achieved by tissue penetration and an atypical lymphatic drainage mechanism that is different from phagocytic uptake mechanisms used by locally injected NPs to accumulate at regional draining lymph nodes. Coating of the NP surface with a lipid bilayer or dextran may contribute to the unusual lymphotropism.
2. Materials and Methods
2.1 Materials
Cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), dioleoylphosphatydic acid (DOPA), 1,2-Dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000) ammonium salt (DSPE-PEG2000), and 1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino] dodecanoyl]-sn-glycero-3-phosphocholine (NBD-PC) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). 111InCl in 0.05 N HCl was purchased from PerkinElmer (Waltham, MA). Double-strand oligo DNA (sense sequence, 5′-CAAGGGACTGGAAGGCTGGG-3′) and Texas-Red labeled sense-strand oligo DNA (sequence: 5′-[TxRd]CAAGGGACTGGAAGGCTGGG-3′) were both synthesized by Sigma-Aldrich (St. Louis, MO). 3H labeling of oligonucleotides was prepared by hydrogen exchange with 3H2O at the C8 positions of the purines [26]. 4T1-luc2-GFP Bioware® Ultra Green) cell line was purchased from Caliper (Hopkinton, MA). CR8C peptide was synthesized by Peptide 2.0 Inc. (Chantilly, VA). Other materials were obtained from Sigma-Aldrich (St. Louis, MO).
2.2 The preparation of 111In-LCP core
111In-LCP core was prepared using a previously described method [15–17] with some modifications (Supplementary Figure 1). Two water-in-oil microemulsions were prepared: 1) a calcium emulsion: 111InCl3 was premixed with CaCl2 to make a final 50 μL of 500 mM CaCl2 in 4 mL of cyclohexane oil phase (cyclohexane/Igepal CO-520 = 71/29, v/v), and 2) a phosphate emulsion: a sufficient amount of 0.05 N NaOH was added to pH 9.0 Na2HPO4 (to neutralize the extra HCl in the calcium emulsion) to make final 50 μL of 100 mM Na2HPO4 also in 4 mL of cyclohexane oil phase. DOPA (92.5 μL 34.6 mM in chloroform) as the inner leaflet lipid was also added in the phosphate emulsion.
After mixing the two microemulsions for 40 min, 8 mL of absolute ethanol was added to the micro-emulsion to break the microemulsion system. The mixture was stirred for another 30 min. Then, the mixture was centrifuged at 12,500× g for 15 min to collect the 111In -LCP cores. The cores were washed once with 10 mL absolute ethanol to remove residual surfactants. Then, the cores were washed with 1.2 mL cyclohexane and mixed with 1.4 mL absolute ethanol to remove any residual DOPA. Finally, the cores were washed with 2 mL of absolute ethanol to ensure the removal of cyclohexane. After all washes, the pellets were dispersed in 250 μL chloroform. The product was centrifuged at 10,000× g for 5min. Precipitate containing excess salts and aggregates was discarded and the supernatant containing LCP cores was collected and stored in a glass vial at −20 °C.
2.3 Outer leaflet coating
For outer leaflet coating, 100 μL of 20 mM cholesterol, 100 μL of 20 mM DOPC, and 50 μL of 20 mM of DSPE-PEG2000 (all in CHCl3) were mixed with the LCP cores in a glass vial. After complete removal of the CHCl3 by using a stream of nitrogen and vacuum desiccation for 1 h, the cores were first suspended in 100 μL of pre-warmed absolute alcohol (55 °C) and then dispersed in total 1 mL pre-warmed aqueous solution containing 5% dextrose.
2.4 Sucrose gradient centrifugation
Sucrose gradient centrifugation assays were performed using NBD-PC, which had a structure similar to that of DOPC, to mix with DOPC at 1% of the total DOPC to label the outer leaflet of LCP with fluorescence. The outer leaflet coated LCP was loaded into a tube containing a sucrose density gradient ranging from 0% to 60% (w/w). After ultra-centrifugation at 337,000× g for 4 h, excess lipids that were not associated with the LCP floated to the upper part of the gradient and were separated from the dense LCP which formed a sharp band above the 60% layer in the gradient.
2.5 Tuning 111In-LCP NP size by adjusting surfactant system
To tune the 111In-LCP core size, a Triton surfactant system (cyclohexane/hexanol/Triton X-100 = 75/10/15, v/v/v) was mixed with the original Igepal surfactant system (cyclohexane/Igepal CO-520 = 71/29, v/v). As the portion of the Triton surfactant system increased, the size of 111In-LCP cores could be enlarged to ~50 nm, making the final larger-LCP (L-LCP) around 65 nm in diameter.
2.6 Particle size and zeta potential analysis
Particle size and zeta potential of LCP NPs were determined using a Malvern ZetaSizer Nano series (Westborough, MA). Before measurement, LCP NPs were purified using sucrose gradient centrifuge method to remove excess lipids. Collected NPs were measured in water using protocols suggested by the manufacturer.
2.7 TEM microscopy and sample preparation
LCP cores suspended in chloroform was carefully applied on the formvar/carbon coated copper 200 mesh grid and allowed to dry for 10 min at room temperature. The volume of chloroform used should be as small as possible (~1 μL) to prevent dissolution of the formvar/carbon film. For outer leaflet coated LCP in 5% dextrose solution, 10 μL of the final LCP solution was applied on the formvar/carbon coated copper 200 mesh grid. The LCP was allowed to settle on the grid for 10 min. Then the remaining LCP solution was carefully removed by using a Kimwipes wiper. Another 10 μL of water was used to rinse the grid to prevent formation of dextrose crystals. After the 10 μL of water was removed carefully by using Kimwipes wiper, the grid was placed in a dust-free area to allow complete dryness for at least 30 min before observation using a TEM microscopy (JEOL 100 CS II).
2.8 Determining siRNA entrapment efficiency
To ensure modifications would not compromise the ability to encapsulate siRNA, 35 μg total double-stranded oligo DNA (as surrogate to siRNA) including 10 μg of 3H-labeled DNA oligo was added to the calcium emulsion during 111In-LCP preparation. After the LCP core preparation, oligo entrapment efficiency was determined using liquid scintillation counting for 3H.
2.9 In vivo SPECT/CT imaging and biodistribution study
All animal work was performed in accordance with and approved by the University of North Carolina Institutional Animal Care and Use Committee guidelines. Athymic nude (nu/nu) mice and wild type C57BL/6 mice purchased from National Cancer Institute were used. SPECT/CT experiments were performed using a GE eXplore speCZT system. A mouse 7-pinhole collimator was used for high resolution SPECT imaging. Each mouse was injected through the tail vein with 200 μL of the final 111In-LCP containing ~0.5 mCi of 111In. Mice were anesthetized with isoflurane and their body temperature was controlled using a water circuit and warm air. Mice were scanned at 2 h, 4h, and 24 h post IV injection. SPECT/CT imaging was performed by Biomedical Research Imaging Center at UNC.
Mice were sacrificed and organs/tissues collected at 24 h post injection for biodistribution studies. Eight lymph nodes, including 2 axillary, 2 brachial, 2 inguinal, and 2 popliteal lymph nodes were collected. Each organ/tissue sample was counted with a gamma counter and the reading was corrected for the 111In decay factor during analysis.
2.10 LCP tissue penetrating study by IM injection
111In loaded S- and L-LCP were coated with or without 20% DSPE-PEG2000. Each mouse received only 10 μL IM injection containing ~5 μCi 111In to the right hind leg muscle to reduce possible tissue damage. After 3 h or 24 h, mice were sacrificed with major organs collected for gamma counting. Draining popliteal lymph nodes was collected and four counter side distal lymph nodes (1 axillary, 1 brachial, 1 inguinal, and 1 popliteal from the counter side of the IM injection) were also collected for comparison. The gamma reading was corrected for the 111In decay factor during analysis. Statistical analysis was performed using a Student’s t-test.
2.11 Loading Texas-Red oligo into LCP NPs
Double-stranded oligo DNA with Texas-Red labeled on the sense strand was added to the calcium emulsion during the preparation of the LCP cores. Following the same LCP preparation protocol, 10 μL IM injection of the Texas-Red oligo loaded S- and L-LCP were used for the experiment examining the distribution patterns in the draining lymph nodes.
2.12 Study of the distribution of LCP in the draining lymph nodes
Mice were sacrificed 24 h after IM injection of the Texas-Red oligo loaded S- or L-LCP with the draining popliteal lymph nodes collected. The lymph nodes were fixed in formalin overnight then put in 30% sucrose solution for another overnight. The lymph nodes were then mounted in OCT (optimum cutting temperature) compound and snap-frozen by liquid nitrogen. Frozen sections were cryo-sectioned at a thickness of 20 μm. FITC labeled antibodies against CD11c or CD11b were diluted to a concentration of 1:500 for immunostaining. After PBS wash, the slides were mounted with DAPI containing mounting medium for confocal microscopy observation using a Leica SP2 confocal microscope.
2.13 Establishing 4T1 tumor metastasis model
The luciferase and GFP double-expressed 4T1 murine breast cancer cell line (4T1-luc2-GFP Bioware® Ultra Green) was used. The tumor model was established by hock injection of 2×105 4T1-Luc2-GFP cells in the right hind leg of 6–8 weeks old female BALB/c mice. Bioluminescence imaging in a Kodak In-Vivo FX PRO system was performed within 15 min after injection with intraperitoneal luciferin to evaluate tumor metastatic progression starting around 10 d after hock inoculation. Fluorescent image of the GFP expression of the dissected lymph nodes were taken using an IVIS Kinetic imaging system.
2.14 Lymph node gene delivery with S-LCP
S-LCP cores were loaded with RFP plasmid and CR8C peptide by mixing 50 μg RFP plasmid and 50 μg CR8C peptide sequentially with 50 μL of 500 mM CaCl2 solution [18] with some 111InCl3 for biodistribution evaluation. The core preparation and coating with an outer leaflet were completed following the same procedure described above. Outer leaflet lipids of 40% DOPC (or DOTAP) plus 40% cholesterol and 20% DSPE-PEG2000 were used for coating. Each C57BL/6 mouse received 200 μL of the final S-LCP containing 10 μg of RFP plasmid and 10 μg of CR8C peptide by IV injection. After 24 h, the mice were sacrificed and major organs, including 8 lymph nodes, were collected for RFP fluorescence imaging using a Carestream In-Vivo Imaging System FX Pro and gamma counting.
3 Results
3.1 Preparation of S-LCP and L-LCP
The general procedure for LCP preparation was originally established and optimized for siRNA encapsulation [15–17]. In order to efficiently formulate 111In into the CaP core, some major adjustments have been made to this procedure. Since 111In will compete with calcium for phosphate, the CaCl2 concentration has been reduced from 2.5 M to 500 mM and the Na2HPO4 concentration (pH 9.0) has been increased from 12.5 mM to 100 mM. For the same LCP preparation size, the working microemulsion was thus reduced to 1/8 of the original volume.
The LCP made with the original Igepal surfactant system (cyclohexane/Igepal CO-520 = 71/29, v/v) was small in size. The CaP core was ~10 nm in diameter and the final LCP NP coated with the outer leaflet lipids was ~25 nm (Figure 1A), consistent with previous observations [15, 16]. These small NPs have been termed S-LCP.
Figure 1.
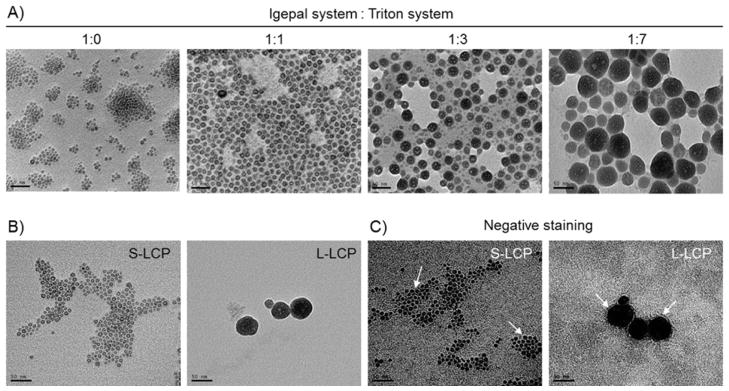
A) TEM images of 111In -LCP core made with different Igepal system to Triton system mixing ratio. S- and L-LCP were made by Igepal system to Triton system ratio 1:0 and 1:7, respectively. B) TEM images of S- and L-LCP after outer leaflet coating. C) Negative staining of S- and L-LCP to show the lipid coating.
By adjusting the microemulsion surfactant system, LCP core size could be tuned between ~10 and ~50 nm in diameter. When mixing the Igepal system with the Triton system at a 1:1 or 1:3 ratio, particles became progressively larger (Figure 1A). When using an Igepal:Triton ratio at 1:7, the 111In-LCP core was significantly enlarged to ~50 nm. This larger LCP with a final size, including the outer-leaflet coating, of ~65 nm have been termed L-LCP. Both S- and L-LCP were coated with dioleoylphosphatidylcholine (DOPC)/Cholesterol/1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-poly(ethylene glycol)2000 (DSPE-PEG2000) at 2/2/1 molar ratio for the experiments described here unless otherwise specified.
TEM images of the final S- and L-LCP after outer leaflet coating showed that both NPs were well-dispersed (Figure 1B). The lipid membrane coating of S- and L-LCP was further characterized by negative staining using uranyl acetate (Figure 1C).
3.2 Characterization of 111In-LCPs
Sucrose-gradient centrifugation was used to purify LCP NPs for characterization described below. DLS analysis revealed a fairly uniform S-LCP size (~25 nm) with a polydispersity index (PDI) below 0.3 and a zeta potential of approximately −20 mV. L-LCP, on the other hand, was around 67 nm in size with a PDI of ~0.4 and a zeta potential around −18 mV (Table 1). The zeta-potentials of LCP with different outer leaflet coatings are listed in Table 2.
Table 1.
Characterization of S-LCP and L-LCP
| DNA oligo entrapment [a] | 111In entrapment [b] | Average size/PDI [c] | Zeta potential [c] (outer leaflet DOPC/Cholesterol + 20% DSPE-PEG) | Calcium/Phosph ate ratio of the LCP core [d] | |
|---|---|---|---|---|---|
| S-LCP | 64.4 ± 4.4% | 30.7 ± 4.1 % | 25.3 ± 1.9 nm/0.3 | −19.9 ± 4.1 mV | 1.01 ± 0.04 |
| L-LCP | 29.2 ± 0.5% | 32.3 ± 2.2 % | 67.2 ± 2.0 nm/0.4 | −18.0 ± 2.0 mV | 0.89 ± 0.03 |
determined by tritium labeled DNA oligo and liquid scintillation counting (N=3)
determined by 111In gamma counting (N=4)
measured by dynamic light scattering (N=3)
analyzed by inductively coupled plasma mass spectrometry. (N=3)
Table 2.
Zeta-potentials of LCP with different outer leaflet coatings
| DOPC/Cholesterol + 20% DSPE-PEG | DOPC/Cholesterol + 0% DSPE-PEG | DOTAP/Cholesterol + 20% DSPE-PEG | |
|---|---|---|---|
| S-LCP | −19.9 ± 4.1 mV | −3.8 ± 2.1 mV | 21.3 ± 2.1 mV |
| L-LCP | −18.0 ± 2.0 mV | −3.3 ± 1.8 mV | N/A |
Measured by dynamic light scattering (N=3)
Using a 3H-labeled oligodeoxynucleotide (oligo) and scintillation counting, it was determined that S-LCP could entrap oligo at an efficiency of ~60%, which was not affected by the presence of 111In. Oligo entrapment by L-LCP was also unaffected by the presence of 111In, but only ~30% of the oligo was entrapped (Table 1). 111In entrapment of both S- and L-LCP was around 30% (Table 1). Inductively coupled plasma mass spectrometry was used to analyze the LCP core composition. For both S- and L-LCP, the Ca/P ratio was about 1:1, which was different from that of the naturally occurring hydroxyapatite (Ca10(PO4)6(OH)2, Ca/P = 1.67) (Table 1). These results indicated that the modifications made for efficient 111In encapsulation did not significantly change any of the properties listed in Table 1 of the LCP made without 111In.
3.3 Accumulation of S-LCP in the lymph nodes
To study the biodistribution of S-LCP, S-LCP containing ~0.5 mCi 111In was tail vein injected into both wild-type C57BL/6 and nude mice. SPECT/CT images were taken at 2, 4 and 24 h post injection. Little accumulation occurred in the mononuclear phagocytic system (MPS) in both the liver and spleen (Figure 2A). The majority of the 111In was found in the heart and vena cava at 2 h and 4 h post injection, indicating that the S-LCP remain in the blood circulation after injection. At 24 h, symmetrical lymph nodes throughout the animal accumulated significant amounts of the S-LCP (Figure 2B). After SPECT/CT imaging, mice were sacrificed and their major organs were collected. Gamma counting was performed on collected organs to determine biodistribution. The collected major lymph nodes had ~70 % ID/g accumulation of S-LCP. Improved S-LCP core washing and coating with the outer leaflet contributed to a reduced uptake of the particles by the MPS, ~25% ID/g in both the liver and spleen (Figure 2C). When mice were injected with L-LCP, higher accumulation in the MPS (~39% ID/g in the liver and ~289% ID/g in the spleen) and lower lymph node accumulation (~23% ID/g) were observed (Figure 2C).
Figure 2.

A) SPECT/CT images of nude mouse taken at 2 h and 4 h post IV injection of S-LCP containing ~0.5 mCi 111In. Strong 111In signals were mainly observed in the heart and vena cava (red arrow). B) SPECT/CT images taken at 24 h post IV injection. Four different horizontal sections were included to show that symmetrical lymph nodes (yellow arrows) throughout the body accumulated significant amount of S-LCP. C) Biodistribution results determined by organ dissection and gamma counting. S-LCP in nude mice and C57BL/6 mice; L-LCP in C57BL/6 mice were shown. Note that only eight major lymph nodes were collected for counting. (N=3 for each group)
3.4 PEGylated S-LCP was more tissue penetrating than L-LCP
To study whether PEGylated S-LCP could penetrate into tissue and lead to lymphatic accumulation, we designed an intramuscular (IM) injection experiment due to the difficulty of directly observing particles penetrating into tissues from circulation. PEGylated S-LCP or L-LCP at 10 μL was IM injected into the muscle of right, hind leg. After 3 h or 24 h, mice were sacrificed and major organs, including lymph nodes and the injected leg, were collected for gamma counting. Our hypothesis was that if S-LCP has the ability to achieve high tissue penetration following local intramuscular injection, they can move more freely in the tissue, enter into circulation in the blood, and accumulate in the distal lymph nodes.
When the NPs were PEGylated, both S- and L-LCP had an early blood distribution (observed at 3 h) that could be explained by the mechanical force created by the injection (Figure 3). In support of our hypothesis, S-LCP exhibited much higher tissue penetration between 3 and 24 h than L-LCP, as illustrated by decreased retention at the injection site (p < 0.01, N=3) and a sustained concentration in the circulation. Although there was some accumulation of S-LCP in the liver, the S-LCP depot at the IM injection site served as a reservoir to provide a continuous supply of NPs to the blood. Distal lymph nodes were able to accumulate ~41% ID/g of IM injected S-LCP.
Figure 3.

Biodistribution comparison after IM injection of 111In loaded S- and L-LCP coated with or without 20% DSPE-PEG2000. Mice were sacrificed at 3 h or 24 h post IM injection.
As the initial blood distribution of PEGylated, L-LCP was cleared by the liver, the L-LCP depot at the IM injection site could not penetrate into the blood to maintain the NP concentration. The L-LCP depot was limited to the injection site and did not decrease significantly between 3 and 24 h post-injection (Figure 3). The larger NPs were favored by the MPS uptake processes; PEGylation in a manner similar to that of the smaller NPs did not alter this pattern [25, 27]. This phenomenon may explain why PEGylated L-LCP accumulated in the draining lymph node more than S-LCP. In either case, however, the effects of PEG protection in reducing their uptake by the MPS (i.e., local macrophages and dendritic cells) were clear.
3.5 Non-PEGylated L-LCP showed highest accumulation in the draining lymph node
Without PEGylation, both S- and L-LCP had limited mobility (though S-LCP was still more mobile), trapping more than 80% of the injected dose at the site of injection after 3 h. Since there was no PEG protection against MPS, the dominant uptake mechanism must be by phagocytosis of NPs by the nearby macrophages. Rapid and efficient accumulation in the draining lymph nodes at both 3 and 24 h post-injection were observed. Larger particles without PEGylation were preferentially taken up by MPS, as demonstrated by their accumulation levels of ~3000% ID/g in the draining lymph nodes after 3 h. A list of zeta-potentials of LCP NPs with different surface coatings can be found in Table 2.
3.6 Different distribution patterns of LCP in the draining lymph node
To study the distribution pattern of LCP in the draining lymph node, S- and L-LCP, both with 20% PEGylation and loaded with Texas-Red labeled DNA oligo, were used to repeat the IM injection experiment. After 24 h, the draining lymph nodes were collected and fixed with formalin for frozen section and observation with confocal microscopy. The results indicated that labeled S-LCP rarely overlapped with CD11c (dendritic cell marker) or CD11b (macrophage marker) staining (Figure 4). Labeled L-LCP, on the contrary, mainly overlapped with CD11c and partially with CD11b.
Figure 4.

S-LCP was largely not co-localized with phagocytic cells in the draining lymph node observed with confocal microscopy. LCP was labeled with Texas-red-labeled oligo and phagocytic cells (CD11c for dendritic cells and CD11b for macrophages) were labeled with green. Nuclei were stained with DAPI in blue. Left panels are for S- and right panels are for L-LCP, respectively.
3.7 PEG coating for S- and L-LCP
To demonstrate the importance of PEGylation to the lymphotropism of S-LCP, an experiment varying the amount of PEG coating on the S- and L-LCP was conducted. S-LCP without PEGylation exhibited high accumulation in the liver and spleen, as predicted. However, there was no difference observed among groups modified with 5, 10, and 20% PEGylation, indicating that S-LCP did not require a high degree of PEGylation (Figure 5A). As little as 5% PEG reduced MPS accumulation and increased lymphotropism at levels comparable to NPs with 20% PEGylation. On the other hand, increased PEGylation on L-LCP was beneficial, as liver accumulation of the NPs decreased with increasing amounts of PEGylation (Figure 5B). When coated with 5% PEG, the accumulation of L-LCP became higher in the spleen compared to NPs without PEG. Increasing the amount of PEG to 10 or 20% can further reduce accumulation of the particles in the liver, but accumulation in the spleen remains unchanged.
Figure 5.

Effect of PEGylation on the biodistribution of S-LCP (A) and L-LCP (B). Results with different degree of PEGylation at 0%, 5%, 10%, and 20% were shown.
3.8 Imaging lymph node metastasis
A variety of human cancers disseminate via regional lymph node metastasis [28]. The ability to image sentinel lymph nodes and evaluate the stage of metastasis is highly desirable. To demonstrate whether S-LCP can be used to detect lymph node metastasis, a 4T1 murine breast cancer cell line expressing both luciferase and green fluorescent protein (4T1-luc2-GFP) was used to establish a lymph node metastasis model [29]. After confirming the lymph node metastasis with luciferase imaging (Figure 6A), the mice were injected through the tail vein with 111In-S-LCP. SPECT/CT imaging taken at 24 h after injection clearly illustrated the enlarged, tumor-loaded, metastatic lymph node (Figure 6B).
Figure 6.

Imaging lymph node metastasis on a 4T1 breast cancer model. A) Bioluminescence image of two BALB/c mice was taken 10 d after hock inoculation. B) SPECT/CT images taken at 24 h post IV injection of 111In-S-LCP. Two horizontal and two vertical sections were shown. The enlarged and tumor loaded sentinel, popliteal lymph node could be directly compared with the counter-side, popliteal lymph node. C) GFP fluorescent images of the metastatic 4T1-luc2-GFP cancer cells in the lymph nodes. Eight lymph nodes (from top to bottom: 2 axillary, 2 brachial, 2 inguinal, and 2 popliteal) from both side of one mouse were shown. D) S-LCP biodistribution 28 h post injection at liver, spleen, 4T1 tumor, and various lymph nodes by gamma counting. (N=3)
After SPECT/CT imaging, the mouse was sacrificed for GFP imaging of metastatic cancer in the lymph nodes (Figure 6C) and organ biodistribution analysis by gamma counting (Figure 6D). The total accumulated dose in the metastatic lymph nodes was ~1.5 times higher than in counter-side, popliteal lymph nodes. Accumulation in metastatic lymph nodes decreased to ~9.1% ID/g, whereas the counter-side, popliteal lymph node remained at 35.4% ID/g. The overall lymph node accumulation level in this BALB/c, 4T1 model was lower than what was observed in the C57BL/6 and nude mice. High tumor accumulation (19.1% ID/g) was observed in the 4T1 tumor.
3.9 Lymph node gene delivery with S-LCP
The ability of S-LCP NPs to deliver genes to the lymph nodes was demonstrated using a plasmid containing RFP cDNA. An oligo-arginine peptide flanked by two cysteines (sequence: CR8C) that significantly enhances the gene-expression level in a study of LCP-mediated delivery of genes to hepatocytes was also used [18]. S-LCP loaded with the RFP plasmid, CR8C peptide, and 111In were prepared for this study. The cationic lipid, DOTAP (1,2-dioleoyl-3-trimethylammonium-propane), was included in this study for outer leaflet coating to compare with DOPC since DOTAP was known for its higher transfection efficiency. PEGylation at 20% was still used for both DOTAP and DOPC coated S-LCP NPs. As demonstrated in Figure 7A, S-LCP-DOTAP had high accumulation in the liver, but 111In gamma counting indicated much lower accumulation in the lymph nodes. On the other hand, S-LCP-DOPC showed accumulation that was low in the liver and spleen, but high in the lymph nodes.
Figure 7.

A) Biodistributions of S-LCP-DOPC and S-LCP-DOTAP with 20% PEGylation at 24 h post IV injection determined by gamma counting. RFP gene expression at the major organs (B) and the eight lymph nodes (C) were shown by RFP fluorescent imaging.
The level of RFP gene expression in liver was also high in mice injected with S-LCP-DOTAP, which correlated well with the accumulation levels (Figure 7B). For gene expression in the lymph nodes, although S-LCP-DOPC had a higher accumulation level, the RFP expression level was lower than that produced by injection with S-LCP-DOTAP (Figure 7C).
4 Discussion
The invention of the LCP illustrated that a small NP with a lipid bilayer coating could be created while also maintaining a well-PEGylated surface. LPD (Lipid/Polycation/DNA) NPs previously developed in our lab, also with a lipid-bilayer coating, had an average size around 100 nm [30]. Although there are extensive studies of NP biodistribution within this small size range, they are focused mainly on iron oxide and gold NPs, polymeric micelles, and quantum dots with different surface protection coatings [19, 27]. There is no literature that characterizes the in vivo biodistribution behavior of well-PEGylated, lipid-bilayer-coated NPs in such a small size range. To conduct these studies, an enlarged version of LCP (L-LCP) was thus developed and the in vivo performance of NPs of both sizes was evaluated for comparison.
When characterizing NP, it is important to ensure that impurities have been properly removed after preparation. Since excess lipid may form liposomes or micelles that could interfere with DLS analysis of LCP, purification using sucrose gradient centrifugation was necessary to ensure accurate characterization of size and zeta-potential. The same LCP purification method was also applied while determining 111In loading and DNA oligo entrapment efficiency.
In the in vivo SPECT/CT imaging study, the reduced accumulation of S-LCP in the MPS is comparable to many other NPs that are similar in size [19, 25, 27]. However, predominant systemic accumulation in the lymph nodes is rare after IV injection of NPs. For L-LCP, there was a small amount of accumulation in the lymph nodes that could be attributed to the presence of smaller NPs generated during the preparation of L-LCP cores (Figure 1A, Supplementary Figure 2). The comparison between S-LCP and L-LCP demonstrated that small size (~25 nm) is an important factor for lymphotropism
Twenty-four hours after the injection of 111In-S-LCP, over 80% of the total injected 111In dose was retained in the mouse (Figure 2C), suggesting elimination of S-LCP from the mouse body was very slow. Thus, the elimination of S-LCP from the blood could be due to redistribution throughout the body, presumably to the lymphatic system (as shown by the high accumulation in the carcass and lymph nodes, Fig 2C). This finding is in contrast to previously mentioned reference NPs [24, 25], which might be excreted after reaching the liver or kidney. Furthermore, these findings support our hypothesis that S-LCP could penetrate tissues, enter the lymphatic system, and efficiently accumulate in the lymph nodes via an atypical lymphatic drainage mechanism due to (1) small size (~25 nm), (2) a well-PEGylated lipid surface, and (3) a slightly negative surface charge. Lymphatic accumulation of the nanoparticles also explains the reduction in their concentration in the blood while maintaining minimal excretion and accumulation in other major organs (liver, spleen, etc.).
Since S-LCP was more able to penetrate tissues and was taken up minimally by the MPS due to their smaller size, one reasonable prediction is that the majority of S-LCP were drained into the lymph nodes as individual particles. On the other hand, the bulk of the accumulation of L-LCP in the lymph nodes after IM injection is most likely MPS mediated [31]. As shown in Figure 4, labeled S-LCP rarely overlapped with CD11c (dendritic cell marker) or CD11b (macrophage marker) staining. Since dendritic cells and macrophages were the two major phagocytic cells in the lymph nodes, our results suggested that S-LCP remained as individual particles in the lymph nodes. In the case of L-LCP, Texas-Red fluorescence overlapped mostly with CD11c and only partially with CD11b. Combined with the observation that IV injected L-LCP had low accumulation in the lymph nodes, this suggests that after IM injection, L-LCP accumulation in the draining lymph nodes was mainly mediated by MPS uptake at the injection site, after which the MPS cells migrated to the draining lymph nodes.
How the degree of PEGylation affects LCP lymphotropism is also interesting. Geometrically, NPs will have increased curvature as they become smaller in size. In order to provide a comparable degree of surface protection, a higher degree of PEGylation would be required by an S-LCP (~25 nm) than an L-LCP (~67 nm). However, smaller NPs might inherently possess a stealth property that allows them to avoid uptake by the MPS and thus require less PEGylation. The results in Figure 5 supported this hypothesis. It also suggested that PEGylation could significantly reduce uptake by the MPS in the liver, but not the spleen. Only when the NP size was reduced, could we achieve significant reduction in the amount of accumulation of the NPs in the spleen. This accumulation pattern also holds true for many other types of NPs in addition to the one described here [25, 27].
Even when PEGylated at 20% PEG, L-LCP exhibited much lower accumulation in the lymph nodes compared to S-LCP, (~21% vs. ~70% ID/g, respectively). The lower degree of accumulation in the lymph nodes could be explained by the population of smaller particles that was generated as a by-product during the process to create the L-LCP cores (Figure 1A). Alternatively, NPs could be carried by MPS cells in the periphery and migrated into the lymph nodes [31].
An uneven distribution of 111In in the metastatic lymph node (Figure 6B) was most likely due to the presence of the tumor mass that excluded lymphatic fluid, an observation that was also reported in other clinical studies using Feridex® [20, 32].
Due to enlargement of the metastatic lymph nodes caused by tumor growth and inflammation, the accumulation level was reduced to ~9.1% ID/g as a result of increased organ weight of the lymph nodes, whereas the counter-side, popliteal lymph node was measured at 35.4% ID/g (Figure 6D). However, the accumulated 111In signal was sufficient for imaging, leading to the observation of the uneven distribution pattern (Figure 6B) [32]. The overall lymph node accumulation in this BALB/c, 4T1 model was lower than what was observed in the C57BL/6 and nude mice. The decrease could be attributed to higher MPS function induced by the 4T1 tumor, as indicated by a significantly larger spleen in this model (data not shown). However, the 4T1 tumor achieved high tumor accumulation (19.1% ID/g), which might also contribute to the overall lower accumulation in the lymph nodes of this model. This work demonstrated the feasibility of using 111In-S-LCP to image metastatic lymph nodes via intravenous injection.
The high accumulation and RFP expression level with S-LCP-DOTAP in the liver (Figure 7A) was due to efficient uptake by hepatocytes but not Kupffer cells (as been discussed by Liu et al, manuscript in submission). For the observation that S-LCP-DOTAP has lower accumulation but higher RFP expression in the lymph node (Figure 7B), there are two possible explanations. The first depends on the high transfection efficiency of positively-charged DOTAP, due to its ability to facilitate endosome escape. The higher expression of RFP despite the lower delivered dose suggests that S-LCP-DOTAP exhibited a higher gene expression activity than S-LCP-DOPC. Alternatively, positively charged DOTAP could also aid in the cellular uptake process after PEG shedding [33–35]. Cellular uptake of S-LCP-DOPC in the lymph nodes might be limited (Figure 4). If cellular uptake was low, the bioavailability of the RFP plasmid is likely to be low as well.
Figure 3 suggests that local injection of non-PEGylated larger particles, such as non-PEGylated L-LCP, would be sufficient for vaccination or sentinel lymph node mapping since systemic accumulation and delivery to the lymph node is not required and local injection of larger particles actually achieved higher levels of local accumulation [36–42]. NPs for vaccination or sentinel lymph node mapping are readily achievable since they do not require a high degree of PEGylation. There is also a fairly broad, acceptable range of particle size. On the other hand, the more sophisticated S-LCP-DOPC would be more effective in achieving systemic lymphatic drug delivery, due to highly specific accumulation in the lymph nodes after IV injection. This characteristic would be desirable in the treatment of diseases such as HIV infection. The HIV virus in the lymph nodes is difficult to treat due at least in part to the limited ability of anti-HIV drugs to access the lymph nodes [43]. Strategies such as formulating anti-HIV drugs into lipid NPs to enhance the lymph-node-specific accumulation of anti-HIV drugs have been reported [44–47]. However, these formulations were administered by subcutaneous injection and could only enhance the concentration of drugs in regional lymph nodes. The lymphotropism of LCP, along with the ability of some anti-viral and anti-tumor nucleoside analogue drugs to be entrapped by a similar strategy [48], supports the use of LCP for delivery of nucleoside-analog drugs in the treatment of metastatic cancer and HIV infection in the lymph nodes.
Targeting ligands could be incorporated to enhance cell-type specific uptake. For instance, CD4-targeting peptides that could enhance cellular uptake by CD4-HIV host cells could be grafted onto S-LCP-DOPC [49]. With the lymphotropism of S-LCP-DOPC, systemic eradication of HIV infection in the lymph nodes might be possible. Future directions to improve the lymphatic drug delivery efficiency of LCP would include: 1) adding an endosome escape enhancer to the S-LCP-DOPC to boost gene expression activity, and 2) adding targeting ligands to enhance cell-type specific uptake in the lymph nodes. If successful, S-LCP-DOPC could serve as a drug delivery formulation that is highly specific to the lymph nodes.
5 Conclusion
We have demonstrated successful loading of 111In into LCP NPs. A rare lymphotropic property of S-LCP was studied. The results supported the hypotheses that S-LCP was able to penetrate into tissues, travel within the lymphatic system, and accumulate in the lymph nodes as individual NP due to 1) small size, 2) a well-PEGylated lipid surface, and 3) a slightly negative surface charge. With SPECT/CT, IV injected, lymphotropic S-LCP could be useful for imaging of systemic lymph node metastasis. Systemic gene/drug delivery to the lymph nodes has been demonstrated with RFP cDNA expression in lymph nodes. These results suggest the potential use of IV injected S-LCP formulation as a theranostic delivery system to the lymphatic system.
Supplementary Material
Acknowledgments
We thank the staff at Biomedical Research Imaging Center, University of North Carolina, Chapel Hill for assisting the SPECT/CT imaging experiment. We also thank Dr. Andrew Wang for insightful discussion and Kelly Racette and Steven Plonk for editing the manuscript. Work supported by NIH grants CA129835, CA149363, and CA151652.
ABBREVIATIONS
- LCP
lipid/calcium/phosphate
- NP
nanoparticle
- Ksp
solubility product constant
- MPS
mononuclear phagocytic system
- ID/g
injected dose per gram tissue
- SPECT
single photon emission computer tomography
- PET
positron emission tomography
- USPIO
ultra-small superparamagnetic iron oxide
- DOPC
dioleoylphosphatidylcholine
- DOPA
dioleoylphosphatidic acid
- CaP
calcium phosphate
- DSPE-PEG2000
(1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-poly(ethylene glycol)2000)
- LPD
Lipid/Polycation/DNA
- CT
computed tomography
- NBD-PC
1-Oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-Glycero-3-Phosphocholine
- DLS
dynamic light scattering
- PDI
polydispersity index
- PK
pharmacokinetics
- GFP
green fluorescent protein
- RFP
red fluorescent protein
Footnotes
Author contributions
YC.T. designed and prepared all the nanoparticles, performed most of the experiments, and wrote the manuscript. Z.X. helped with the lymph node RFP cDNA delivery experiment. K.G. performed SPEC/CT imaging. H.Y. provided advice on the SPECT/CT imaging study. L.H. supervised the project and contributed to writing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xie Y, Bagby TR, Cohen MS, Forrest ML. Drug delivery to the lymphatic system: importance in future cancer diagnosis and therapies. Expert Opin Drug Deliv. 2009;6:785–92. doi: 10.1517/17425240903085128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 3.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 4.Sleeman JP. The relationship between tumors and the lymphatics: what more is there to know? Lymphology. 2006;39:62–8. [PubMed] [Google Scholar]
- 5.Porter CJ. Drug delivery to the lymphatic system. Crit Rev Ther Drug Carrier Syst. 1997;14:333–93. [PubMed] [Google Scholar]
- 6.Reddy LH, Murthy RS. Pharmacokinetics and biodistribution studies of Doxorubicin loaded poly(butyl cyanoacrylate) nanoparticles synthesized by two different techniques. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2004;148:161–6. doi: 10.5507/bp.2004.029. [DOI] [PubMed] [Google Scholar]
- 7.Kaminskas LM, Porter CJ. Targeting the lymphatics using dendritic polymers (dendrimers) Adv Drug Deliv Rev. 2011;63:890–900. doi: 10.1016/j.addr.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev. 2011;63:901–8. doi: 10.1016/j.addr.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erogbogbo F, Yong KT, Roy I, Hu R, Law WC, Zhao W, et al. In vivo targeted cancer imaging, sentinel lymph node mapping and multi-channel imaging with biocompatible silicon nanocrystals. ACS Nano. 2011;5:413–23. doi: 10.1021/nn1018945. [DOI] [PubMed] [Google Scholar]
- 10.Tang L, Yang X, Dobrucki LW, Chaudhury I, Yin Q, Yao C, et al. Aptamer-functionalized, ultra-Small, monodisperse silica nanoconjugates for targeted dual-modal imaging of lymph nodes with metastatic tumors. Angew Chem Int Ed Engl. 2012;51:12721–6. doi: 10.1002/anie.201205271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Psimadas D, Georgoulias P, Valotassiou V, Loudos G. Molecular nanomedicine towards cancer: (111) In-labeled nanoparticles. J Pharm Sci. 2012;101:2271–80. doi: 10.1002/jps.23146. [DOI] [PubMed] [Google Scholar]
- 12.Kelkar SS, Reineke TM. Theranostics: combining imaging and therapy. Bioconjug Chem. 2011;22:1879–903. doi: 10.1021/bc200151q. [DOI] [PubMed] [Google Scholar]
- 13.Doane TL, Burda C. The unique role of nanoparticles in nanomedicine: imaging, drug delivery and therapy. Chem Soc Rev. 2012;41:2885–911. doi: 10.1039/c2cs15260f. [DOI] [PubMed] [Google Scholar]
- 14.Nam J, Won N, Bang J, Jin H, Park J, Jung S, et al. Surface engineering of inorganic nanoparticles for imaging and therapy. Adv Drug Deliv Rev. 2012;65:622–48. doi: 10.1016/j.addr.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Controlled Release. 2012;158:108–14. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Hu Y, Wang Y, Li J, Liu F, Huang L. Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung caner. Mol Pharm. 2012;9:2280–9. doi: 10.1021/mp300152v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Li J, Liu F, Huang L. Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Mol Ther. 2012;20:609–15. doi: 10.1038/mt.2011.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Y, Haynes MT, Wang Y, Liu F, Huang L. A highly efficient synthetic vector: nonhydrodynamic delivery of DNA to hepatocyte nuclei in vivo. ACS Nano. 2013;7:5376–84. doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almeida JP, Chen AL, Foster A, Drezek R. In vivo biodistribution of nanoparticles. Nanomedicine (Lond) 2011;6:815–35. doi: 10.2217/nnm.11.79. [DOI] [PubMed] [Google Scholar]
- 20.Harisinghani MG, Barentsz J, Hahn PF, Deserno WM, Tabatabaei S, van de Kaa CH, et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N Engl J Med. 2003;348:2491–9. doi: 10.1056/NEJMoa022749. [DOI] [PubMed] [Google Scholar]
- 21.Harisinghani MG, Saksena MA, Hahn PF, King B, Kim J, Torabi MT, et al. Ferumoxtran-10-enhanced MR lymphangiography: does contrast-enhanced imaging alone suffice for accurate lymph node characterization? AJR Am J Roentgenol. 2006;186:144–8. doi: 10.2214/AJR.04.1287. [DOI] [PubMed] [Google Scholar]
- 22.Nakai G, Matsuki M, Harada T, Tanigawa N, Yamada T, Barentsz J, et al. Evaluation of axillary lymph nodes by diffusion-weighted MRI using ultrasmall superparamagnetic iron oxide in patients with breast cancer: initial clinical experience. J Magn Reson Imaging. 2011;34:557–62. doi: 10.1002/jmri.22651. [DOI] [PubMed] [Google Scholar]
- 23.Xiong L, Shuhendler AJ, Rao J. Self-luminescing BRET-FRET near-infrared dots for in vivo lymph-node mapping and tumour imaging. Nat Commun. 2012;3:1193. doi: 10.1038/ncomms2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong H, Dube N, Shu JY, Seo JW, Mahakian LM, Ferrara KW, et al. Long-circulating 15 nm micelles based on amphiphilic 3-helix peptide-PEG conjugates. ACS Nano. 2012;6:5320–9. doi: 10.1021/nn301142r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Liu Y, Luehmann H, Xia X, Brown P, Jarreau C, et al. Evaluating the pharmacokinetics and in vivo cancer targeting capability of au nanocages by positron emission tomography imaging. ACS Nano. 2012;6:5880–8. doi: 10.1021/nn300464r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham MJ, Freier SM, Crooke RM, Ecker DJ, Maslova RN, Lesnik EA. Tritium labeling of antisense oligonucleotides by exchange with tritiated water. Nucleic Acids Res. 1993;21:3737–43. doi: 10.1093/nar/21.16.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, et al. Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials. 2009;30:1928–36. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–76. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Zhang F, Lee S, Swierczewska M, Kiesewetter DO, Lang L, et al. Long-term multimodal imaging of tumor draining sentinel lymph nodes using mesoporous silica-based nanoprobes. Biomaterials. 2012;33:4370–8. doi: 10.1016/j.biomaterials.2012.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li SD, Chono S, Huang L. Efficient gene silencing in metastatic tumor by siRNA formulated in surface-modified nanoparticles. J Controlled Release. 2008;126:77–84. doi: 10.1016/j.jconrel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mou Y, Hou Y, Chen B, Hua Z, Zhang Y, Xie H, et al. In vivo migration of dendritic cells labeled with synthetic superparamagnetic iron oxide. Int J Nanomedicine. 2011;6:2633–40. doi: 10.2147/IJN.S24307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang F, Zhu L, Huang X, Niu G, Chen X. Differentiation of reactive and tumor metastatic lymph nodes with diffusion-weighted and SPIO-enhanced MRI. Mol Imaging Biol. 2012;15:40–7. doi: 10.1007/s11307-012-0562-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romberg B, Hennink WE, Storm G. Sheddable coatings for long-circulating nanoparticles. Pharm Res. 2008;25:55–71. doi: 10.1007/s11095-007-9348-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Controlled Release. 2010;145:178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721–31. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keidar Z, Israel O, Krausz Y. SPECT/CT in tumor imaging: technical aspects and clinical applications. Semin Nucl Med. 2003;33:205–18. doi: 10.1053/snuc.2003.127310. [DOI] [PubMed] [Google Scholar]
- 37.Kim W, Menda Y, Willis J, Bartel TB, Graham MM. Use of lymphoscintigraphy with SPECT/CT for sentinel node localization in a case of vaginal melanoma. Clin Nucl Med. 2006;31:201–2. doi: 10.1097/01.rlu.0000204201.69676.31. [DOI] [PubMed] [Google Scholar]
- 38.Ishihara T, Kaguchi A, Matsushita S, Shiraishi S, Tomiguchi S, Yamashita Y, et al. Management of sentinel lymph nodes in malignant skin tumors using dynamic lymphoscintigraphy and the single-photon-emission computed tomography/computed tomography combined system. Int J Clin Oncol. 2006;11:214–20. doi: 10.1007/s10147-005-0554-1. [DOI] [PubMed] [Google Scholar]
- 39.Khafif A, Schneebaum S, Fliss DM, Lerman H, Metser U, Ben-Yosef R, et al. Lymphoscintigraphy for sentinel node mapping using a hybrid single photon emission CT (SPECT)/CT system in oral cavity squamous cell carcinoma. Head Neck. 2006;28:874–9. doi: 10.1002/hed.20434. [DOI] [PubMed] [Google Scholar]
- 40.Even-Sapir E, Lerman H, Lievshitz G, Khafif A, Fliss DM, Schwartz A, et al. Lymphoscintigraphy for sentinel node mapping using a hybrid SPECT/CT system. J Nucl Med. 2003;44:1413–20. [PubMed] [Google Scholar]
- 41.Kizu H, Takayama T, Fukuda M, Egawa M, Tsushima H, Yamada M, et al. Fusion of SPECT and multidetector CT images for accurate localization of pelvic sentinel lymph nodes in prostate cancer patients. J Nucl Med Technol. 2005;33:78–82. [PubMed] [Google Scholar]
- 42.Sherif A, Garske U, de la Torre M, Thorn M. Hybrid SPECT-CT: an additional technique for sentinel node detection of patients with invasive bladder cancer. Eur Urol. 2006;50:83–91. doi: 10.1016/j.eururo.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Cavert W, Notermans DW, Staskus K, Wietgrefe SW, Zupancic M, Gebhard K, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science. 1997;276:960–4. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 44.Desormeaux A, Bergeron MG. Lymphoid tissue targeting of anti-HIV drugs using liposomes. Methods Enzymol. 2005;391:330–51. doi: 10.1016/S0076-6879(05)91019-7. [DOI] [PubMed] [Google Scholar]
- 45.Kinman L, Brodie SJ, Tsai CC, Bui T, Larsen K, Schmidt A, et al. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr. 2003;34:387–97. doi: 10.1097/00126334-200312010-00005. [DOI] [PubMed] [Google Scholar]
- 46.Gagne JF, Desormeaux A, Perron S, Tremblay MJ, Bergeron MG. Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. Biochim Biophys Acta. 2002;1558:198–210. doi: 10.1016/s0005-2736(01)00432-1. [DOI] [PubMed] [Google Scholar]
- 47.Kinman L, Bui T, Larsen K, Tsai CC, Anderson D, Morton WR, et al. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr. 2006;42:155–61. doi: 10.1097/01.qai.0000214822.33905.87. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Schwerbrock NM, Rogers AB, Kim WY, Huang L. Codelivery of VEGF siRNA and gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol Ther. 2013;21:1559–69. doi: 10.1038/mt.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Endsley AN, Ho RJ. Enhanced anti-HIV efficacy of indinavir after inclusion in CD4 targeted lipid nanoparticles. J Acquir Immune Defic Syndr. 2012;61:417–24. doi: 10.1097/QAI.0b013e3182653c1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.