

Abstract
Chronic T cell responses, as they occur in rheumatoid arthritis, are complex and are likely to involve many mechanisms. There is a growing body of evidence that, in concert with the T cell antigen receptor signal, CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4; CD152) are the primary regulators of T cell responses. Whereas CD28 primarily activates T cell processes, CTLA-4 inhibits them. The mechanism for this dichotomy is not fully understood, especially as CD28 and CTLA-4 recruit similar signalling molecules. In addition, recent studies demonstrate that CD28 and CTLA-4 have multiple functions during T cell responses. In particular, CTLA-4 exerts independent distinct effects during different phases of T cell responses that could be exploited for the treatment of rheumatoid arthritis.
Keywords: CD152, costimulation, CTLA-4Ig, inflammation, polymorphism, signal transduction
Introduction
A broad repertoire of mature effector T cells with specific, diverse functional capabilities is generated in adaptive immune responses. The differentiation and regulation of these diverse effector cells have to be tightly regulated and controlled to avoid unwanted immune responses. The T cell antigen receptor (TCR) alone provides insufficient signals for optimal T cell stimulation. A second costimulatory signal for optimal T cell stimulation is needed. Critical for the generation and control of functional diversity are the costimulatory signals provided during the antigen-specific stimulation of T cells by antigen-presenting cells (APCs) [1]. The quality and magnitude of an antigen-specific immune response are determined not only by the quality of positive costimulation but also by the integration of the absence of positive costimulatory signals and the presence of negative costimulatory signals. CD28 and cytotoxic T lymphocyte antigen-4 (CTLA-4; CD152), two homologous members of the immunoglobulin superfamily, are the key receptors for this regulation via positive and negative costimulation [2]. These receptors and their pathways therefore provide promising therapeutic targets for modulating immune responses.
Expression of CD28, CTLA-4 and their ligands
CD28 and CTLA-4 bind to the same ligands, CD86 (B7-2) and CD80 (B7-1), which are expressed almost exclusively on bone marrow-derived APCs. This restricted expression of the ligands ensures that regulation of T-lymphocyte responses by the T cell molecules CD28 and CTLA-4 is exerted only by specialized, professional APCs. CD80 and CD86 have different kinetics of expression. CD86 is constitutively expressed on dendritic cells, macrophages, and B cells and is further upregulated upon activation [3] (D Gärtner and MC Brunner-Weinzierl, unpublished observation). CD80 is absent from resting cells and is expressed only upon activation of the APC. Because only CD86 is expressed on the cell surface early after the activation of APCs, it is indispensable during the initiation phase of the immune response, which is demonstrated by data showing altered immune regulation in CD86 knockout mice [4]. CD80 and CD86 have overlapping functions [5], despite different binding determinants, different dissociation kinetics, and different binding affinities for CD28 and CTLA-4. Differences in their functions seem to be due to different kinetics of expression on different cell types. The crystal structures and an analysis of the binding affinities and kinetics of CTLA-4 with its ligands suggest that CD86 monomers bind to CTLA-4 dimers, whereas CD80 dimers bind two adjacent bivalent CTLA-4 dimers, building a lattice-like network (Fig. 1) [6-8]. In contrast, CD28 is monovalent and only able to bind a single CD80 or CD86 molecule. Interestingly, CTLA-4 has a much higher binding affinity for CD86 and CD80 than CD28 [9]. At 0.2 μM, the affinity of the CTLA-4-CD80 interaction is one of the highest described for surface molecules.
Figure 1.
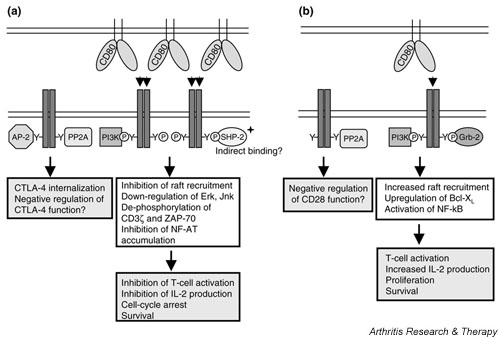
CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) recruit similar and distinct signalling molecules. (a) The unphosphorylated CTLA-4 molecule binds the medium-chain subunit of the clathrin adaptor AP-2. This interaction leads to a rapid internalization of CTLA-4 and tight regulation of surface CTLA-4. CTLA-4 is also able to bind the serine/threonine phosphatase protein phosphatase 2A (PP2A). PP2A seems to act as a negative regulator of CTLA-4 function and dissociates from CTLA-4 upon ligand binding. The ligands are the dimeric CD80 and the monomeric CD86. Binding of CD80 to the divalent CTLA-4 leads to the formation of a lattice-like structure on the cell surface. This pattern formation cannot occur by the interaction of CD86 with CTLA-4. Activation of CTLA-4 by binding to its ligands leads to the phosphorylation of tyrosine residues in the cytoplasmic tail of CTLA-4 and its association with phosphoinositide 3-kinase (PI-3K) and (perhaps indirectly) the tyrosine phosphatase SHP-2. The immediate consequences of these interactions are unclear but eventually lead to an inhibition of T cell activation. This includes decreased raft recruitment to the plasma membrane, decreased phosphorylation of CD3-ζ and ZAP-70, downregulation of mitogen-activated protein kinases such as extracellular signal-related kinase and c-Jun N-terminal kinase, and inhibition of the nuclear translocation of the transcription factors AP-1 and nuclear factor of activated T-cells (NFAT). This results in decreased interleukin (IL)-2 production and cell-cycle arrest. (b) In its unphosphorylated state CD28 binds the serine/threonine phosphatase PP2A. CD28 shares the same ligands, CD80 and CD86, as CTLA-4. However, because CD28 is monovalent it is not able to form higher-order structures after interaction with CD80. The tyrosine phosphorylation of CD28 after stimulation by CD80 or CD86 is followed by the association of PI-3K and Grb-2 to the cytoplasmic tail of CD28. This leads to increased T cell activation, indicated by enhanced raft expression and upregulated production of IL-2. The increased survival is a consequence of upregulated Bcl-XL and the activation of nuclear factor (NF)-κB
CD28 is constitutively expressed on naive CD4+ T cells and is slightly upregulated after T cell activation. The expression of CTLA-4 mRNA is detectable in naive T cells within 1 hour after activation [10]. After activation of the T cell, intracellular CTLA-4 protein increases steadily in concentration and is stored in vesicles. Intracellular CTLA-4 protein is still detectable after a resting period of a week [11]. Because CTLA-4 protein can be detected intracellularly 24–48 hours after the onset of T cell activation, it has been suggested that the molecule is probably also expressed on the cell surface of these cells and is functional. Surface expression of CTLA-4 does not peak until 48–72 hours after T cell stimulation, and it has recently been shown that only a fraction of activated T cells express it on the cell surface [11]. The localization of CTLA-4 on the cell surface is regulated by the association of clathrin-coated pit adaptor protein AP-2 with the intracellular tyrosine-based motif of CTLA-4 (Fig. 1) [12,13]. CTLA-4 molecules are mobilized toward the sites of antigen receptor engagement and are probably displayed at the immunological synapse [14,15].
The highly restricted regulation of CTLA-4 localization in a cell suggests that the restricted surface expression of CTLA-4 is a major control point for the regulation of the inhibitory function of CTLA-4 on T cells. A quantitative increase in the surface expression of CTLA-4 has been suggested to correlate with the number of cell cycles [16]. This would imply that the transcription machinery of the CTLA-4 gene would have the ability to count cell cycles and convert this information into expression. However, using a sensitive detection method to detect CTLA-4 on the surface of activated T cells with a sensitivity of less than 200 molecules per cell, it can be shown that expression is independent of the proliferative history of the cell and is exclusively dependent on the time elapsed since the onset of activation of the T cell [11]. The expression of surface CTLA-4 is not correlated with the proliferative history, nor is proliferation a mandatory prerequisite for CTLA-4 expression. Furthermore, the instruction for a T cell to express surface CTLA-4 2 days after the onset of the activation requires less than 12 hours of T cell stimulation, implying that the induction of CTLA-4 surface expression and its function can happen at distinct sites in the body. This result has major implications for the response of activated T cells, because the cells that receive the instruction to express surface CTLA-4 in this time window will eventually express CTLA-4, with all the consequences.
Surface CTLA-4 is rarely expressed on activated primary CD4+ T cells and is expressed at higher frequencies after restimulation [11], indicating that it is an important regulator of responses of antigen-experienced T cells. This was confirmed by comparing the T cell responses of monospecific CTLA-4-/- and CTLA-4+/+ cells [17]. No difference between these populations was detectable during a primary response, but there was enhanced expansion of CTLA-4-/- T cells in a secondary response. After initial activation, naive CD4+ cells differentiate into Th1 and Th2 cells, which secrete distinct sets of cytokines. Studies on CTLA-4 expression of differentiated Th1 and Th2 cells have been performed mostly in Th1 and Th2 long-term T cell clones [18]. With the use of a conventional detection method, it could be shown that Th2 clones express surface CTLA-4, whereas the protein was undetectable on Th1 clones. Hence, the differentiation history of an activated naive T cell apparently correlates with CTLA-4 surface expression after re-encountering an antigen. This also implies that mainly antigen-experienced T cells express surface CTLA-4 and are probably regulated by it [17-19]. This fact is of particular interest for already established T cell responses driven by antigen-experienced T cells as they occur during chronic immunopathology.
Controlled T cell activation by CD28 and CTLA-4
CD28 and CTLA-4 have distinct functions during T cell activation. Triggering of CD28 enhances raft accumulation and the accumulation of transcription factors, such as AP-1 and nuclear factor of activated T cells (NFAT), in the nucleus; this strongly upregulates the initiation of interleukin (IL)-2 transcription [20]. CD28 also enhances the mRNA stability of cytokine genes, for example IL-2 and interferon-γ [21], as well as the expression of G1-kinases, a prerequisite for cell cycling. In addition, the induction of IL-2 leads to autocrine support for the activation of the cell cycle machinery; the IL-2 signal induces the degradation of the cell cycle inhibitor p27 and expression of G1-kinases, which ultimately leads to T cell proliferation [22].
CTLA-4 seems to be an important downregulator of T cell activation. As early as 4 hours after the onset of T cell activation, crosslinking of CTLA-4 by specific antibodies shows that it is expressed functionally by at least some T cells and prevents complete T cell activation [22]. The main effect of CTLA-4 engagement during T cell activation is probably the inhibition of transcription of the IL-2 gene by preventing NFAT translocation to the nucleus [22]. This might just be a consequence of the prevention of T cell activation in general; nevertheless, CTLA-4 also directly inhibits the expression of key components of the cell cycle machinery such as cyclin D3, cyclin-dependent kinase (Cdk)4, and Cdk6, which are partly IL-2 dependent and partly upregulated independently. The expression of activation-induced molecules such as CD69 and CD25 is also prevented by crosslinking of CTLA-4 [23].
So far, CTLA-4 protein has not been detected on the cell surface of naive or resting CD4+ T cells. But even small amounts of CTLA-4 could potentially inhibit T cell activation when CD80/CD86 molecules are expressed at low levels because CTLA-4 has a much higher affinity than CD28 for CD80/CD86 [2]; moreover, it is preferentially localized in lipid rafts [24]. According to earlier results, CTLA-4 mRNA was detectable in naive CD4+ T cells [25], but enhanced surface staining for CTLA-4 performed on naive T cells did not detect CTLA-4 protein [11]. The surface CTLA-4 is therefore expressed at very low concentrations (fewer than 100–200 CTLA-4 molecules per cell) on naive T cells and functional at this expression level during early T cell receptor triggering, or surface CTLA-4 is quickly and shortly upregulated after CD4+ T cell activation, at least from some cells, which has been reported for other molecules such as IL-4 [26-30].
Signal transduction of CD28 and CTLA-4
The mechanisms by which CD28 and CTLA-4 transmit their respective signals are not well understood. Despite their opposing roles in T cell function, both molecules share some basic features. It has been shown that not only do CD28 and CTLA-4 compete for the ligands CD80 and CD86 [22,23] but both also initiate signalling pathways. However, both molecules lack intrinsic catalytic activity in their cytoplasmic tails and they therefore require association with further signalling molecules. Despite their opposing functions during T cell responses, CD28 and CTLA-4 interact with identical signalling molecules: the phosphoinositide 3-kinase (PI-3K) and the protein phosphatase 2A (PP2A) (Fig. 1) [31-34]. However, the functional relevance and consequences of these shared properties are not well understood.
It is still controversial whether CD28 transmits a unique signal or only amplifies TCR signals. After engagement of CD28 by its ligand, tyrosine residues in the cytoplasmic tail of CD28 become phosphorylated by Src-family kinases [35], leading to the binding of PI-3K to CD28 [31,32]. Additionally, CD28 triggering induces the phosphorylation and activation of the kinases Tec and Itk [36,37] as well as other signalling molecules such as the guanine-nucleotide-exchange factor Vav-1 or phospholipase Cγ1 [38]. All of these molecules are also activated by TCR signalling, so CD28 might only be an amplifier. A unique signal could arise from the dependence of full phospholipase Cγ1 activation on a signal provided by CD28 that involves PI-3K, Vav-1, and the adapter molecule SLP-76 [39].
In another model, CD28 sets the threshold for T cell activation and amplifies the TCR signal by enhancing the recruitment of lipid rafts to the plasma membrane [40,41]. In resting/naive cells, lipid rafts are stored in intracellular vesicles and are redistributed to the plasma membrane after stimulation. This redistribution is strongly enhanced by CD28 and facilitates the full signal leading to T cell activation. However, the signal required for raft relocalization is unknown at present.
Indications for both the quantitative and the qualitative signal mediated by CD28 can be derived from the analysis of gene expression after stimulation with TCR alone, CD28 alone, or a combination of TCR and CD28 [42]. This study shows that CD28 acts primarily as a signal amplifier of TCR signalling but also leads to the activation of a few, though important, distinct genes (such as CD69 and tumor necrosis factor).
Like CD28, CTLA-4 becomes phosphorylated on tyrosine residues after stimulation, which is mediated by Src-family kinases, JAK-2 or Rlk [43-45]. The tyrosine residue is located within a YVKM motif and this has been shown to serve as the binding site for several molecules (Fig. 1). In its unphosphorylated state this motif is bound to the medium-chain subunit AP-50 of the AP-2 clathrin adapter [12,13], leading to the rapid endocytosis of CTLA-4. In contrast, tyrosine phosphorylation results in the surface retention of CTLA-4 and the binding of PI-3K to the YVKM motif [33]. It has been also described that CTLA-4 can be found in a complex together with CD3ζ and the tyrosine phosphatase SHP-2 [46-48]. The direct interaction between SHP-2 and the signalling molecule CD3ζ is thought to be a mechanism by which CTLA-4 downregulates TCR signalling. This could also explain the observation that CD3ζ is hyperphosphorylated in CTLA-4 knockout mice [46]. However, the crosslinking of CTLA-4 in combination with TCR and CD28 did not lead to a decreased phosphorylation of CD3ζ [49]. In addition, our own results, gained by the retroviral transduction of SHP-2 mutants into primary T cells, do not support the idea of a prominent contribution of SHP-2 in CTLA-4 signalling (H Hoff and MC Brunner-Weinzierl, unpublished observation).
A second phosphatase that has been shown to interact with CTLA-4 is the serine/threonine phosphatase PP2A [34,50]. Because PP2A has been described as a negative regulator for the mitogen-activated protein kinases extracellular signal-related kinase and c-Jun N-terminal kinase, and these molecules are downregulated after CTLA-4 engagement [49], PP2A might serve as the mediator for these downstream effects of CTLA-4. However, so far only the opposite role for PP2A as a negative regulator for CTLA-4 function has been described [50].
CTLA-4 is also able to interfere with raft recruitment to the plasma membrane. It has been shown that CTLA-4 can be found in lipid rafts [24] and is able to suppress raft aggregation mediated by TCR and CD28 [51]. This mechanism would account for a general downregulation of early T cell activation events by CTLA-4, such as a lack of NFAT translocation to the nucleus and IL-2 gene transcription but would dismiss further downstream specific CTLA-4 signals [22,42]. The nature of this specific signal is still unknown. Further studies should seek to analyze the integration of the CTLA-4 signal into the cell signalling machinery [11] on cells that have already formed rafts. We have recently reported that already upregulated molecules such as the α-chain of the IL-2 receptor cannot be downregulated by CTLA-4 on activated T cells [11], suggesting that the gene transcription of activated T cells, rather than the regulation of proteins, is altered by CTLA-4.
It is not yet clear whether CTLA-4 interferes with CD28 costimulation or with TCR stimulation. Most probably it interferes with both via the inhibition of raft accumulation, because it inhibits TCR-mediated effects such as the upregulation of cyclin-dependent kinases and CD28-mediated effects such as enhanced accumulation of NFAT in the nucleus [22]. However, the engagement of CTLA-4 does not interfere with the CD28-mediated stabilization of IL-2 mRNA [22].
Responses of already activated T cells
The control of T cells after a successful stimulation – whereby T cells accumulate rafts at the cell surface, produce growth factors such as IL-2, and proliferate – is still a matter of debate. CD28 does not exclusively provide costimulatory function on already activated T cells, because activated T cells also express other costimulatory molecules such as ICOS. However, constitutively expressed CD28 on T cells is needed to prolong T cell responses. This is indicated by data from CD28 knockout mice [52] in which immunization can initiate, but not sustain, T cell responses.
Detectable CTLA-4 surface expression does not peak until 48–72 hours after the onset of T cell activation, when it probably exerts its main function. Most studies indicate that triggering of CTLA-4 downregulates the proliferation and cytokine production of the entire T cell population, but this conclusion is probably due to difficulties in detecting surface-expressed CTLA-4 [22,23,47,53-55]. Applying a highly sensitive detection method for surface molecules, we showed recently that at all time points after the onset of an antigen-specific T cell response, CTLA-4 expression was limited to a minority of activated cells with a maximum frequency of surface CTLA-4+ T cells at 48 hours [11]. It has been shown that CTLA-4 expression needs TCR signaling and is synergistically enhanced by CD28 and IL-2 signals, which are undoubtedly stochastic components of the strength of activation of T cells likely to be involved [14,56]. In addition, Allison's group has shown by microscopy that CTLA-4 traffics differentially to the immunological synapse depending on the strength of the signal [15], suggesting that CTLA-4 inhibits some T cell clones with a high-affinity TCR by decreasing their competitive advantage over clones with a low-affinity TCR [57]. Preferential inhibition of T cells with a high-affinity TCR would prevent these clones from dominating the response during early stages and would thereby help to maintain the diversity of antigen-specific cells.
The functional consequence of the heterogeneous surface expression of CTLA-4 was demonstrated only recently when highly activated proliferating T cell populations were separated on the basis of surface CTLA-4 expression and restimulated [11]. The CTLA-4-expressing cells did not divide at all, whereas all CTLA-4- cells went through at least one more cell cycle. The inhibition of proliferation was mediated by CTLA-4 engagement during restimulation of the CTLA-4+ T cells as shown by CTLA-4 blockade with specific Fab fragments. No difference in the proliferative response was seen when CTLA-4 was blocked in isolated restimulated CTLA-4- T cells. Thus, the diversity of clonal T cell proliferation is mediated by the differential expression of CTLA-4 on the cell surface of activated individual T lymphocytes. This raises the possibility that surface CTLA-4-expressing cells might also have heterogeneous fates. It will be important to determine whether the surface expression of CTLA-4 restricts only the expansion of T cells that receive a strong signal or whether surface CTLA-4-expressing cells represent a distinct pool of memory T cells [11,15].
Control of apoptosis by CD28 and CTLA-4
The decision between the survival and apoptosis of T cells is of particular importance for adaptive immune responses to ensure that a defined number of specialized T cells remain in the organism, thus maintaining memory and homeostasis. The primary form of apoptosis of clonally expanded T cells is activation-induced cell death (AICD), which is controlled mainly by the Fas (CD95) system [58,59]. Despite the apparently opposing roles of CD28 and CTLA-4 on T cell functions, synergistic signal transduction is still a possibility because of their similar recruitment of signalling molecules such as PI-3K as described above [33]. PI-3K is an important signalling node for activating survival pathways via Akt activation [60,61]. CD28-mediated inhibition of AICD has been associated with the upregulation of cellular FLICE-inhibitory protein (c-FLIP) and Bcl-xL and with the inhibition of FasL expression [62]. Because the upregulation of apoptosis-inducing molecules is activation dependent, CTLA-4 crosslinking during T cell activation prevents T cell activation rather than terminating AICD. Thus, these unactivated or incompletely activated T cells are not prone to AICD and do not upregulate FasL [63].
CTLA-4 ligation in previously activated concanavalin A-induced blasts or anti-CD3-stimulated T cells has been suggested to induce apoptosis, thus terminating the T cell response [16,64]. This would mean that activated T cells are stopped by CTLA-4 from proliferating just to be eliminated by apoptosis, which would happen anyway by AICD. We observed that resistance to AICD is mediated by CTLA-4 on already activated Th cells. This CTLA-4-induced resistance is dependent on the suppression of the Fas system and is mediated by PI-3K [65]. This activity of CTLA-4 could explain the observation that Rag2-deficient mice reconstituted with a mixture of CTLA-4+/+ and CTLA-4-/- T cells do not show enhanced, but rather decreased, total numbers of lymphocytes after infection with lymphocytic choriomeningitis virus and Leishmania major [66]. This surprising observation indicates that CTLA-4 affects T cell survival not only in a non-autonomous fashion but eventually also by modulating the expression of a proapoptotic factor [66].
Indirect inhibitory effects of CTLA-4
The CTLA-4 knockout mouse shows a dramatic phenotype [2,54]. It develops a lymphoproliferative disease and dies at 4–5 weeks of age. But bone marrow chimeras derived from CTLA-4+/+ and CTLA-4-/- cells do not show the lymphoproliferative disorder known from CTLA-4 knockout mice, suggesting that CTLA-4-mediated inhibition is at least not only cell autonomous [67]. Non-autonomous indirect effects of CTLA-4 have been suggested, such as the possibility that tolerance induction by CTLA-4 might actually work via the APC. CTLA-4 crosslinking of its ligands CD80/CD86 on the surface of dendritic cells makes them the principal mediator of inhibition [68]. Ligation of CD80/CD86 induces the production of indoleamine 2,3-dioxygenase, which breaks down tryptophan. The absence of tryptophan mediates the downregulation of T cell activation. This mechanism is not completely understood; for example, interferon-γ is obligatory for its induction, which would mean that any Th1 response could initiate similar effects. Indirect inhibitory effects have been described involving the induction of transforming growth factor-β expression by CTLA-4; this has not been confirmed by others [69,70].
Other indirect inhibitory effects mediated by CTLA-4 are attributed to Treg cells, which express large amounts of intracellular CTLA-4 concomitantly with CD25 and show prolonged surface expression of CTLA-4 after activation [71]. It is still controversial whether CTLA-4 is needed for Treg cell effector function. On the one hand, CTLA-4-/- Treg cells are able to downregulate the activation of target cells; on the other, blockade of CTLA-4 abrogates the inhibitory function of Treg cells [72]. Interestingly, naive T cells, converted to Treg cells by retroviral transduction with the transcription factor FoxP3, show high expression of CTLA-4 [73]. Because CD25 is apparently only a surrogate marker for Treg cells, and the transcription marker FoxP3 is expressed only intracellularly, prolonged expression of surface CTLA-4 could be a good marker for identifying Treg cells viable for the autologous cell therapy of chronic inflammations.
Polymorphisms of CTLA-4 in rheumatoid arthritis (RA) and other autoimmune diseases
The human CD28 and CTLA-4 genes map to chromosome 2q33 and are separated by about 60 kilobases. The homologies between CD28 and CTLA-4 strongly suggest that both genes arose by gene duplication. High evolutionary pressure, especially on the CTLA-4 gene, is demonstrated by comparing human and mouse sequences: the homology of the DNA sequence is 78%, and that of the protein sequence is 74%. Nevertheless, four polymorphisms of the CTLA-4 gene have been identified in humans. There is a C→T transition at position -318 of the promoter sequence and a G→A transition at position +49 of exon 1, resulting in an alanine to threonine amino acid substitution in codon 17 of the leader peptide. A third polymorphism is a dinucleotide repeat of about 7–32 ATs in exon 3, and a fourth has been mapped to the 3' untranslated region of the CTLA-4 gene. All four polymorphisms have been investigated for linkage with autoimmune diseases.
The functional consequences have been described for some of these polymorphisms. For example, T cells from people carrying the G allele at position +49 showed increased proliferation in combination with a lower expression of CTLA-4 on T cells [74,75], whereas people carrying the protective A allele of the CTLA-4 gene have an increased expression of CTLA-4 on T cells and decreased proliferative capacity [76]. This suggests that carrying the susceptible G allele of CTLA-4 will result in a loss of peripheral tolerance, leading to autoimmune pathology.
The studies analyzing the possible association between a CTLA-4 polymorphism and an autoimmune disease vary greatly in their outcome. In type 1 diabetes, most studies indicate that the occurrence of the G allele in position +49 constitutes a risk factor, whereas the AA genotype is protective [77-80]. This linkage to disease was found in Italian, Romanian, Chinese, and German people. In contrast, others did not find a linkage between CTLA-4 polymorphisms and type 1 diabetes in French and Czech populations [81,82]. For multiple sclerosis, most studies showed no indication for a contribution of CTLA-4 polymorphisms at positions -318 and +49 as a disease risk factor in Canadian, Polish, Finnish, and Dutch populations [83-86], whereas others found an association between the G allele of the CTLA-4 gene at position +49 and the severity of multiple sclerosis in Swedish and German patients [87,88].
A recent study in mice identified a disease-susceptibility polymorphism of the CTLA-4 gene affecting CTLA-4 splicing in exon 2. An A→G transition leads to the skipping of exon 2, resulting in an increase in the expression of a ligand-independent isoform of CTLA-4 [89]. In humans, a new CTLA-4 polymorphism was found in the 3' untranslated region 2 kilobases upstream of the stop codon of CTLA-4 [88]. An A→G transition is associated with autoimmune diseases such as Grave's disease, type 1 diabetes, and autoimmune hypothyroidism [89]. This polymorphism of the CTLA-4 gene affects the splicing of CTLA-4 mRNA; interestingly, this results in a lower expression of the soluble form of CTLA-4 mRNA. The authors of the study speculate that a reduced interaction of B7 and soluble CTLA-4 might lead to enhanced T cell stimulation. However, the functional consequences of these findings are still unknown.
The contribution of CTLA-4 polymorphisms to the risk of developing RA is still controversial. Whereas some studies show no association of the CTLA-4 polymorphism in people from Spain, the UK, and Korea [90-92], others show CTLA-4 as a disease risk factor in Spanish and Chinese populations [93-95]. More detailed studies combining the CTLA-4 polymorphisms with the HLA genotype of patients found a correlation between the G allele of CTLA-4 (+49) and the HLA genotype HLA-DRB1, known to be a susceptibility gene for RA [96-99]. This correlation was found in German, Japanese, French, Italian, and Portuguese populations. This finding stresses the point that the inheritance of autoimmune diseases are most probably due to multiple susceptible genes and also to environmental factors. Thus, minor susceptibility loci are difficult to identify but still modify risk.
The multiple-function model
Taken together, the new insights into the functional consequences of CTLA-4 engagement allow the proposal of a new model of three distinct functions of CTLA-4 that might be relevant under different circumstances (Fig. 2). First, CTLA-4 sets the threshold for T cell activation, and thus probably contributes to maintenance of peripheral tolerance [100]. However, the observation that the expression of surface CTLA-4 after the activation of T cells is detectable on proliferating cells with an activated phenotype indicates that during an optimal T cell response the CTLA-4-mediated inhibition of early T cell activation is dispensable [11]. Second, whereas only a fraction of activated T cells express CTLA-4 at the cell surface, CTLA-4 has additional functions in already activated T cells: (1) to restrain T cell proliferation and (2) to initiate the survival of T cells. Cells that express a CTLA-4 signal will be inhibited in their proliferation and survive, whereas cells that do not express CTLA-4 will exhibit a brief spurt of enhanced proliferation to eliminate foreign pathogens and will then die, ensuring that the response is stopped. A fraction of surviving cells at the end of the immune response are potential progenitors for memory cells.
Figure 2.
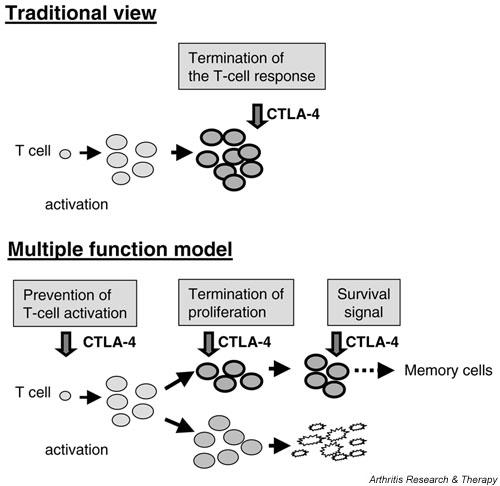
Multiple-function model for cytotoxic T-lymphocyte antigen-4 (CTLA-4). The traditional view of the function of CTLA-4 is that it is upregulated upon stimulation of the T cells and attenuates the response (top). The newly proposed model puts together new insights into CTLA-4 functions (bottom). (1) During suboptimal T cell activation, CTLA-4 sets the threshold for activation. (2) Already activated T cells are inhibited in their proliferation by CTLA-4. (3) CTLA-4 signalling enhances PI-3K function, triggering cell-autonomous survival signals in already activated T cells. Surviving cells at the end of an immune response could be prone to differentiation into memory cells.
Blockade of CD28 and CTLA-4 ligands by CTLA-4Ig during chronic immune responses
CTLA-4Ig is constructed by genetically fusing the external domain of human CTLA-4 to the heavy-chain constant region of human IgG1. CTLA-4Ig binds CD80 and CD86 on APCs, interfering with B7/CTLA-4 and B7/CD28 ligation. In collagen-induced arthritis in rats, CTLA4Ig has prevented disease induction via a blockade of costimulation by CD28 during T cell activation [101]. However, the prevention of T cell activation by CTLA-4Ig is not complete, as shown by the finding that co-administration of CTLA-4Ig with adoptively transferred TCRtg T cells into primary immunized mice resulted in reduced, but not completely abolished, expansion of antigen-specific T cells [102]. However, the prevention of CD28 signals could also block the activation of beneficial T cells, which has been suggested for transplantation [103]. Administration of CTLA-4Ig at the time of transplantation enhances transplant rejection, presumably by preventing the induction of regulatory T cells. In addition, the function of CTLA-4Ig is very probably more complex when administered during continuing responses, because such responses consist of several individual T cell responses at different stages running simultaneously (Fig. 2). In addition, the ligands for both receptors, CD28 and CTLA-4, are blocked by CTLA-4Ig, thus leaving different distinct differentiation processes and effector functions of newly recruited and activated T cells uncontrolled.
During chronic inflammation, the stimulation of antigen-experienced T cells is, at least partly, independent of CD28 signalling, putting CTLA-4/CD80 and CTLA-4/ CD86 into the spotlight of the CTLA-4Ig treatment. Furthermore, it has been shown that CD28- T cells, which upregulate CTLA-4, contribute to the immunopathology of RA or might even drive it [104]. Blocking CD28 and CTLA-4 signals could lead to either enhanced apoptosis by reduced CD28 and CTLA-4 signals or enhanced expansion and thus more cytokine production by reduced CTLA-4 signals. However, we feel that under some circumstances the T cell proliferation of activated CTLA-4-/-T cells in vitro is overemphasized, because no difference in proliferation could be detected in bone marrow chimeras generated from a mixture of wild-type and CTLA-4-/-cells [66,67]. Thus, the third function of CTLA-4 that we propose here, namely the control of survival and apoptosis, might be more relevant [65,66]. This mechanism could contribute to the success of the treatment of RA patients with CTLA-4Ig (see below).
However, whatever mechanisms are acting during the CTLA-4Ig treatment of RA, a recent double-blind study on 339 RA patients receiving treatment with 10 mg/kg CTLA4Ig concomitant with methotrexane showed significant improvement over the placebo group from month 2 to month 6 [105]. Only a very slight increase in infections was observed in comparison with methotrexate alone, but health-related quality of life and both clinical and laboratory markers of disease activity were significantly improved. The significance of the finding that two patients developed seroconversion for CTLA-4-specific antibodies means that autoimmunity needs to be further investigated.
Conclusion
In several studies, the use of CTLA-4Ig to treat patients with RA and other inflammatory diseases was shown to be successful, pinpointing T cells and their costimulation as an important target for therapy. However, the precise mechanism is not yet fully understood, because costimulation is very complex. The precise function of distinct costimulatory molecules depends on the differentiation and activation status of the T cells as well as the immunological microenvironment. Thus, a better understanding of costimulation is of great importance and might lead to even more specific strategies for novel immunotherapy of RA and other autoimmune diseases.
Competing interests
None declared.
Abbreviations
AICD = activation-induced cell death; APC = antigen-presenting cell; CTLA-4 = cytotoxic T-lymphocyte antigen-4; IL = interleukin; NFAT = nuclear factor of activated T cells; PI-3K = phosphoinositide 3-kinase; PP2A = protein phosphatase 2A; TCR = T cell antigen receptor.
Acknowledgments
Acknowledgement
We thank Susanne Schneider for support.
References
- Brunner MC. Costimulatory molecules and modulation. Immunologist. 1999;7:9–12. [Google Scholar]
- Chambers CA, Krummel MF, Boitel B, Hurwitz A, Sullivan TJ, Fournier S, Cassell D, Brunner M, Allison JP. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol Rev. 1996;153:27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Su GH, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, Miller J, Bluestone JA. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci USA. 1993;90:11054–11058. doi: 10.1073/pnas.90.23.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- McAdam AJ, Schweitzer AN, Sharpe AH. The role of B7 costimulation in activation and differentiation of CD4+ and CD8+T cells. Immunol Rev. 1998;165:231–247. doi: 10.1111/j.1600-065x.1998.tb01242.x. [DOI] [PubMed] [Google Scholar]
- Ikemizu S, Gilbert RJ, Fennelly JA, Collins AV, Harlos K, Jones EY, Stuart DI, Davis SJ. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12:51–60. doi: 10.1016/s1074-7613(00)80158-2. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for costimulation by the human CTLA-4/B7-2 complex. Nature. 2001;410:604–608. doi: 10.1038/35069112. [DOI] [PubMed] [Google Scholar]
- Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS, Mosyak L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 2001;410:608–611. doi: 10.1038/35069118. [DOI] [PubMed] [Google Scholar]
- Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, Davis SJ. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Lee KP, Harris ES, Petryniak B, Craighead N, Reynolds PJ, Lombard DB, Freeman GJ, Nadler LM, Gray GS. Characterization of CTLA-4 structure and expression on human T cells. J Immunol. 1993;151:3489–3499. [PubMed] [Google Scholar]
- Maszyna F, Hoff H, Kunkel D, Radbruch A, Brunner-Weinzierl MC. Diversity of clonal T cell proliferation is mediated by differential expression of CD152 (CTLA-4) on the cell surface of activated individual T lymphocytes. J Immunol. 2003;171:3459–3466. doi: 10.4049/jimmunol.171.7.3459. [DOI] [PubMed] [Google Scholar]
- Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol. 1997;159:144–151. [PubMed] [Google Scholar]
- Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, Saito T. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6:583–589. doi: 10.1016/s1074-7613(00)80346-5. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35. doi: 10.1016/s1074-7613(01)00259-x. [DOI] [PubMed] [Google Scholar]
- Doyle AM, Mullen AC, Villarino AV, Hutchins AS, High FA, Lee HW, Thompson CB, Reiner SL. Induction of cytotoxic T lymphocyte antigen 4 (CTLA-4) restricts clonal expansion of helper T cells. J Exp Med. 2001;194:893–902. doi: 10.1084/jem.194.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol. 1998;28:3137–3143. doi: 10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.3.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Alegre ML, Shiels H, Thompson CB, Gajewski TF. Expression and function of CTLA-4 in Th1 and Th2 cells. J Immunol. 1998;161:3347–3356. [PubMed] [Google Scholar]
- Lühders F, Chambers C, Allison JP, Benoist C, Matthis D. Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. PNAS. 2000;97:12204–12209. doi: 10.1073/pnas.200348397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Ragheb JA, Kitagawa-Sakakida S, Schwartz RH. Molecular regulation of interleukin-2 expression by CD28 costimulation and anergy. Immunol Rev. 1998;165:287–300. doi: 10.1111/j.1600-065x.1998.tb01246.x. [DOI] [PubMed] [Google Scholar]
- Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–5820. [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington PJ, Baroja ML, Chau TA, Siu E, Ling V, Carreno BM, Madrenas J. Surface cytotoxic T lymphocyte-associated antigen 4 partitions within lipid rafts and relocates to the immunological synapse under conditions of inhibition of T cell activation. J Exp Med. 2002;195:1337–1347. doi: 10.1084/jem.20011868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner-Weinzierl MC, Maszyna F, Hoff H. Mechanisms of T-cell activation. In: Mackiewicz A, Kurpisz M, Zeromski J, editor. In Proceedings of the 14th European Immunology Meeting – EFIS. Bologna: Monduzzi editore; 2001. pp. 676–681. [Google Scholar]
- Brunner MC, Mitchison NA. Regulation by non-major histocompatibility complex genes of the allo-4-hydroxy-phenylpyruvate dioxygenase (F liver protein) response. Immunology. 1996;88:452–455. doi: 10.1046/j.1365-2567.1996.d01-670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth N, Hu-Li J, Paul WE. Conventional, naive CD4+ T cells provide an initial source of IL-4 during Th2 differentiation. J Immunol. 2000;165:3620–3625. doi: 10.4049/jimmunol.165.7.3620. [DOI] [PubMed] [Google Scholar]
- Schuler T, Kammertoens T, Preiss S, Debs P, Noben-Trauth N, Blankenstein T. Generation of tumor-associated cytotoxic T lymphocytes requires interleukin 4 from CD8+ T cells. J Exp Med. 2001;194:1767–1775. doi: 10.1084/jem.194.12.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, Larsen S, Sette A, Mitchison A. Altered Th1/Th2 balance associated with the immunosuppressive/protective effect of the H-2Ab allele on the response to allo-4-hydroxyphenylpyruvate dioxygenase. Eur J Immunol. 1995;25:3285–3289. doi: 10.1002/eji.1830251213. [DOI] [PubMed] [Google Scholar]
- Mitchison NA, Brunner MC. Association of H2Ab with resistance to collagen-induced arthritis in H2-recombinant mouse strains: an allele associated with reduction of several apparently unrelated responses. Immunogenetics. 1995;41:239–245. doi: 10.1007/BF00172065. [DOI] [PubMed] [Google Scholar]
- Pages F, Ragueneau M, Rottapel R, Truneh A, Nunes J, Imbert J, Olive D. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369:327–329. doi: 10.1038/369327a0. [DOI] [PubMed] [Google Scholar]
- Prasad KV, Cai YC, Raab M, Duckworth B, Cantley L, Shoelson SE, Rudd CE. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc Natl Acad Sci USA. 1994;91:2834–2838. doi: 10.1073/pnas.91.7.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H, Prasad KV, Shoelson SE, Rudd CE. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J Exp Med. 1995;181:351–355. doi: 10.1084/jem.181.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ, Thompson CB. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. 2000;13:313–322. doi: 10.1016/s1074-7613(00)00031-5. [DOI] [PubMed] [Google Scholar]
- Raab M, Cai YC, Bunnell SC, Heyeck SD, Berg LJ, Rudd CE. p56Lck and p59Fyn regulate CD28 binding to phosphatidylinositol 3-kinase, growth factor receptor-bound protein GRB-2, and T cell-specific protein-tyrosine kinase ITK: implications for T-cell costimulation. Proc Natl Acad Sci USA. 1995;92:8891–8895. doi: 10.1073/pnas.92.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August A, Gibson S, Kawakami Y, Kawakami T, Mills GB, Dupont B. CD28 is associated with and induces the immediate tyrosine phosphorylation and activation of the Tec family kinase ITK/EMT in the human Jurkat leukemic T-cell line. Proc Natl Acad Sci USA. 1994;91:9347–9351. doi: 10.1073/pnas.91.20.9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WC, Olive D. Tec kinase is involved in transcriptional regulation of IL-2 and IL-4 in the CD28 pathway. Eur J Immunol. 1999;29:1842–1849. doi: 10.1002/(SICI)1521-4141(199906)29:06<1842::AID-IMMU1842>3.3.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ward SG. CD28: a signalling perspective. Biochem J. 1996;318:361–377. doi: 10.1042/bj3180361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S, Acuto O. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity. 2001;15:935–945. doi: 10.1016/s1074-7613(01)00244-8. [DOI] [PubMed] [Google Scholar]
- Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci USA. 2002;99:11790–11795. doi: 10.1073/pnas.162359999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake S, Nakaseko C, Umemori H, Yamamoto T, Saito T. Src family tyrosine kinases associate with and phosphorylate CTLA-4 (CD152) Biochem Biophys Res Commun. 1998;249:444–448. doi: 10.1006/bbrc.1998.9191. [DOI] [PubMed] [Google Scholar]
- Schneider H, Schwartzberg PL, Rudd CE. Resting lymphocyte kinase (Rlk/Txk) phosphorylates the YVKM motif and regulates PI 3-kinase binding to T-cell antigen CTLA-4. Biochem Biophys Res Commun. 1998;252:14–19. doi: 10.1006/bbrc.1998.9559. [DOI] [PubMed] [Google Scholar]
- Chikuma S, Murakami M, Tanaka K, Uede T. Janus kinase 2 is associated with a box 1-like motif and phosphorylates a critical tyrosine residue in the cytoplasmic region of cytotoxic T lymphocyte associated molecule-4. J Cell Biochem. 2000;78:241–250. doi: 10.1002/(SICI)1097-4644(20000801)78:2<241::AID-JCB7>3.3.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272:1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA. Molecular basis of T cell inactivation by CTLA-4. Science. 1998;282:2263–2266. doi: 10.1126/science.282.5397.2263. [DOI] [PubMed] [Google Scholar]
- Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun. 2000;269:279–283. doi: 10.1006/bbrc.2000.2234. [DOI] [PubMed] [Google Scholar]
- Calvo CR, Amsen D, Kruisbeek AM. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J Exp Med. 1997;186:1645–1653. doi: 10.1084/jem.186.10.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, Collins M, Carreno BM, Madrenas J, Kuchroo VK. Inhibition of CTLA-4 function by the regulatory subunit of serine/ threonine phosphatase 2A. J Immunol. 2002;168:5070–5078. doi: 10.4049/jimmunol.168.10.5070. [DOI] [PubMed] [Google Scholar]
- Martin M, Schneider H, Azouz A, Rudd CE. Cytotoxic T lymphocyte antigen 4 and CD28 modulate cell surface raft expression in their regulation of T cell function. J Exp Med. 2001;194:1675–1681. doi: 10.1084/jem.194.11.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PJ, Negishi I, Nakayama K, Fields LE, Loh DY. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J Immunol. 1995;154:5757–5768. [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Finn PW, He H, Wang Y, Wang Z, Guan G, Listman J, Perkins DL. Synergistic induction of CTLA-4 expression by costimulation with TCR plus CD28 signals mediated by increased transcription and messenger ribonucleic acid stability. J Immunol. 1997;158:4074–4081. [PubMed] [Google Scholar]
- Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- Lynch DH, Watson ML, Alderson MR, Baum PR, Miller RE, Tough T, Gibson M, Davis-Smith T, Smith CA, Hunter K. The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity. 1994;1:131–136. doi: 10.1016/1074-7613(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Flavell RA. Apoptosis genes and autoimmunity. Curr Opin Immunol. 2000;12:719–724. doi: 10.1016/S0952-7915(00)00168-0. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S, Muller WW, Li-Weber M, Krammer PH. Up-regulation of c-FLIPshort and reduction of activation-induced cell death in CD28-costimulated human T cells. Eur J Immunol. 2000;30:2765–2774. doi: 10.1002/1521-4141(200010)30:10<2765::AID-IMMU2765>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- da Rocha DS, Rudd CE. CTLA-4 blockade of antigen-induced cell death. Blood. 2001;97:1134–1137. doi: 10.1182/blood.V97.4.1134. [DOI] [PubMed] [Google Scholar]
- Scheipers P, Reiser H. Fas-independent death of activated CD4+ T lymphocytes induced by CTLA-4 crosslinking. Proc Natl Acad Sci USA. 1998;95:10083–10088. doi: 10.1073/pnas.95.17.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Gärtner D, Soezeri O, Radbruch A, Schulze-Osthoff K, Brunner-Weinzierl MC. CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J Exp Med. 2004;199:1–13. doi: 10.1084/jem.20031058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Gallimore A, Jones E, Ecabert B, Acha-Orbea H, Kopf M. Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur J Immunol. 2001;31:450–458. doi: 10.1002/1521-4141(200102)31:2<450::AID-IMMU450>3.3.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163:1128–1131. [PubMed] [Google Scholar]
- Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- Chen W, Jin W, Wahl SM. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J Exp Med. 1998;188:1849–1857. doi: 10.1084/jem.188.10.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan TJ, Letterio JJ, van Elsas A, Mamura M, van Amelsfort J, Sharpe S, Metzler B, Chambers CA, Allison JP. Lack of a role for transforming growth factor-β in cytotoxic T lymphocyte antigen-4-mediated inhibition of T cell activation. Proc Natl Acad Sci USA. 2001;98:2587–2592. doi: 10.1073/pnas.051632398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+ CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Kouki T, Sawai Y, Gardine CA, Fisfalen ME, Alegre ML, DeGroot LJ. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves' disease. J Immunol. 2000;165:6606–6611. doi: 10.4049/jimmunol.165.11.6606. [DOI] [PubMed] [Google Scholar]
- Maurer M, Loserth S, Kolb-Maurer A, Ponath A, Wiese S, Kruse N, Rieckmann P. A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics. 2002;54:1–8. doi: 10.1007/s00251-002-0429-9. [DOI] [PubMed] [Google Scholar]
- Ligers A, Teleshova N, Masterman T, Huang WX, Hillert J. CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun. 2001;2:145–152. doi: 10.1038/sj.gene.6363752. [DOI] [PubMed] [Google Scholar]
- Cosentino A, Gambelunghe G, Tortoioli C, Falorni A. CTLA-4 gene polymorphism contributes to the genetic risk for latent autoimmune diabetes in adults. Ann N Y Acad Sci. 2002;958:337–340. doi: 10.1111/j.1749-6632.2002.tb03000.x. [DOI] [PubMed] [Google Scholar]
- Guja C, Marshall S, Welsh K, Merriman M, Smith A, Todd JA, Ionescu-Tirgoviste C. The study of CTLA-4 and vitamin D receptor polymorphisms in the Romanian type 1 diabetes population. J Cell Mol Med. 2002;6:75–81. doi: 10.1111/j.1582-4934.2002.tb00312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Tang X, Chang W, Gao L, Li M, Yan W. CTLA-4 gene A/G polymorphism associated with diabetes mellitus in Han Chinese. Chin Med J (Engl) 2002;115:1248–1250. [PubMed] [Google Scholar]
- Wood JP, Pani MA, Bieda K, Meyer G, Usadel KH, Badenhoop K. A recently described polymorphism in the CD28 gene on chromosome 2q33 is not associated with susceptibility to type 1 diabetes. Eur J Immunogenet. 2002;29:347–349. doi: 10.1046/j.1365-2370.2002.00328.x. [DOI] [PubMed] [Google Scholar]
- Fajardy I, Vambergue A, Stuckens C, Weill J, Danze PM, Fontaine P. CTLA-4 49 A/G dimorphism and type 1 diabetes susceptibility: a French case-control study and segregation analysis. Evidence of a maternal effect. Eur J Immunogenet. 2002;29:251–257. doi: 10.1046/j.1365-2370.2002.00309.x. [DOI] [PubMed] [Google Scholar]
- Cinek O, Drevinek P, Sumnik Z, Bendlova B, Kolouskova S, Snajderova M, Vavrinec J. The CTLA4 +49 A/G dimorphism is not associated with type 1 diabetes in Czech children. Eur J Immunogenet. 2002;29:219–222. doi: 10.1046/j.1365-2370.2002.00292.x. [DOI] [PubMed] [Google Scholar]
- Dyment DA, Steckley JL, Willer CJ, Armstrong H, Sadovnick AD, Risch N, Ebers GC. No evidence to support CTLA-4 as a susceptibility gene in MS families: the Canadian Collaborative Study. J Neuroimmunol. 2002;123:193–198. doi: 10.1016/S0165-5728(01)00493-3. [DOI] [PubMed] [Google Scholar]
- Bocko D, Bilinska M, Dobosz T, Zoledziewska M, Suwalska K, Tutak A, Gruszka E, Frydecka I. Lack of association between an exon 1 CTLA-4 gene polymorphism A49G and multiple sclerosis in a Polish population of the Lower Silesia region. Arch Immunol Ther Exp (Warsz) 2003;51:201–205. [PubMed] [Google Scholar]
- Luomala M, Lehtimaki T, Huhtala H, Ukkonen M, Koivula T, Hurme M, Elovaara I. Promoter polymorphism of IL-10 and severity of multiple sclerosis. Acta Neurol Scand. 2003;108:396–400. doi: 10.1034/j.1600-0404.2003.00165.x. [DOI] [PubMed] [Google Scholar]
- van Veen T, Crusius JB, van Winsen L, Xia B, Barkhof F, Salvador PA, Polman CH, Uitdehaag BM. CTLA-4 and CD28 gene polymorphisms in susceptibility, clinical course and progression of multiple sclerosis. J Neuroimmunol. 2003;140:188–193. doi: 10.1016/S0165-5728(03)00184-X. [DOI] [PubMed] [Google Scholar]
- Ligers A, Xu C, Saarinen S, Hillert J, Olerup O. The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol. 1999;97:182–190. doi: 10.1016/S0165-5728(99)00072-7. [DOI] [PubMed] [Google Scholar]
- Maurer M, Ponath A, Kruse N, Rieckmann P. CTLA4 exon 1 dimorphism is associated with primary progressive multiple sclerosis. J Neuroimmunol. 2002;131:213–215. doi: 10.1016/S0165-5728(02)00275-8. [DOI] [PubMed] [Google Scholar]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C, Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- Barton A, Myerscough A, John S, Gonzalez-Gay M, Ollier W, Worthington J. A single nucleotide polymorphism in exon 1 of cytotoxic T-lymphocyte-associated-4 (CTLA-4) is not associated with rheumatoid arthritis. Rheumatology (Oxford) 2000;39:63–66. doi: 10.1093/rheumatology/39.1.63. [DOI] [PubMed] [Google Scholar]
- Milicic A, Brown MA, Wordsworth BP. Polymorphism in codon 17 of the CTLA-4 gene (+49 A/G) is not associated with susceptibility to rheumatoid arthritis in British Caucasians. Tissue Antigens. 2001;58:50–54. doi: 10.1034/j.1399-0039.2001.580110.x. [DOI] [PubMed] [Google Scholar]
- Lee YH, Choi SJ, Ji JD, Song GG. No association of polymorphisms of the CTLA-4 exon 1(+49) and promoter(-318) genes with rheumatoid arthritis in the Korean population. Scand J Rheumatol. 2002;31:266–270. doi: 10.1080/030097402760375142. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Escribano MF, Rodriguez R, Valenzuela A, Garcia A, Garcia-Lozano JR, Nunez-Roldan A. CTLA4 polymorphisms in Spanish patients with rheumatoid arthritis. Tissue Antigens. 1999;53:296–300. doi: 10.1034/j.1399-0039.1999.530311.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez MR, Nunez-Roldan A, Aguilar F, Valenzuela A, Garcia A, Gonzalez-Escribano MF. Association of the CTLA4 3' untranslated region polymorphism with the susceptibility to rheumatoid arthritis. Hum Immunol. 2002;63:76–81. doi: 10.1016/S0198-8859(01)00358-5. [DOI] [PubMed] [Google Scholar]
- Lee CS, Lee YJ, Liu HF, Su CH, Chang SC, Wang BR, Chen TL, Liu TL. Association of CTLA4 gene A-G polymorphism with rheumatoid arthritis in Chinese. Clin Rheumatol. 2003;22:221–224. doi: 10.1007/s10067-003-0720-7. [DOI] [PubMed] [Google Scholar]
- Seidl C, Donner H, Fischer B, Usadel KH, Seifried E, Kaltwasser JP, Badenhoop K. CTLA4 codon 17 dimorphism in patients with rheumatoid arthritis. Tissue Antigens. 1998;51:62–66. doi: 10.1111/j.1399-0039.1998.tb02947.x. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Tsuchiya N, Shiota M, Komata T, Matsuta K, Zama K, Oka T, Juji T, Yamane A, Tokunaga K. Lack of a strong association of CTLA-4 exon 1 polymorphism with the susceptibility to rheumatoid arthritis and systemic lupus erythematosus in Japanese: an association study using a novel variation screening method. Tissue Antigens. 1999;54:578–584. doi: 10.1034/j.1399-0039.1999.540607.x. [DOI] [PubMed] [Google Scholar]
- Yanagawa T, Gomi K, Nakao EI, Inada S. CTLA-4 gene polymorphism in Japanese patients with rheumatoid arthritis. J Rheumatol. 2000;27:2740–2742. [PubMed] [Google Scholar]
- Alizadeh M, Babron MC, Birebent B, Matsuda F, Quelvennec E, Liblau R, Cournu-Rebeix I, Momigliano-Richiardi P, Sequeiros J, Yaouanq J, Genin E, Vasilescu A, Bougerie H, Trojano M, Martins SB, Maciel P, Clerget-Darpoux F, Clanet M, Edan G, Fontaine B, Semana G. Genetic interaction of CTLA-4 with HLA-DR15 in multiple sclerosis patients. Ann Neurol. 2003;54:119–122. doi: 10.1002/ana.10617. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Ijima K, Murakami M, Okamoto H, Inobe M, Chikuma S, Saito I, Kanegae Y, Kawaguchi Y, Kitabatake A, Uede T. Successful gene therapy via intraarticular injection of adenovirus vector containing CTLA4IgG in a murine model of type II collagen-induced arthritis. Hum Gene Ther. 2001;12:1063–1077. doi: 10.1089/104303401750214285. [DOI] [PubMed] [Google Scholar]
- Judge TA, Tang A, Spain LM, Deans-Gratiot J, Sayegh MH, Turka LA. The in vivo mechanism of action of CTLA4Ig. J Immunol. 1996;156:2294–2299. [PMC free article] [PubMed] [Google Scholar]
- Sayegh MH, Turka LA. The role of T-cell costimulatory activation pathways in transplant rejection. N Engl J Med. 1998;338:1813–1821. doi: 10.1056/NEJM199806183382506. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ. T-cell responses in rheumatoid arthritis: systemic abnormalities – local disease. Curr Opin Rheumatol. 1999;11:210–217. doi: 10.1097/00002281-199905000-00010. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–1915. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]